
The Role of Mathematical Models in Immuno-Oncology: Challenges and Future Perspectives




Abstract
1. Introduction
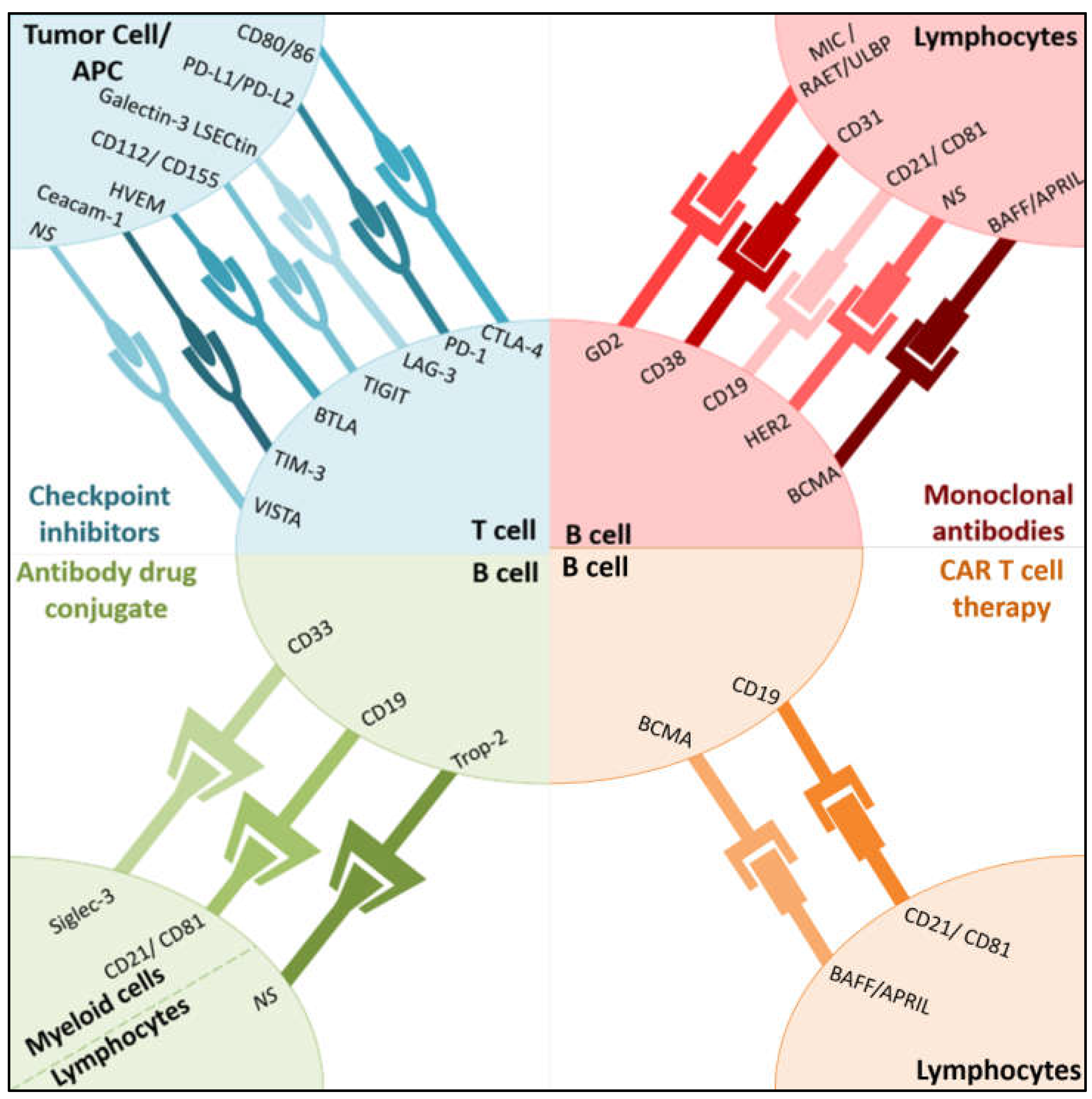
2. Current and Emerging Targets in Immuno-Oncology
2.1. Current Immune Checkpoint Inhibitors
2.2. Novel Immune Checkpoint Inhibitors
2.3. Adoptive Cellular Immunotherapy
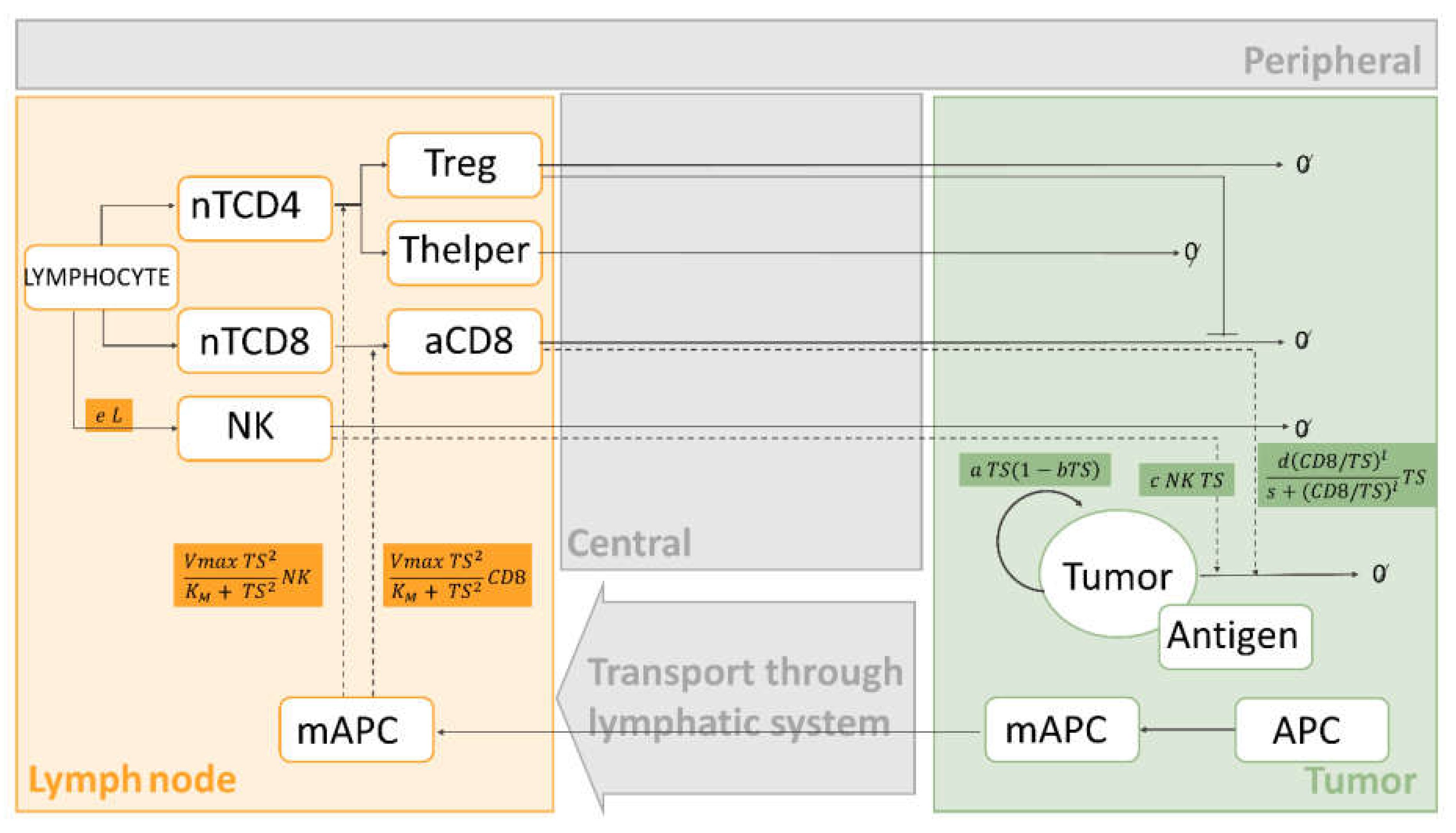
3. Mathematical Approaches Integrating Cancer Immunity Cycle with Immuno-Oncology Therapies
3.1. Top-Down Modelling and Simulation Approaches


{kind=link}
{kind=link}
{kind=link}
| Parameter | Value | Units | Estimation | Indication | Treatment | Ref. |
|---|---|---|---|---|---|---|
| Tumor | ||||||
| Tumor growth | ||||||
| Lineal: a | Colon cancer | Colon cancer | IL-2 | [84] | ||
| Melanoma patients [80] | Melanoma | Anti-PD1 | [80] | |||
| Melanoma patients [81] | Melanoma | Anti-PD1 | [81] | |||
| Renal carcinoma [86,87] | Renal carcinoma | Sunitinib | [88] | |||
| AD: AI: | Prostatic cancer [89] | Prostate | Intermittent ADT + DC vaccine | [90] | ||
| Tumor cell kill by CD8 | ||||||
None None | 3 × 105 B16-BL6 cells [91]/Human [92] | Metastatic melanoma | Chemotherapy + TIL | [93] | ||
None None | 3 × 105 B16-BL6 cells [91]/Human [92] | Metastatic melanoma | Chemotherapy + TIL + IL2 + cancer vaccine | [94,95] | ||
| High grade gliomas patients [96] | Bladder | IL2 + BCG | [97,98] | |||
| Equation in [99] | Assumed [99] | NSCLC | Anti-PD1 | [99] | ||
| Tumor cell kill by NK cells | ||||||
| 3 × 105 B16-BL6 cells [91]/Human [92] | Metastatic melanoma | Chemotherapy + TIL | [93] | |||
| 3 × 105 B16-BL6 cells [91]/Human [92] | Metastatic melanoma | Chemotherapy + TIL + IL2 | [100] | |||
| 3 × 105 B16-BL6 cells [91]/Human [92] | Metastatic melanoma | Chemotherapy + TIL + IL2 + IFNα | [95] | |||
| CD8 cells | ||||||
| Number of CD8 per microliter of blood | ||||||
| 1000 | - | CD4+ count of 640-1175/µL humans | Melanoma | Pembrolizumab | [80] | |
| CD8 recruitment by tumor | ||||||
| BCL 1 lymphoma of chimeric mice [91,92,93,94,95,96,97,98,99,100,101]/Human [92] | Metastatic melanoma | Chemotherapy + TIL | [93] | |||
| BCL 1 lymphoma of chimeric mice [91,101]/Human [92] | Metastatic melanoma | Chemotherapy + TIL + IL2 + IFNα | [95] | |||
| BCL 1 lymphoma of chimeric mice [91,92,93,94,95,96,97,98,99,100,101]/Human [92] | Metastatic melanoma | Chemotherapy + TIL + IL2 | [100] | |||
| In vitro/Estimated bladder cancer patients [102] In vitro/Estimated bladder cancer patients [103] | Bladder | IL2 + BCG | [97] | |||
| CD8 activation by APCs | ||||||
| Preclinical experiments [104] Prostate cancer [105] | Prostate | Intermittent ADT + DC vaccine | [90] | |||
| NK cells | ||||||
| Production rate NK | ||||||
| Preclinical experiments renal carcinoma [86,87] | Renal carcinoma | Sunitinib | [88] | |||
| NK recruitment | ||||||
| BCL 1 lymphoma of chimeric mice [91,92,93,94,95,96,97,98,99,100,101]/Human [92] | Metastatic melanoma | Chemotherapy + TIL + IFNα | [93,94,95] | |||
3.2. Middle-Out Modelling and Simulation Approaches
3.3. Bottom-Up Modelling and Simulation Approaches
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- How Immunotherapy Is Used to Treat Cancer. Available online: https://www.cancer.org/content/dam/CRC/PDF/Public/6678.00.pdf (accessed on 19 April 2021).
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol. Cancer 2019, 18, 1–17. [Google Scholar] [CrossRef]
- European Medicines Agency. Available online: https://www.ema.eu,ropa.eu/en (accessed on 8 June 2021).
- Drugs@FDA: FDA-Approved Drugs. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/ (accessed on 8 June 2021).
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 165. [Google Scholar] [CrossRef] [PubMed]
- Netterberg, I.; Li, C.C.; Molinero, L.; Budha, N.; Sukumaran, S.; Stroh, M.; Jonsson, E.N.; Friberg, L.E. A PK/PD Analysis of Circulating Biomarkers and Their Relationship to Tumor Response in Atezolizumab-Treated non-small Cell Lung Cancer Patients. Clin. Pharmacol. Ther. 2019, 105, 486–495. [Google Scholar] [CrossRef]
- Bradshaw, E.L.; Spilker, M.E.; Zang, R.; Bansal, L.; He, H.; Jones, R.D.O.; Le, K.; Penney, M.; Schuck, E.; Topp, B.; et al. Applications of Quantitative Systems Pharmacology in Model-Informed Drug Discovery: Perspective on Impact and Opportunities. CPT Pharmacometrics Syst. Pharmacol. 2019, 8, 777–791. [Google Scholar] [CrossRef]
- Bender, B.C.; Schindler, E.; Friberg, L.E. Population pharmacokinetic-pharmacodynamic modelling in oncology: A tool for predicting clinical response. Br. J. Clin. Pharmacol. 2015, 79, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Valentinuzzi, D.; Jeraj, R. Computational modelling of modern cancer immunotherapy. Phys. Med. Biol. 2020, 65, 24TR01. [Google Scholar] [CrossRef]
- Bekisz, S.; Geris, L. Cancer modeling: From mechanistic to data-driven approaches, and from fundamental insights to clinical applications. J. Comput. Sci. 2020, 46, 101198. [Google Scholar] [CrossRef]
- Peskov, K.; Azarov, I.; Chu, L.; Voronova, V.; Kosinsky, Y.; Helmlinger, G. Quantitative mechanistic modeling in support of pharmacological therapeutics development in immuno-oncology. Front. Immunol. 2019, 10, 924. [Google Scholar] [CrossRef]
- Sové, R.J.; Jafarnejad, M.; Zhao, C.; Wang, H.; Ma, H.; Popel, A.S. QSP-IO: A Quantitative Systems Pharmacology Toolbox for Mechanistic Multiscale Modeling for Immuno-Oncology Applications. CPT Pharmacometrics Syst. Pharmacol. 2020, 9, 484–497. [Google Scholar] [CrossRef]
- Chelliah, V.; Lazarou, G.; Bhatnagar, S.; Gibbs, J.P.; Nijsen, M.; Ray, A.; Stoll, B.; Thompson, R.A.; Gulati, A.; Soukharev, S.; et al. Quantitative Systems Pharmacology Approaches for Immuno-Oncology: Adding Virtual Patients to the Development Paradigm. Clin. Pharmacol. Ther. 2021, 109, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Della Gravara, L.; Battiloro, C.; Cantile, R.; Letizia, A.; Vitiello, F.; Montesarchio, V.; Rocco, D. Chemotherapy and/or immune checkpoint inhibitors in NSCLC first-line setting: What is the best approach? Lung Cancer Manage. 2020, 9, LMT22. [Google Scholar] [CrossRef]
- Quinn, C.; Garrison, L.P.; Pownell, A.K.; Atkins, M.B.; De Pouvourville, G.; Harrington, K.; Ascierto, P.A.; McEwan, P.; Wagner, S.; Borrill, J.; et al. Current challenges for assessing the long-term clinical benefit of cancer immunotherapy: A multi-stakeholder perspective. J. Immunother. Cancer 2020, 8, e000648. [Google Scholar] [CrossRef] [PubMed]
- Gevaert, T.; Van Eycke, Y.R.; Broeck, T.V.; Van Poppel, H.; Salmon, I.; Rorive, S.; Muilwijk, T.; Claessens, F.; De Ridder, D.; Joniau, S.; et al. The potential of tumour microenvironment markers to stratify the risk of recurrence in prostate cancer patients. PLoS ONE 2020, 15, e0244663. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Na, K.J.; Choi, H. Differences in Tumor Immune Microenvironment in Metastatic Sites of Breast Cancer. Front. Oncol. 2021, 11, 722. [Google Scholar] [CrossRef] [PubMed]
- Cocco, C.; Morandi, F.; Airoldi, I. Immune Checkpoints in Pediatric Solid Tumors: Targetable Pathways for Advanced Therapeutic Purposes. Cells 2021, 10, 927. [Google Scholar] [CrossRef] [PubMed]
- Marshall, H.T.; Djamgoz, M.B.A. Immuno-oncology: Emerging targets and combination therapies. Front. Oncol. 2018, 8, 315. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.L.; Jin, L.P. Immune cell population in ovarian tumor microenvironment. J. Cancer 2017, 8, 2915–2923. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Heong, V.; Ngoi, N.; Peng Tan, D.S. Update on immune checkpoint inhibitors in gynecological cancers. J. Gynecol. Oncol. 2017, 28, e20. [Google Scholar] [CrossRef]
- Martinez-Bosch, N.; Vinaixa, J.; Navarro, P. Immune evasion in pancreatic cancer: From mechanisms to therapy. Cancers 2018, 10, 6. [Google Scholar] [CrossRef]
- Strasner, A.; Karin, M. Immune infiltration and prostate cancer. Front. Oncol. 2015, 5, 128. [Google Scholar] [CrossRef] [PubMed]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 pathways similarities, differences, and implications of their inhibition. Am. J. Clin. Oncol. Cancer Clin. Trials 2016, 39, 98–106. [Google Scholar]
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. Fc-dependent depletion of tumor-infiltrating regulatory t cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med. 2013, 210, 1695–1710. [Google Scholar] [CrossRef] [PubMed]
- Pol, J.; Kroemer, G. Anti-CTLA-4 immunotherapy: Uncoupling toxicity and efficacy. Cell Res. 2018, 28, 501–502. [Google Scholar] [CrossRef] [PubMed]
- Yusa, S.; Campbell, K.S. Src Homology Region 2-Containing Protein Tyrosine Phosphatase-2 (SHP-2) Can Play a Direct Role in the Inhibitory Function of Killer Cell Ig-Like Receptors in Human NK Cells. J. Immunol. 2003, 170, 4539–4547. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Zappasodi, R.; Merghoub, T.; Wolchok, J.D. Emerging Concepts for Immune Checkpoint Blockade-Based Combination Therapies. Cancer Cell 2018, 33, 581–598. [Google Scholar] [CrossRef]
- Du, W.; Yang, M.; Turner, A.; Xu, C.; Ferris, R.L.; Huang, J.; Kane, L.P.; Lu, B. Tim-3 as a target for cancer immunotherapy and mechanisms of action. Int. J. Mol. Sci. 2017, 18, 645. [Google Scholar] [CrossRef]
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 2010, 207, 2175–2186. [Google Scholar] [CrossRef]
- Sakuishi, K.; Apetoh, L.; Sullivan, J.M.; Blazar, B.R.; Kuchroo, V.K.; Anderson, A.C. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 2010, 207, 2187–2194. [Google Scholar] [CrossRef]
- Ngiow, S.F.; Von Scheidt, B.; Akiba, H.; Yagita, H.; Teng, M.W.L.; Smyth, M.J. Anti-TIM3 antibody promotes T cell IFN-γ-mediated antitumor immunity and suppresses established tumors. Cancer Res. 2011, 71, 3540–3551. [Google Scholar] [CrossRef]
- Kon, E.; Benhar, I. Immune checkpoint inhibitor combinations: Current efforts and important aspects for success. Drug Resist. Updat. 2019, 45, 13–29004. [Google Scholar] [CrossRef]
- Dougall, W.C.; Kurtulus, S.; Smyth, M.J.; Anderson, A.C. TIGIT and CD96: New checkpoint receptor targets for cancer immunotherapy. Immunol. Rev. 2017, 276, 112–120. [Google Scholar] [CrossRef]
- He, Y.; Rivard, C.J.; Rozeboom, L.; Yu, H.; Ellison, K.; Kowalewski, A.; Zhou, C.; Hirsch, F.R. Lymphocyte-activation gene-3, an important immune checkpoint in cancer. Cancer Sci. 2016, 107, 1193–1197. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.T.; Workman, C.J.; Flies, D.; Pan, X.; Marson, A.L.; Zhou, G.; Hipkiss, E.L.; Ravi, S.; Kowalski, J.; Levitsky, H.I.; et al. Role of LAG-3 in regulatory T cells. Immunity 2004, 21, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, S.D.; Shin, H.; Haining, W.N.; Zou, T.; Workman, C.J.; Polley, A.; Betts, M.R.; Freeman, G.J.; Vignali, D.A.A.; Wherry, E.J. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 2009, 10, 29–37. [Google Scholar] [CrossRef]
- Wang-Gillam, A.; Plambeck-Suess, S.; Goedegebuure, P.; Simon, P.O.; Mitchem, J.B.; Hornick, J.R.; Sorscher, S.; Picus, J.; Suresh, R.; Lockhart, A.C.; et al. A phase i study of IMP321 and gemcitabine as the front-line therapy in patients with advanced pancreatic adenocarcinoma. Invest. New Drugs 2013, 31, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Brignone, C.; Gutierrez, M.; Mefti, F.; Brain, E.; Jarcau, R.; Cvitkovic, F.; Bousetta, N.; Medioni, J.; Gligorov, J.; Grygar, C.; et al. First-line chemoimmunotherapy in metastatic breast carcinoma: Combination of paclitaxel and IMP321 (LAG-3Ig) enhances immune responses and antitumor activity. J. Transl. Med. 2010, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Legat, A.; Maby-El Hajjami, H.; Baumgaertner, P.; Cagnon, L.; Maillard, S.A.; Geldhof, C.; Iancu, E.M.; Lebon, L.; Guillaume, P.; Dojcinovic, D.; et al. Vaccination with LAG-3Ig (IMP321) and peptides induces specific CD4 and CD8 T-cell responses in metastatic melanoma patients-report of a phase I/IIa clinical trial. Clin. Cancer Res. 2016, 22, 1330–1340. [Google Scholar] [CrossRef] [PubMed]
- Lines, J.L.; Sempere, L.F.; Broughton, T.; Wang, L.; Noelle, R. VISTA Is a novel broad-spectrum negative checkpoint regulator for cancer immunotherapy. Cancer Immunol. Res. 2014, 2, 510–517. [Google Scholar] [CrossRef]
- Wang, L.; Rubinstein, R.; Lines, J.L.; Wasiuk, A.; Ahonen, C.; Guo, Y.; Lu, L.F.; Gondek, D.; Wang, Y.; Fava, R.A.; et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J. Exp. Med. 2011, 208, 577–592. [Google Scholar] [CrossRef]
- Mulati, K.; Hamanishi, J.; Matsumura, N.; Chamoto, K.; Mise, N.; Abiko, K.; Baba, T.; Yamaguchi, K.; Horikawa, N.; Murakami, R.; et al. VISTA expressed in tumour cells regulates T cell function. Br. J. Cancer 2019, 120, 115–127. [Google Scholar] [CrossRef]
- Wang, H.; Milberg, O.; Bartelink, I.H.; Vicini, P.; Wang, B.; Narwal, R.; Roskos, L.; Santa-Maria, C.A.; Popel, A.S. In silico simulation of a clinical trial with anti-CTLA-4 and anti-PD-L1 immunotherapies in metastatic breast cancer using a systems pharmacology model. R. Soc. Open Sci. 2019, 6, 190366. [Google Scholar] [CrossRef]
- Wang, L.; Jia, B.; Claxton, D.F.; Ehmann, W.C.; Rybka, W.B.; Mineishi, S.; Naik, S.; Khawaja, M.R.; Sivik, J.; Han, J.; et al. VISTA is highly expressed on MDSCs and mediates an inhibition of T cell response in patients with AML. Oncoimmunology 2018, 7, e1469594. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Ward, J.F.; Pettaway, C.A.; Shi, L.Z.; Subudhi, S.K.; Vence, L.M.; Zhao, H.; Chen, J.; Chen, H.; Efstathiou, E.; et al. VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat. Med. 2017, 23, 551–555. [Google Scholar] [CrossRef]
- Wu, L.; Deng, W.-W.; Huang, C.-F.; Bu, L.-L.; Yu, G.-T.; Mao, L.; Zhang, W.-F.; Liu, B.; Sun, Z.-J. Expression of VISTA correlated with immunosuppression and synergized with CD8 to predict survival in human oral squamous cell carcinoma. Cancer Immunol. Immunother. 2017, 66, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Vendel, A.C.; Calemine-Fenaux, J.; Izrael-Tomasevic, A.; Chauhan, V.; Arnott, D.; Eaton, D.L. B and T Lymphocyte Attenuator Regulates B Cell Receptor Signaling by Targeting Syk and BLNK. J. Immunol. 2009, 182, 1509–1517. [Google Scholar] [CrossRef]
- Derré, L.; Rivals, J.P.; Jandus, C.; Pastor, S.; Rimoldi, D.; Romero, P.; Michielin, O.; Olive, D.; Speiser, D.E. BTLA mediates inhibition of human tumor-specific CD8+ T cells that can be partially reversed by vaccination. J. Clin. Invest. 2010, 120, 157–167. [Google Scholar] [CrossRef]
- Han, P.; Goularte, O.D.; Rufner, K.; Wilkinson, B.; Kaye, J. An Inhibitory Ig Superfamily Protein Expressed by Lymphocytes and APCs Is Also an Early Marker of Thymocyte Positive Selection. J. Immunol. 2004, 172, 5931–5939. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, J.A.; Reidy, A.; Mirandola, L.; Trotter, K.; Suvorava, N.; Figueroa, A.; Konala, V.; Aulakh, A.; Littlefield, L.; Grizzi, F.; et al. Chimeric antigen receptor engineering: A right step in the evolution of adoptive cellular immunotherapy. Int. Rev. Immunol. 2015, 34, 154–187. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Coulie, P.G.; Van Den Eynde, B.J.; Van Der Bruggen, P.; Boon, T. Tumour antigens recognized by T lymphocytes: At the core of cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Shank, B.R.; Do, B.; Sevin, A.; Chen, S.E.; Neelapu, S.S.; Horowitz, S.B. Chimeric Antigen Receptor T Cells in Hematologic Malignancies. Pharmacotherapy 2017, 37, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Knochelmann, H.M.; Smith, A.S.; Dwyer, C.J.; Wyatt, M.M.; Mehrotra, S.; Paulos, C.M. CAR T Cells in Solid Tumors: Blueprints for Building Effective Therapies. Front. Immunol. 2018, 9, 1740. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Maude, S.L.; Teachey, D.T.; Rheingold, S.R.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Barker, C.S.; Callahan, C.; Frey, N.V.; Nazimuddin, F.; et al. Sustained remissions with CD19-specific chimeric antigen receptor (CAR)-modified T cells in children with relapsed/refractory ALL. J. Clin. Oncol. 2016, 34, 3011. [Google Scholar] [CrossRef]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 2014, 6, 224ra25. [Google Scholar] [CrossRef]
- Bonifant, C.L.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics 2016, 3, 16011. [Google Scholar] [CrossRef]
- Lamers, C.H.J.; Sleijfer, S.; Vulto, A.G.; Kruit, W.H.J.; Kliffen, M.; Debets, R.; Gratama, J.W.; Stoter, G.; Oosterwijk, E. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: First clinical experience. J. Clin. Oncol. 2006, 24, e20–e22. [Google Scholar] [CrossRef]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of t cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef] [PubMed]
- D’Aloia, M.M.; Zizzari, I.G.; Sacchetti, B.; Pierelli, L.; Alimandi, M. CAR-T cells: The long and winding road to solid tumors review-article. Cell Death Dis. 2018, 9, 282. [Google Scholar] [CrossRef]
- Xu, X.; Sun, Q.; Liang, X.; Chen, Z.; Zhang, X.; Zhou, X.; Li, M.; Tu, H.; Liu, Y.; Tu, S.; et al. Mechanisms of Relapse After CD19 CAR T-Cell Therapy for Acute Lymphoblastic Leukemia and Its Prevention and Treatment Strategies. Front. Immunol. 2019, 10, 2664. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yannelli, J.R.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Weber, J.S.; Parkinson, D.R.; Seipp, C.A.; Einhorn, J.H.; White, D.E. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. J. Natl. Cancer Inst. 1994, 86, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Wrzesinski, C.; Paulos, C.M.; Kaiser, A.; Muranski, P.; Palmer, D.C.; Gattinoni, L.; Yu, Z.; Rosenberg, S.A.; Restifo, N.P. Increased intensity lymphodepletion enhances tumor treatment efficacy of adoptively transferred tumor-specific T cells. J. Immunother. 2010, 33, 1–7. [Google Scholar] [CrossRef]
- Dudley, M.E.; Wunderlich, J.R.; Yang, J.C.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Sherry, R.M.; Marincola, F.M.; Leitman, S.F.; Seipp, C.A.; et al. A Phase I Study of Nonmyeloablative Chemotherapy and Adoptive Transfer of Autologous Tumor Antigen-Specific T Lymphocytes in Patients With Metastatic Melanoma. J. Immunother. 2002, 25, 243–251. [Google Scholar] [CrossRef]
- Kvistborg, P.; Shu, C.J.; Heemskerk, B.; Fankhauser, M.; Thrue, C.A.; Toebes, M.; Van Rooij, N.; Linnemann, C.; Van Buuren, M.M.; Urbanus, J.H.M.; et al. TIL therapy broadens the tumor-reactive CD8+ T cell compartment in melanoma patients. Oncoimmunology 2012, 1, 409–418851. [Google Scholar] [CrossRef]
- Coulie, P.G.; Brichard, V.; Van Pel, A.; Wolfel, T.; Schneider, J.; Traversari, C.; Mattei, S.; De Plaen, E.; Lurquin, C.; Szikora, J.P.; et al. A new gene coding for a differentiation antigen recognized by autologous cytolytic T lymphocytes on HLA-A2 melanomas. J. Exp. Med. 1994, 180, 35–42. [Google Scholar] [CrossRef]
- Kawakami, Y.; Eliyahu, S.; Delgado, C.H.; Robbins, P.F.; Sakaguchi, K.; Appella, E.; Yannelli, J.R.; Adema, G.J.; Miki, T.; Rosenberg, S.A. Identification of a human melanoma antigen recognized by tumor- infiltrating lymphocytes associated with in vivo tumor rejection. Proc. Natl. Acad. Sci. USA 1994, 91, 6458–6462. [Google Scholar] [CrossRef]
- AACR Publications. An Overview of the MAGE Gene Family with the Identification of All Human Members of the Family | Cancer Research. Available online: https://cancerres.aacrjournals.org/content/61/14/5544.long (accessed on 19 April 2021).
- US National Library of Medicine. Home—ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ (accessed on 19 April 2021).
- Rohaan, M.W.; Van Den Berg, J.H.; Kvistborg, P.; Haanen, J.B.A.G. Adoptive transfer of tumor-infiltrating lymphocytes in melanoma: A viable treatment option 11 Medical and Health Sciences 1107 Immunology 11 Medical and Health Sciences 1112 Oncology and Carcinogenesis. J. Immunother. Cancer 2018, 6, 102. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Clinical Trials Using Tumor Infiltrating Lymphocyte Therapy. Available online: https://www.cancer.gov/about-cancer/treatment/clinical-trials/intervention/tumor-infiltrating-lymphocyte-therapy (accessed on 14 April 2021).
- de Greef, R.; Elassaiss-Schaap, J.; Chatterjee, M.; Turner, D.; Ahamadi, M.; Forman, M.; Cutler, D.; de Alwis, D.; Kondic, A.; Stone, J. Pembrolizumab: Role of Modeling and Simulation in Bringing a Novel Immunotherapy to Patients With Melanoma. CPT Pharmacometrics Syst. Pharmacol. 2017, 6, 5–7. [Google Scholar] [CrossRef][Green Version]
- Elassaiss-Schaap, J.; Rossenu, S.; Lindauer, A.; Kang, S.; de Greef, R.; Sachs, J.; de Alwis, D. Using Model-Based “Learn and Confirm” to Reveal the Pharmacokinetics-Pharmacodynamics Relationship of Pembrolizumab in the KEYNOTE-001 Trial. CPT Pharmacometrics Syst. Pharmacol. 2017, 6, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Lindauer, A.; Valiathan, C.; Mehta, K.; Sriram, V.; De Greef, R.; Elassaiss-Schaap, J.; De Alwis, D.P. Translational Pharmacokinetic/Pharmacodynamic Modeling of Tumor Growth Inhibition Supports Dose-Range Selection of the Anti–PD-1 Antibody Pembrolizumab. CPT Pharmacometrics Syst. Pharmacol. 2017, 6, 11–20. [Google Scholar] [CrossRef]
- Chatterjee, M.; Elassaiss-Schaap, J.; Lindauer, A.; Turner, D.; Sostelly, A.; Freshwater, T.; Mayawala, K.; Ahamadi, M.; Stone, J.; De Greef, R.; et al. Population Pharmacokinetic/Pharmacodynamic Modeling of Tumor Size Dynamics in Pembrolizumab-Treated Advanced Melanoma. CPT Pharmacometrics Syst. Pharmacol. 2017, 6, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Ribba, B.; Boetsch, C.; Nayak, T.; Grimm, H.P.; Charo, J.; Evers, S.; Klein, C.; Tessier, J.; Charoin, J.E.; Phipps, A.; et al. Prediction of the optimal dosing regimen using a mathematical model of tumor uptake for immunocytokine-based cancer immunotherapy. Clin. Cancer Res. 2018, 24, 3325–3333. [Google Scholar] [CrossRef]
- Parra-Guillen, Z.P.; Berraondo, P.; Ribba, B.; Trocóniz, I.F. Modeling Tumor Response after Combined Administration of Different Immune-Stimulatory Agents s. J. Pharmacol. Exp. Ther. J Pharmacol Exp Ther 2013, 346, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Parra-Guillen, Z.P.; Berraondo, P.; Grenier, E.; Ribba, B.; Troconiz, I.F. Mathematical model approach to describe tumour response in mice after vaccine administration and its applicability to immune-stimulatory cytokine-based strategies. AAPS J. 2013, 15, 797–807. [Google Scholar] [CrossRef]
- Betts, A.; Clark, T.; Jasper, P.; Tolsma, J.; van der Graaf, P.H.; Graziani, E.I.; Rosfjord, E.; Sung, M.; Ma, D.; Barletta, F. Use of translational modeling and simulation for quantitative comparison of PF-06804103, a new generation HER2 ADC, with Trastuzumab-DM1. J. Pharmacokinet. Pharmacodyn. 2020, 47, 513–526. [Google Scholar] [CrossRef]
- Gao, P.; Ding, Q.; Wu, Z.; Jiang, H.; Fang, Z. Therapeutic potential of human mesenchymal stem cells producing IL-12 in a mouse xenograft model of renal cell carcinoma. Cancer Lett. 2010, 290, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Doehn, C.; Esser, N.; Pauels, H.G.; Kießig, S.T.; Stelljes, M.; Grossmann, A.; Jocham, D.; Drevs, J. Mode-of-Action, Efficacy, and Safety of a Homologous Multi-Epitope Vaccine in a Murine Model for Adjuvant Treatment of Renal Cell Carcinoma. Eur. Urol. 2009, 56, 123–133. [Google Scholar] [CrossRef]
- De Pillis, L.; Caldwell, T.; Sarapata, E.; Williams, H. Mathematical modeling of regulatory T cell effects on renal cell carcinoma treatment. Discret. Contin. Dyn. Syst. B 2013, 18, 915–943. [Google Scholar]
- Ideta, A.M.; Tanaka, G.; Takeuchi, T.; Aihara, K. A mathematical model of intermittent androgen suppression for prostate cancer. J. Nonlinear Sci. 2008, 18, 593–614. [Google Scholar] [CrossRef]
- Portz, T.; Kuang, Y. A Mathematical Model for the Immunotherapy of Advanced Prostate Cancer; World Scientific Pub Co Pte Ltd.: Singapore, 2013; pp. 70–85. [Google Scholar]
- Diefenbach, A.; Jensen, E.R.; Jamieson, A.M.; Raulet, D.H. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature 2001, 413, 165–171. [Google Scholar] [CrossRef]
- Dudley, M.E.; Wunderlich, J.R.; Robbins, P.F.; Yang, J.C.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Sherry, R.; Restifo, N.P.; Hubicki, A.M.; et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 2002, 298, 850–854. [Google Scholar] [CrossRef] [PubMed]
- De Pillis, L.G.; Radunskaya, A.E.; Wiseman, C.L. A validated mathematical model of cell-mediated immune response to tumor growth. Cancer Res. 2005, 65, 7950–7958. [Google Scholar] [CrossRef] [PubMed]
- De Pillis, L.G.; Gu, W.; Radunskaya, A.E. Mixed immunotherapy and chemotherapy of tumors: Modeling, applications and biological interpretations. J. Theor. Biol. 2006, 238, 841–862. [Google Scholar] [CrossRef]
- Mamat, M.; Kartono, A. Mathematical Model of Cancer Treatments Using Immunotherapy, Chemotherapy and Biochemotherapy. Appl. Math. Sci. 2013, 7, 247–261. [Google Scholar] [CrossRef]
- Kogan, Y.; Fory´s, U.; Fory´s, F.; Shukron, O.; Kronik, N. CELLULAR IMMUNOTHERAPY FOR HIGH GRADE GLIOMAS: MATHEMATICAL ANALYSIS DERIVING EFFICACIOUS INFUSION RATES BASED ON PATIENT REQUIREMENTS *. Soc. Ind. Appl. Math. 2010, 70, 1953–1976. [Google Scholar] [CrossRef]
- Bunimovich-Mendrazitsky, S.; Halachmi, S.; Kronik, N. Improving Bacillus Calmette-Guérin (BCG) immunotherapy for bladder cancer by adding interleukin 2 (IL-2): A mathematical model. Math. Med. Biol. 2016, 33, 159–188. [Google Scholar] [CrossRef]
- Bunimovich-Mendrazitsky, S.; Shochat, E.; Stone, L. Mathematical model of BCG immunotherapy in superficial bladder cancer. Bull. Math. Biol. 2007, 69, 1847–1870. [Google Scholar] [CrossRef]
- Jafarnejad, M.; Gong, C.; Gabrielson, E.; Bartelink, I.H.; Vicini, P.; Wang, B.; Narwal, R.; Roskos, L.; Popel, A.S. A Computational Model of Neoadjuvant PD-1 Inhibition in Non-Small Cell Lung Cancer. AAPS J. 2019, 21, 79. [Google Scholar] [CrossRef] [PubMed]
- de Pillis, L.; Fister, K.R.; Gu, W.; Collins, C.; Daub, M.; Gross, D.; Moore, J.; Preskill, B. Mathematical model creation for cancer chemo-immunotherapy. Comput. Math. Methods Med. 2009, 10, 165–184. [Google Scholar] [CrossRef]
- KUZNETSOV, V.; MAKALKIN, I.; TAYLOR, M.; PERELSON, A. Nonlinear dynamics of immunogenic tumors: Parameter estimation and global bifurcation analysis. Bull. Math. Biol. 1994, 56, 295–321. [Google Scholar] [CrossRef]
- Fikri, Y.; Pastoret, P.-P.; Nyabenda, J. Costimulatory Molecule Requirement for Bovine WC1 + γδ T Cells’ Proliferative Response to Bacterial Superantigens. Scand. J. Immunol. 2002, 55, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Kronin, V.; Fitzmaurice, C.J.; Caminschi, I.; Shortman, K.; Jackson, D.C.; Brown, L.E. Differential effect of CD8+ and CD8- dendritic cells in the stimulation of secondary CD4+ T cells. Int. Immunol. 2001, 13, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, D.; Panetta, J.C. Modeling immunotherapy of the tumor-immune interaction. J. Math. Biol. 1998, 37, 235–252. [Google Scholar] [CrossRef] [PubMed]
- Small, E.J.; Fratesi, P.; Reese, D.M.; Strang, G.; Laus, R.; Peshwa, M.V.; Valone, F.H. Immunotherapy of hormone-refractory prostate cancer with antigen-loaded dendritic cells. J. Clin. Oncol. 2000, 18, 3894–3903. [Google Scholar] [CrossRef] [PubMed]
- Chan Kwong, A.H.X.P.; Calvier, E.A.M.; Fabre, D.; Gattacceca, F.; Khier, S. Prior information for population pharmacokinetic and pharmacokinetic/pharmacodynamic analysis: Overview and guidance with a focus on the NONMEM PRIOR subroutine. J. Pharmacokinet. Pharmacodyn. 2020, 47, 431–446. [Google Scholar] [CrossRef] [PubMed]
- Isaeva, O.G.; Osipov, V.A. Different strategies for cancer treatment: Mathematical modelling. Comput. Math. Methods Med. 2009, 10, 253–272. [Google Scholar] [CrossRef]
- Perlstein, D.; Shlagman, O.; Kogan, Y.; Halevi-Tobias, K.; Yakobson, A.; Lazarev, I.; Agur, Z. Personal response to immune checkpoint inhibitors of patients with advanced melanoma explained by a computational model of cellular immunity, tumor growth, and drug. PLoS ONE 2019, 14, e0226869. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xu, J.X. A mathematical prognosis model for pancreatic cancer patients receiving immunotherapy. J. Theor. Biol. 2016, 406, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Mahasa, K.J.; Ouifki, R.; Eladdadi, A.; de Pillis, L. Mathematical model of tumor–immune surveillance. J. Theor. Biol. 2016, 404, 312–330. [Google Scholar] [CrossRef]
- Milberg, O.; Gong, C.; Jafarnejad, M.; Bartelink, I.H.; Wang, B.; Vicini, P.; Narwal, R.; Roskos, L.; Popel, A.S. A QSP Model for Predicting Clinical Responses to Monotherapy, Combination and Sequential Therapy Following CTLA-4, PD-1, and PD-L1 Checkpoint Blockade. Sci. Rep. 2019, 9, 1–17. [Google Scholar]
- Wang, H.; Sové, R.J.; Jafarnejad, M.; Rahmeh, S.; Jaffee, E.M.; Stearns, V.; Torres, E.T.R.; Connolly, R.M.; Popel, A.S. Conducting a Virtual Clinical Trial in HER2-Negative Breast Cancer Using a Quantitative Systems Pharmacology Model with an Epigenetic Modulator and Immune Checkpoint Inhibitors. Front. Bioeng. Biotechnol. 2020, 8, 141. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Wang, H.; Sové, R.J.; Wang, J.; Giragossian, C.; Popel, A.S. Combination therapy with T cell engager and PD-L1 blockade enhances the antitumor potency of T cells as predicted by a QSP model. J. Immunother. Cancer 2020, 8, e001141. [Google Scholar] [CrossRef] [PubMed]
- Hardiansyah, D.; Ng, C.M. Quantitative Systems Pharmacology Model of Chimeric Antigen Receptor T-Cell Therapy. Clin. Transl. Sci. 2019, 12, 343–349. [Google Scholar] [CrossRef]


| Therapy | Date | Active Principle | Commercial Name | Company | Indication | Agency | ||
|---|---|---|---|---|---|---|---|---|
| Checkpoint Inhibitor | 2020 | January | Pembrolizumab | Keytruda | MSD | Bacillus Calmette–Guerin (BCG)-unresponsive, high-risk, non-muscle invasive bladder cancer (NMIBC) with carcinoma in situ (CIS) with or without papillary tumors who are ineligible for or have elected not to undergo cystectomy | FDA | |
| March | Durvalumab | Imfinzi | AstraZeneca | First-line treatment of patients with extensive-stage small cell lung cancer (ES-SCLC) | FDA | |||
| Nivolumab + Ipilimumab | Opdivo/ Yervoy | Bristol-Myers Squibb | Hepatocellular carcinoma (HCC) who have been previously treated with sorafenib | FDA | ||||
| May | Nivolumab | Opdivo | Bristol-Myers Squibb | Metastatic non-small cell lung cancer (NSCLC) with epidermal growth factor receptor (EGFR) exon 19 deletions or exon 21 (L858R) mutations | FDA | |||
| Atezolizumab | Tecentriq | Genentech | Unresectable or metastatic hepatocellular carcinoma who have not received prior systemic therapy | FDA | ||||
| Nivolumab + Ipilimumab | Opdivo/ Yervoy | Bristol-Myers Squibb | First-line treatment for patients with metastatic or recurrent NSCLC, with no epidermal growth factor receptor (EGFR) or anaplastic lymphoma kinase (ALK) genomic tumor aberrations | FDA | ||||
| Atezolizumab | Tecentriq | Genentech | First-line treatment of adult patients with metastatic NSCLC whose tumors have high PD-L1 expression with no EGFR or ALK genomic tumor aberrations | FDA | ||||
| Nivolumab + Ipilimumab | Opdivo/ Yervoy | Bristol-Myers Squibb | First-line treatment for patients with metastatic NSCLC whose tumors express PD-L1(≥1%) with EGFR or ALK genomic tumor aberrations | FDA | ||||
| June | Avelumab | Bavencio | EMD Serono | Maintenance treatment of patients with locally advanced or metastatic urothelial carcinoma (UC) that has not progressed with first-line platinum-containing chemotherapy | FDA | |||
| Pembrolizumab | Keytruda | MSD | First-line treatment of patients with unresectable or metastatic microsatellite instability-high (MSI-H) or mismatch repair deficient (dmmr) colorectal cancer | FDA | ||||
| Recurrent or metastatic cutaneous squamous cell carcinoma (cscc) that is not curable by surgery or radiation | FDA | |||||||
| Pembrolizumab | Keytruda | MSD | Unresectable or metastatic tumor mutational burden-high (TMB H) [≥10 mutations/megabase (mut/Mb)] solid tumors | FDA | ||||
| July | Atezolizumab | Tecentriq | Genentech | BRAF V600 mutation-positive unresectable or metastatic melanoma | FDA | |||
| September | Nivolumab | Opdivo | Bristol-Myers Squibb | Nd | EMA | |||
| Ipilimumab | Yervoy | Bristol-Myers Squibb | Nd | EMA | ||||
| Atezolizumab | Tecentriq | Roche | Nd | EMA | ||||
| October | Pembrolizumab | Keytruda | MSD | Relapsed or refractory classical Hodgkin lymphoma (chl) | FDA | |||
| Pembrolizumab | Keytruda | MSD | Pediatric patients with refractory chl, or chl that has relapsed after 2 or more lines of therapy | FDA | ||||
| Nivolumab + Ipilimumab | Opdivo/ Yervoy | Bristol-Myers Squibb | First-line treatment for adult patients with unresectable malignant pleural mesothelioma | FDA | ||||
| November | Pembrolizumab | Keytruda | MSD | Locally recurrent unresectable or metastatic triple-negative breast cancer (TNBC) whose tumors express PD-L1 (CPS ≥ 10) | FDA | |||
| 2021 | January | Nivolumab + Cabozantinib | Opdivo/ Cabometyx | Bristol-Myers Squibb/Exelixis | First-line treatment for patients with advanced renal cell carcinoma | FDA | ||
| February | Cemiplimab | Libtayo | Regeneron Pharmaceuticals | First-line treatment of patients with advanced NSCLC whose tumors have high PD-L1 expression | FDA | |||
| Cemiplimab | Libtayo | Regeneron Pharmaceuticals | Locally advanced and metastatic basal cell carcinoma | FDA | ||||
| Dostarlimab | Jemperli | GSK | Treatment of certain types of recurrent or advanced endometrial cancer | EMA | ||||
| Nivolumab | Opdivo | Bristol-Myers Squibb | Nd | EMA | ||||
| March | Atezolizumab | Tecentriq | Roche | First-line treatment of adult patients with metastatic NSCLC whose tumours have a PD-L1 expression ≥ 50% tumour cells or ≥ 10% tumour-infiltrating immune cells and who do not have EGFR mutant or ALK-positive NSCLC | EMA | |||
| Pembrolizumab | Keytruda | MSD | Metastatic or locally advanced esophageal or gastroesophageal carcinoma who are not candidates for surgical resection or definitive chemoradiation | FDA | ||||
| Dostarlimab | Jemperli | GSK | Mismatch repair deficient recurrent or advanced endometrial cancer | FDA | ||||
| Nivolumab | Opdivo | Bristol-Myers Squibb | Advanced or metastatic gastric cancer, gastroesophageal junction cancer, and esophageal adenocarcinoma | FDA | ||||
| Nivolumab | Opdivo | Bristol-Myers Squibb | Malignant pleural mesothelioma | EMA | ||||
| Ipilimumab | Yervoy | Bristol-Myers Squibb | Malignant pleural mesothelioma | EMA | ||||
| Monoclonal Antibody | 2020 | March | Isatuximab-irfc | Sarclisa | Sanofi | Multiple myeloma who have received at least two prior therapies including lenalidomide and a proteasome inhibitor | FDA | |
| Isatuximab-irfc | Sarclisa | Sanofi | Multiple myeloma | EMA | ||||
| May | Daratumumab + hyaluronidase-fihj | Darzalex Faspro | Janssen Biotech | Newly diagnosed or relapsed/refractory multiple myeloma | FDA | |||
| July | Tafasitamab-cxix | Monjuvi | MorphoSys US | Relapsed or refractory diffuse large B-cell lymphoma not otherwise specified, including DLBCL arising from low grade lymphoma, and who are not eligible for autologous stem cell transplant | FDA | |||
| August | Belantamab mafodotin-blmf | Blenrep | GSK | Relapsed or refractory multiple myeloma who have received at least 4 prior therapies, including an anti-CD38 monoclonal antibody, a proteasome inhibitor, and an immunomodulatory agent | FDA | |||
| November | Naxitamab | Danyelza | Y-mAbs Therapeutics | Pediatric patients one year of age and older and adult patients with relapsed or refractory high-risk neuroblastoma in the bone or bone marrow demonstrating a partial response, minor response, or stable disease to prior therapy | FDA | |||
| December | Margetuximab-cmkb | Margenza | MacroGenics | Metastatic HER2-positive breast cancer who have received two or more prior anti-HER2 regimens, at least one of which was for metastatic disease | FDA | |||
| 2021 | March | Isatuximab-irfc | Sarclisa | Sanofi | Relapsed or refractory multiple myeloma who have received one to three prior lines of therapy | FDA | ||
| Antibody Drug Conjugate | 2020 | April | Sacituzumab govitecan-hziy | Trodelvy | Immunomedics | Metastatic TNBC who received at least two prior therapies for metastatic disease | FDA | |
| 2021 | April | Loncastuximab tesirine-lpyl | Zynlonta | ADC Therapeutics | Relapsed or refractory large B-cell lymphoma after two or more lines of systemic therapy, including DLBCL not otherwise specified, DLBCL arising from low grade lymphoma, and high-grade B-cell lymphoma | FDA | ||
| Sacituzumab govitecan | Trodelvy | Immunomedics | Advanced urothelial cancer | FDA | ||||
| Sacituzumab govitecan | Trodelvy | Immunomedics | Unresectable locally advanced or metastatic TNBC who have received two or more prior systemic therapies, at least one of them for metastatic disease | FDA | ||||
| CAR T-Cell Therapy | 2020 | June | Gemtuzumab ozogamicin | Mylotarg | Wyeth | Newly-diagnosed CD33-positive acute myeloid leukemia (AML) to include pediatric patients 1 month and older | FDA | |
| July | Brexucabtagene autoleucel | Tecartus | Gilead | Relapsed or refractory mantle cell lymphoma | FDA | |||
| 2021 | January | Daratumumab + Hyaluronidase | Darzalex Faspro | Janssen Biotech | Newly diagnosed light chain (AL) amyloidosis | FDA | ||
| February | Lisocabtagene maraleucel | Breyanzi | Juno | Relapsed or refractory large B-cell lymphoma after two or more lines of systemic therapy | FDA | |||
| Isatuximab | Sarclisa | Sanofi | Multiple myeloma who have received at least one prior therapy | EMA | ||||
| March | Idecabtagene vicleucel | Abecma | Bristol-Myers Squibb | Relapsed or refractory multiple myeloma after four or more prior lines of therapy, including an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 monoclonal antibody | FDA | |||
| Axicabtagene ciloleucel | Yescarta | Kite Pharma | Relapsed or refractory follicular lymphoma (FL) after two or more lines of systemic therapy | FDA | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sancho-Araiz, A.; Mangas-Sanjuan, V.; Trocóniz, I.F. The Role of Mathematical Models in Immuno-Oncology: Challenges and Future Perspectives. Pharmaceutics 2021, 13, 1016. https://doi.org/10.3390/pharmaceutics13071016
Sancho-Araiz A, Mangas-Sanjuan V, Trocóniz IF. The Role of Mathematical Models in Immuno-Oncology: Challenges and Future Perspectives. Pharmaceutics. 2021; 13(7):1016. https://doi.org/10.3390/pharmaceutics13071016
Chicago/Turabian StyleSancho-Araiz, Aymara, Victor Mangas-Sanjuan, and Iñaki F. Trocóniz. 2021. "The Role of Mathematical Models in Immuno-Oncology: Challenges and Future Perspectives" Pharmaceutics 13, no. 7: 1016. https://doi.org/10.3390/pharmaceutics13071016
APA StyleSancho-Araiz, A., Mangas-Sanjuan, V., & Trocóniz, I. F. (2021). The Role of Mathematical Models in Immuno-Oncology: Challenges and Future Perspectives. Pharmaceutics, 13(7), 1016. https://doi.org/10.3390/pharmaceutics13071016