Abstract
Microwave-induced in situ amorphization of a drug into a polymeric amorphous solid dispersion (ASD) has been suggested to follow a dissolution process of the drug into the polymeric network, at temperatures above the glass transition temperature (Tg) of the polymer. Thus, increasing the compact temperature, above the Tg of the polymer, is expected to increase the rate of drug dissolution in the mobile polymer, i.e., the rate of amorphization, in a direct proportional fashion. To test this hypothesis, the present study aimed at establishing a linear correlation between the compact temperature and the rate of drug amorphization using celecoxib (CCX) and the polymers polyvinylpyrrolidone (PVP) 12 and PVP17 as the model systems. Water sorbed into the drug–polymer compacts during 2 weeks of storage at 75% relative humidity was used as the dielectric heating source for the present drug amorphization process, and therefore directly affected the compact temperature during exposure to microwave radiation; the loss of water during heating was also studied. For this, compacts prepared with 30 wt% CCX, 69.5 wt% PVP12 or PVP17 and 0.5 wt% magnesium stearate (lubricant) were conditioned to have a final water content of approx. 20 wt%. The conditioned compacts were exposed to microwave radiation for 10 min at variable power outputs to achieve different compact temperatures. For compacts containing CCX in both PVP12 and PVP17, a linear correlation was established between the measured compact end temperature and the rate of drug amorphization during 10 min of exposure to microwave radiation. For compacts containing CCX in PVP12, a fully amorphous ASD was obtained after 10 min of exposure to microwave radiation with a measured compact end temperature of 71 °C. For compacts containing CCX in PVP17, it was not possible to obtain a fully amorphous ASD. The reason for this is most likely that a fast evaporation of the sorbed water increased the Tg of the conditioned drug–polymer compacts to temperatures above the highest reachable compact temperature during exposure to microwave radiation in the utilized experimental setup. Supporting this conclusion, evaporation of the sorbed water was observed to be faster for compacts containing PVP17 compared to compacts containing PVP12.
1. Introduction
In situ amorphization is a novel approach to increase the oral bioavailability of poorly water-soluble drugs [1,2,3]. In situ amorphization describes the process of transforming a crystalline state of a drug into an amorphous form by forming an amorphous solid dispersion (ASD) within the final dosage form (e.g., a tablet/compact) either in the final manufacturing step or immediately prior to administration [2,4,5,6]. Utilizing the concept of in situ amorphization, a physical mixture of a crystalline drug and a polymer can be manufactured into a final dosage form by using cheap, fast and standard direct tableting protocols. It is advantageous to use a crystalline form of the drug, compared to an amorphous form, in terms of physical stability and manufacturability. For example, the poor flowability of an amorphous powder is not of concern for a crystalline powder [7]. After manufacturing, the crystalline drug can be transformed into the amorphous form, allowing the final dosage form to have the characteristic advantages of the amorphous drug such as, e.g., higher solubility and faster dissolution rate compared to the crystalline form [8,9].
In situ amorphization has been demonstrated by immersion in water, heat convection, microwave radiation and laser radiation [2,4,5,6,10,11,12,13]. Presently, microwave or laser radiation appear to be the most promising approaches to achieve in situ amorphization, as these, so far, are the only approaches for which complete amorphization has been reported [2,13]. Microwave-induced in situ amorphization relies on the absorption of microwave radiation by dielectric excipients inside the final dosage form. The absorption of microwave radiation by a substance is specified by the dielectric properties of the substance. A common dielectric molecule is water, as the dipoles of water can easily adapt to the alternating electromagnetic field at 2.45 GHz, which is the fixed frequency most household microwave ovens work at [14,15,16,17]. The movement of the dielectric molecules, aligning to the electromagnetic field, causes friction which results in the generation of heat. Usually, the absorption of microwave radiation by drugs and polymers inside compacts can be neglected with respect to heat generation, as their dielectric properties are poor [16,17]. Examples of dielectric molecules that have been used for in situ amorphization are water (including inorganic crystal hydrate salts), glycerol and polyethylene glycol [5,6,12,16,17,18,19].
Several studies in the field of microwave-induced in situ amorphization have shown that water, sorbed into compacts, acts both as a plasticizer (i.e., a mobility enhancer), and an enabling dielectric excipient (i.e., a dielectric heating source) [1,2,4,20,21]. The plasticizer function is vital as the rate of amorphization has been shown to increase significantly at temperatures above the glass transition temperature (Tg) of the pure polymer and the forming ASD, when the mobility of polymer increases and its viscosity decreases [6,21].
In previous studies, several factors have been shown to influence the rate of the microwave-induced in situ amorphization. For example, it was shown that reducing the particle size of the drug and the polymer (i.e., increasing the surface area of both the drug and polymer particles), and increasing the temperature above the Tg of the polymer, increased the rate of amorphization [2]. Furthermore, it has been shown that decreasing the viscosity of the polymer, e.g., by using a lower molecular weight polymer or higher temperatures obtained during exposure, increases the rate of in situ amorphization [6]. All this data supports the hypothesis that microwave-induced in situ amorphization can be described as a dissolution process of the drug into the mobile polymer at temperatures above the Tg of the polymer. Accordingly, the rate of amorphization may theoretically be described by the Noyes–Whitney equation (Equation (1)) [22].
- dm/dt = solute dissolution rate;
- m = mass of dissolved material;
- t = time;
- A = surface area of the solute particle;
- h = thickness of the diffusion layer;
- Cs = particle saturation concentration;
- Cb = concentration in the bulk solution;
- D = Diffusion coefficient of the solute in solution;
- k = Boltzmann constant;
- T = absolute temperature;
- η = viscosity of the solvent;
- r = radius of the solute molecule.
From the Noyes–Whitney equation, it can be seen that the rate of dissolution (i.e., the rate of in situ amorphization of the drug into the mobile polymer) is linearly dependent on the diffusion coefficient, which according to the Stokes–Einstein equation (Equation (2)) is linearly correlated with temperature.
Based on the hypothesis that the rate of in situ amorphization can be described by the Noyes–Whitney equation (Equation (1)), the first aim of the present study was to study the relation between the compact temperature (at temperatures above the Tg of the polymer) and the rate of in situ amorphization upon exposure of compacts to microwave radiation. For this, compacts containing 30 wt% CCX, 69.5 wt% PVP12 or PVP17 and 0.5 wt% magnesium stearate (lubricant) were exposed to microwave radiation after a conditioning period of 2 weeks at 75% relative humidity and room temperature. A drug load of 30 wt% CCX was chosen for the study, as a previous study had shown complete amorphization of compacts containing 30 wt% CCX and PVP12 after exposure to microwave radiation for 10 min using a household microwave oven [2]. Additionally, the drug load of 30 wt% CCX in PVP (12 and 17) is below the solubility of CCX in PVP at room temperature, which means that the formation of a fully amorphous and physically stable ASD (upon cooling) should be possible [23].
The role of water evaporation from the conditioned compacts during exposure to microwave radiation was also studied. As previously mentioned, water acts not only as a plasticizer, lowering the Tg of the components in the compacts, but also as a dielectric heating source. However, water will evaporate during exposure to microwave radiation with increasing compact temperature. Depending on the degree of interaction between the sorbed water and the polymers (PVP12 and PVP17), the compact temperature profile, as well as the evaporation of the water, is expected to vary. Free water is not restricted in its mobility, which results in fast heating rates upon exposure to microwave radiation. Sorbed water of a polymer, here PVP, is bound and therefore restricted in its mobility [16,24,25]. As tightly bound water is more mobility restricted compared to loosely bound water, exposing a polymer with tightly bound water to microwave radiation is expected to result in a lower heating rate, but also a lower rate of water evaporation, which means that the Tg of this polymer will increase at a slower rate, as compared to a polymer with loosely bound water.
The degree of interaction between the sorbed water and the polymers PVP12 and PVP17 was determined by the Gordon–Taylor equation (Equation (3)). According to the Gordon–Taylor equation, a difference between the measured and the theoretical Tg indicates an interaction between the two components of the system [26].
 Table 1. Glass transition temperatures of the polymers PVP12 and PVP17; mean ± SD (n = 2).
Table 1. Glass transition temperatures of the polymers PVP12 and PVP17; mean ± SD (n = 2).- Tg2 = Tg of water = 135 K
- p1 = amorphous density of PVP = 1.18 g/cm3 [27,28]
- p2 = density of water = 0.999 g/cm3
- w1 = weight fraction of PVP
- w2 = weight fraction of water
Based on the interaction between the sorbed water and PVP (12 or 17), the second aim of the study was to investigate the role of the compact water with respect to in situ amorphization and the interplay between polymer plasticization, dielectric heating source and evaporation. For this, the evaporation of sorbed water from the polymers PVP12 and PVP17 was determined by thermogravimetric analysis, and the results were related to the observed in situ amorphization of CCX in PVP12 and PVP17.
2. Materials and Methods
2.1. Materials
Celecoxib (CCX, Mw = 381.37 g/mol) and magnesium stearate were purchased from Fagron Nordic A/S (Copenhagen, Denmark). Kollidon® 12PF (PVP12, Mw = 2000–3000 g/mol) and Kollidon® 17PF (PVP 17, Mw = 7000–11,000 g/mol) were kindly supplied by BASF (Ludwigshafen, Germany). Sodium chloride (NaCl, Mw = 58.44 g/mol) was purchased from Sigma-Aldrich (St. Louis, MO, USA). All chemicals were used as received.
2.2. Compact Preparation and Storage
CCX, PVP12 and PVP17 were sieved using a 125 µm sieve. Only the particle fraction with a diameter <125 µm was used, as a previous study showed that a small particle size of drug and polymer is advantageous for fast microwave-induced in situ amorphization [2]. Subsequently, physical mixtures containing 30 wt% CCX, 69.5 wt% PVP12 or PVP17 and 0.5 wt% magnesium stearate were prepared using mortar and pestle. Using 100 ± 2 mg of the physical mixtures, flat-faced cylindrical compacts (Ø 6 mm) were prepared using an instrumented single punch tablet press GTP-1 from Gamlen Instruments (Nottingham, UK) fitted with a 500 kg load cell (CT6–500-022) at a compaction pressure of 35 MPa. After preparation, the compacts were conditioned for 2 weeks at 75% relative humidity (over a saturated NaCl solution) at room temperature. The compacts were weighted before and after the conditioning period to assess the weight gain due to the sorbed water.
2.3. In Situ Drug Amorphization Using a Microwave Oven
In order to induce in situ amorphization, a Synthos 3000 microwave oven from Anton Paar GmbH (Graz, Austria) was used. The Synthos 3000 has two magnetrons, which radiate unpulsed microwaves at a frequency of 2.45 GHz. The microwave oven is equipped with a 64 MG5 rotor, which allows 3 rpm and the holding of up to 64 samples. Additionally, the microwave oven has a built-in infrared probe for in situ temperature measurements. The temperature of each compact was approx. measured every 20th second. As the temperature was constantly monitored, it was adjusted by a feedback mechanism regulating the power output, i.e., the temperature monitoring allowed the setting of a maximum temperature of the microwave oven, which was kept constant. The maximal power output was set to 1000 W. The microwave oven adjusted the power output from 0–1000 W to reach the compact temperature set by measuring the compact temperature as described above. Three compacts were placed in individual sealed glass vials in temperature-recording positions. Empty glass vials were placed in the remaining 13 temperature-recording positions. Additionally, 16 sealed glass vials, each containing 1.5 mL of demineralized water, were placed in non-temperature recording positions to absorb residual microwave radiation. The water-containing vials had an offset to the glass vials containing compacts to avoid passive heating by conduction. Compacts containing PVP12 were continuously exposed to microwave radiation for 10 min to reach compact temperatures of 60, 70, 80 and 82 °C. Compacts containing PVP17 were continuously exposed to microwave radiation for 10 min to reach temperatures of 60, 70 and 80 °C. Each experiment was conducted with triplicate measurements for each compact composition—temperature combination simultaneously (n = 3), using the same batch of compacts. The recorded temperature data were interpolated and the mean and standard deviations were calculated. The temperature data were corrected by a technical factor of 1.214 according to the manufacturer; i.e., the technical factor corrects for the difference between the compact temperature and the temperature of the glass vial (bottom), whereas the temperature of the glass vial is measured by the in-built infrared thermometer. The set compact temperatures of 60, 70, 80 and 82 °C (already corrected by the technical factor) responded to average measured compact end temperatures of 51, 59, 70 and 71 °C, respectively.
2.4. Quantification of the Degree of Amorphization
The degree of amorphization, before and after exposure to microwave radiation, was quantified using transmission Raman spectroscopy. For this, a Kaiser RXN1 Microprobe from Kaiser Optical Systems (Ann Arbor, MI, USA) was equipped with a PhaT-probe (Pharmaceutical Area Testing) and used in a transmission Raman configuration setup, as described previously by Edinger et al. (2018) [20]. In short, the compacts were placed on a Thorlabs AD127NT adaptor (Newton, NJ, US), which was situated on the excitation fiber. Using a 5× objective connected to the PhaT-probe, the inelastically scattered lights were collected at a distance of 20 mm to the adaptor. Each spectrum had a total acquisition time of 20 s, which equals an average of 5 measurements with an exposure time of 4 s each. The dark frames were subtracted for each measurement. At the excitation fiber output, the wavelength was 785 nm with an excitation power of 200 mW. At a resolution of 5 cm−1, the Raman shift was measured from 150 to 1900 cm−1. The analysis was conducted using a calibration space and a partial least-squares regression (PLS) model, which was kindly supplied by Edinger et al. (2018) [20]. Distinguishing features present in the spectral region from 705 to 845 cm−1 were used to differentiate between the crystalline and the amorphous form of CCX. The calibration space for the PLS model was obtained from 17 different mixtures containing crystalline CCX, amorphous CCX and PVP17. Preprocessing of the data was performed by Savitzky–Golay smoothing and standard normal variate transformation; all data processing was performed in MatLab from MathWorks (Natick, MA, USA) using the PLS toolbox 8.1.1 from Eigenvector Research Inc. (Manson, WA, USA).
2.5. Thermal Analysis
Thermal analysis was conducted using a Discovery differential scanning calorimeter (DSC) from TA Instruments (New Castle, DE, USA) at a nitrogen gas purge of 50 mL/min. Compacts exposed to microwave radiation at the highest temperature were ground using mortar and pestle. A total of 2–6 mg of powder was weighed into Tzero aluminum pans, which were closed with hermetically sealing lids.
Determination of the Tg values was performed using modulated DSC (mDSC) at a heating rate of 3 °C/min with an amplitude of 1 °C/50 s. The Tg values of the formed ASD and the polymers containing bulk water were determined by heating from −20 °C to 120 °C. The Tg values of the pure conditioned polymers were determined from −60 °C to 50 °C for PVP12, and −60 °C to 70 °C for PVP17. The Tg values of the water-free polymers were determined by applying a heat–cool–heat cycle. For the water to evaporate during the first heating, the lid was perforated. The polymers were initially equilibrated for 10 min at 120 °C and subsequently equilibrated to −20 °C. Afterwards, a heating to 170 °C was applied. The Tg was defined as the midpoint over the change in heat capacity using the TRIOS software (version 5.1.1) from TA Instruments (New Castle, DE, USA). All Tg values were determined in duplicates (n = 2).
The water content of the pure polymers (n = 3) and the water loss of the conditioned polymers (n = 1) were determined using a Discovery thermogravimetric analyzer 1 (TGA) from TA Instruments Inc. (New Castle, DE, USA) using a nitrogen gas purge of 25 mL/min. Approx. 10 mg of the pure polymers were placed in an open platinum pan and analyzed at a heating rate of 10 °C/min up to 150 °C.
Using the TGA, the conditioned polymers were also placed in an open platinum pan (approx. 50 mg) and exposed to the heating rate obtained for compacts containing PVP12 or PVP17 that were exposed to microwave radiation at a measured compact end temperature of 70 °C, to study the water evaporation process. Conditioned PVP12 was analyzed at a heating rate of 30.8 °C/min to 60 °C, followed by a heating rate of 1.2 °C/min to 70 °C. Conditioned PVP17 was analyzed at a heating rate of 38.8 °C/min to 60 °C, followed by a heating rate of 1.1 °C/min to 70 °C. The weight loss was determined using the TRIOS software (version 5.1.1).
2.6. Solid State Characterization
The solid state characteristics of the compacts, before and after exposure to microwave radiation, were confirmed qualitatively using X-Ray powder diffraction (XRPD). For this, an X’Pert Pro diffractometer from PANalytical (Eindhoven, the Netherlands) using Cu Kα radiation (λ = 1.54187 Å) was used. The diffractograms were recorded at 45 kV and 40 mA from 5 to 30° 2θ, and were analyzed using the X’Pert Data viewer software (version 1.2) from PANalytical (Eindhoven, the Netherlands). Each sample was analyzed once (n = 1).
3. Results and Discussion
As previously mentioned, it has been suggested that microwave-induced in situ amorphization can be described as a dissolution process, which means that the rate of amorphization can be described by the Noyes–Whitney equation (Equation (1)). To further support this hypothesis, the present study aimed to investigate the relationship between the compact temperature and rate of drug amorphization. Furthermore, the role of the evaporation of the sorbed water inside the compact was studied, as the dehydration affects the Tg of the polymer PVP and the forming ASD as well as the dielectric heating.
3.1. Characterization of the Compacts after Conditioning
During the conditioning period of 2 weeks at 75% relative humidity, the compacts gained weight due to water sorption of the hygroscopic polymer PVP. The weight gain was determined to be 15.3 ± 0.5 wt% (n = 15) for compacts containing PVP12 and 12.8 ± 1.1 wt% (n = 12) for compacts containing PVP17. Including the water content of the bulk PVP, 4.1 ± 0.0 wt% for PVP12 and 7.7 ± 0.1 wt% for PVP17 (n = 3), the total water content of all the compacts after storage was approx. 20 wt% (19.4 ± 0.5 wt% for PVP12, 20.5 ± 1.2 wt% for PVP17).
As the polymers inside the compacts had a Tg below room temperature after conditioning (Table 1), minor amorphization occurred during the conditioning period, which is in line with findings reported in previous publications [2,4,20]. In the present study, the initial amorphization was found to be 5.0 ± 0.4% (n = 3) for compacts containing PVP12 and 0.2 ± 0.1% (n = 3) for compacts containing PVP17. The observed difference in the initial degree of amorphization for compacts containing PVP12 and PVP17 is suggested to be due to the fact that PVP12 has a lower Tg, due to lower molecular weight and a lower viscosity at room temperature, as compared to PVP17. Upon conditioning, all the compacts significantly softened due to the high amount of sorbed water.
In order to determine the degree of interaction between the sorbed water and the polymer, the theoretical Tg values were calculated using the Gordon–Taylor equation (Equation (3)) [29]. The measured and the theoretical Tg values of the polymers PVP12 and PVP17, prior to-, and after conditioning, can be found in Table 1.
As can be seen in Table 1, it is apparent that the sorbed water, acting as a plasticizer, significantly lowered the Tg of the two polymers. For both PVP12 and PVP17, the Tg was approx. lowered by 65 °C with the sorption of water in the bulk PVP. For PVP12, it can be seen that a Tg below 0 °C was obtained after conditioning.
When comparing the experimental Tg values with the theoretical Tg values obtained using the Gordon–Tayler equation, it is apparent that the experimentally determined Tg values are lower than the theoretical values, in all cases except for PVP17 after conditioning (Table 1). As a negative deviation from the Gordon–Tayler equation is commonly interpreted as an increase in the mobility of the system, i.e., in this case as the water of the bulk polymer having a highly plasticizing effect on PVP12 and PVP17, the degree of plasticization originating from the water of the bulk PVP was found to be higher for PVP12 compared to PVP17. The different degree of (negative) deviation of the experimentally determined Tg values from the theoretical Tg values suggests differences in the degree of interaction of the water molecules with PVP12 and PVP17. Based on these results (Table 1), it is hypothesized that the ratio between tightly bound and loosely bound water is higher for PVP12 compared to PVP17 [16,25].
For both polymers, the negative deviation between the measured and the theoretical Tg value became smaller after the conditioning period, suggesting a higher degree of loosely bound water in both conditioned polymers as compared to the bulk polymers (Table 1).
3.2. Exposure to Microwave Radiation of Compacts Containing PVP12
Compacts containing PVP12 were initially exposed to 10 min of microwave radiation resulting in measured average compact end temperatures of 51, 59 and 70 °C. Immediately after exposure to microwave radiation, the degree of amorphization was determined by transmission Raman spectroscopy. Figure 1a shows the obtained degree of amorphization for CCX plotted as a function of the measured average compact end temperature. The experimentally determined data points for the degree of amorphization of CCX, at the three different temperatures, were linearly correlated with a coefficient of determination of (R2) > 0.974. Based on the high R2 value and the assumption that the rate of amorphization can be described by the Noyes–Whitney equation (Equation (1)), the linear correlation was extrapolated to estimate the compact end temperature at which 100% amorphization of CCX would be achieved. The extrapolation of the linear correlation predicted that compacts containing PVP12 could be fully amorphized when exposed to microwave radiation for 10 min with a measured average compact end temperature of 71 °C. Hence, the set temperature of the microwave oven was increased and compacts containing PVP12 were exposed to microwave radiation for 10 min to experimentally validate the predicted data point. It was found that the exposure of compacts containing PVP12 to microwave radiation for 10 min with a measured average compact end temperature of 71 °C did indeed result in complete amorphization (99.7 ± 0.8%, n = 3). These results support the hypothesis of a linear correlation between temperature and rate of amorphization upon exposure to microwave radiation, at temperatures above the Tg of the polymer.
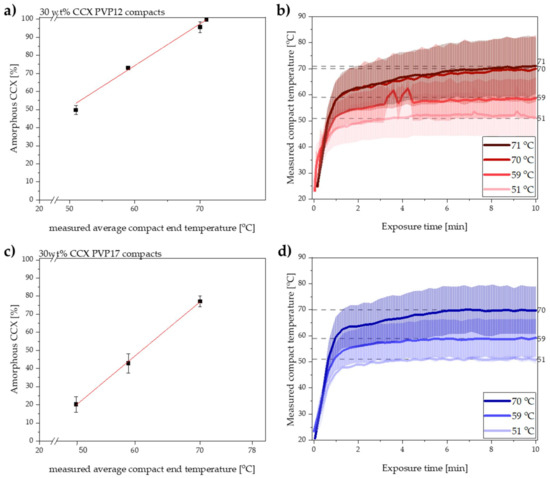
Figure 1.
(a) Degree of amorphization for CCX [%] plotted as a function of the measured average compact end temperature [°C] after 10 min of microwave radiation of compacts containing PVP12. The red line indicates the linear extrapolation to obtain 100% amorphization. Mean ± SD (n = 3). (b) Measured compact temperature of the compacts containing PVP12 exposed to microwave radiation for 10 min at the measured average compact end temperature of 51, 59, 70 and 71 °C. Interpolated mean ± SD (n = 3). The dotted grey lines indicate the measured average compact end temperature. (c) Degree of amorphization for CCX [%] plotted as a function of the measured average compact end temperature [°C] after 10 min of radiation of compacts containing PVP17. The red line indicates the linear relationship. Mean ± SD (n = 3). (d) Measured compact temperature of the compacts containing PVP17 exposed to microwave radiation for 10 min at the measured average compact end temperatures of 51, 59, 70 °C. Interpolated mean ± SD (n = 3). The dotted grey lines indicate the measured average compact end temperature.
The degree of amorphization determined by transmission Raman spectroscopy was qualitatively confirmed by X-ray powder diffraction. From the diffractograms shown in Figure 2a, it can be seen that complete amorphization was obtained with a measured average compact end temperature of 71 °C, as the diffractogram showed a halo.
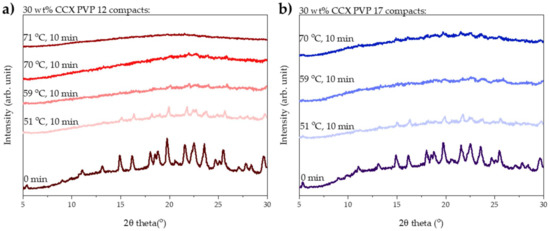
Figure 2.
XRPD diffractograms of compacts before and after exposure to 10 min of microwave radiation. (a) Compacts containing 30 wt% CCX and PVP12 exposed to microwave radiation at a measured average compact end temperature of 51, 59, 70 and 71 °C. (b) Compacts containing 30 wt% CCX and PVP17 exposed to microwave radiation at a measured average compact end temperature of 51, 59 and 70 °C.
The temperature recordings obtained during exposure to microwave radiation of compacts containing PVP12 are shown in Figure 1b. It can be seen that the measured average end temperature of the compacts was reached already after 4–8 min of exposure to microwave radiation.
It is suggested that the rate of amorphization follows the trend of the temperature recording; i.e., the rate of amorphization is expected to increase relatively fast during the initial steep temperature increase and subsequently settles at constant level, depending on the reached end compact temperature (Figure 1b) [2]. With respect to changes in rate of amorphization during the exposure to microwave radiation, it is also important to mention that the viscosity of the polymer will change as a function of temperature [6], but that it is also dependent on the degree of amorphization, as more drug dissolved into the polymer will increase its viscosity [6]. Here, however, it is suggested that the change in the viscosity of each polymer, i.e., PVP12 and PVP17, during exposure to microwave radiation had a minimal effect on the rate of amorphization, as the temperatures were above the Tg of the polymer (throughout the study), and the largest change in viscosity is observed at temperatures around the Tg value. Furthermore, the temperatures chosen in the present study were relatively close to each other (max. 20 °C apart), which means that the viscosity difference was minimal (logarithmic dependency of the viscosity on the temperature) and therefore is expected to have had a non-significant effect on the rate of amorphization [30].
Thermal analysis was used to characterize the ASD formed upon exposure of compacts containing PVP12 to microwave radiation for 10 min at a measured average compact end temperature of 71 °C. Based on the Tg measurement, it was found that the ASD was homogenous, as characterized by a single Tg at 55.1 ± 1.1 °C, (n = 2).
3.3. Exposure to Microwave Radiation of Compacts Containing PVP17
Compacts containing PVP17 were exposed to microwave radiation at measured average compact end temperatures of 51, 59 and 70 °C. In contrast to the compacts containing PVP12, complete amorphization has so far not been described for compacts containing the higher molecular weight/viscosity grades of PVP, i.e., PVP17. As the Tg and the viscosity of PVP17 are higher as compared to PVP12, it was suggested that a higher temperature was needed to reach complete amorphization for compacts prepared with PVP17 compared to PVP12.
After exposing the compacts containing PVP17 to microwave radiation, the degree of amorphization was directly determined using transmission Raman spectroscopy. The results for the degree of amorphization after exposure to microwave radiation for 10 min at the three different measured average compact end temperatures are shown in Figure 1c with the temperature profiles depicted in Figure 1d. As for the PVP12 compacts, the experimentally determined degree of amorphization was linearly correlated with the measured average compact end temperature for the compacts containing PVP17 (R2 > 0.999). Again, the linear relationship was extrapolated to estimate at which temperature 100% amorphization could be achieved.
The calculated temperature to reach the complete amorphization was at a compact temperature of 78 °C, which was above the maximum compact temperature reachable with the present experimental setup and compact composition. Hence, the predicted compact temperature value to obtain a fully amorphous ASD for compacts containing PVP17 could not be experimentally validated with the current experimental setup.
As for the compacts prepared with PVP12, the degree of amorphization was qualitatively confirmed using XRPD. From Figure 2b, it can be seen that no fully amorphous solid dispersion was obtained from compacts containing PVP17.
For the PVP17 compacts, the compact end temperature was reached within 3–6 min of microwave radiation. Comparing the temperature profiles obtained for compacts containing PVP17 (Figure 1d) with compacts containing PVP12 (Figure 1b), it is suggested that differences in the initial average heating rates (t~0–1 min) can be seen. Despite the similar, or even faster, initial heating rate at a measured average compact end temperature of 70 °C, the rate of amorphization for compacts containing PVP17 was slower than for compacts containing PVP12 at similar temperatures (compare Figure 1a,c). As water acted not only as the dielectric heating source inside the compacts upon exposure to microwave radiation, but also as a plasticizer, it is suggested that the different degree of interaction of the water with the polymer played a crucial role here. As suggested above, loosely bound water, which is more mobile compared to tightly bound water, will lead to a faster heating rate; however, it also leads to a faster dehydration process, which in turn will increase the Tg of PVP17 and the forming ASD faster as compared to PVP12 and the forming ASD. It is suggested that this increase in the Tg of the polymer resulted in a slower amorphization for compacts containing PVP17 compared to PVP12.
Following the same procedure as for the PVP12 compacts, thermal analysis of compacts containing PVP17 exposed to microwave radiation at 70 °C for 10 min (highest measured average compact end temperature) was performed. It was revealed that a non-homogenous two-phase system was obtained, with two Tgs, i.e., one for a drug-rich phase and one for a polymer-rich phase. The Tg for the drug-rich phase and polymer-rich phase was 60.1 ± 1.1 °C (n = 2) and 97.4 ± 15.2 °C (n = 2), respectively. The detection of a Tg above 70 °C for the polymer-rich phase indicates that complete amorphization was not possible with the present settings, as this compact temperature would not be reached.
3.4. Influence of Water Evaporation on the Compact Temperature during Exposure to Microwave Radiation, and the Tg of the Polymer
Even though a fast heating rate is beneficial for in situ amorphization, as the rate of amorphization increases with increasing temperature (Equation (1)), a fast heating rate will also cause the compact water to evaporate faster. As a result of water evaporation, the Tg of the polymer and forming ASD will increase (Table 1 and Section 3.3), the mobility of the polymer will decrease and eventually the dielectric heating source will be lost, which is suggested to ultimately limit the in situ amorphization process.
As described in Section 3.1, it is suggested that the two polymers, PVP12 and PVP17, interacted differently with sorbed water, i.e., the water was bound to a different degree, which would lead to an initial higher heating rate for compacts containing PVP17, as the water was less tightly bound in these compacts as compared to compacts containing PVP12. When comparing the initial heating rates (t~0–1 min) observed for compacts containing PVP12 and PVP17, at the measured average compact end temperature of 70 °C (Figure 1b,d), a higher initial average heating rate for compacts containing PVP17 was observed (though determined to not be statistically different, p > 0.05).
Following the initial steep compact temperature increase, the heating rate decreased for both compact compositions, and the compact temperature approached a plateau. It was found that after the initial heating, the heating rate slowed down to 1.1 °C/min for compacts containing PVP17 and to 1.2 °C/min for compacts containing PVP12 (t~1–10 min). This shift in the observed heating rates is suggested to be caused by water evaporation, indirectly indicating that water evaporated faster from the PVP17 compacts. These results correlate well with the suggested degree of interaction of water with PVP12 and PVP17, as PVP12 showed a higher degree of tightly bound water compared to PVP17 (see Section 3.1).
In order to determine how the different heating rates affected the water evaporation from compacts containing PVP12 and PVP17, at the highest common measured average compact end temperature (70 °C), the pure conditioned polymers were exposed to the same heating rates obtained during exposure to microwave radiation on the TGA (Figure 3). It can be seen in Figure 3 that PVP17 did indeed lose the water faster, as compared to PVP12. Hence, it is suggested that complete amorphization could not be achieved for compacts containing PVP17 using sorbed water as a dielectric heating source, as the sorbed water evaporated too fast. Due to a low degree of interactions between the sorbed water and PVP17, the sorbed water evaporated quickly during exposure to microwave radiation, which resulted in a partial loss of the plasticizer, which increased the Tg of the polymer and forming ASD to temperatures which could not be reached, as also the dielectric heating source was partly lost. Based on these results, it is suggested that, when using water as a dielectric heating source for microwave-induced in situ amorphization, some polymers are less suitable than others. Still, it is important to remember that this study only presents a case study based on one model drug and two different grades of PVP, PVP12 and PVP17, i.e., further studies are needed to investigate if the observed behavior also applies for other polymers, including higher molecular weight polymers.
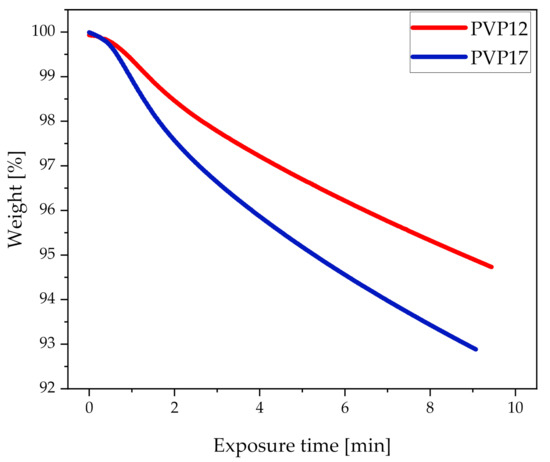
Figure 3.
Pure conditioned polymers, PVP12 (red) and PVP17 (blue) exposed to TGA heating with the heating rates obtained during exposure of compacts containing 30 wt% CCX and PVP12 or PVP17 to microwave radiation for 10 min at a measured average compact end temperature of 70 °C (n = 1).
4. Conclusions
This study showed that during microwave-induced in situ drug amorphization of compacts containing 30 wt% CCX and PVP12 or PVP17, the rate of amorphization was linearly dependent on the compact end temperature in the range 51–70 °C. For PVP12 compacts, a fully amorphous ASD was obtained following exposure to microwave radiation for 10 min at a measured average compact end temperature of 71 °C. Compacts containing PVP17 could not be fully amorphized upon exposure to microwave radiation with the present experimental setup and compact composition. The limitation for in situ amorphization for compacts containing PVP17 was found to be the (sorbed) water evaporating so fast that complete amorphization was not achieved before the Tg of the polymer and forming ASD had increased above the measured average compact end temperature. Hence, using sorbed water as a dielectric heating source, it is recommended to use a polymer with an initial low Tg and a low molecular weight, as well as a high degree of tightly bound water to achieve 100% amorphization upon exposure to microwave radiation.
Author Contributions
Conceptualization, N.-J.H., M.M.K., K.L., R.B.; methodology, N.-J.H., M.M.K., K.L., R.B.; investigation, N.-J.H.; data curation, N.-J.H.; writing—original draft preparation, N.-J.H.; writing—review and editing, N.-J.H., M.M.K., K.L., R.B.; visualization, N.-J.H.; supervision, M.M.K., R.B., K.L.; project administration, K.L.; funding acquisition, K.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Independent Research Fund Denmark, grant number DFF-7026-00052B.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data are available upon request from the authors.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Doreth, M.; Hussein, M.A.; Priemel, P.A.; Grohganz, H.; Holm, R.; Lopez de Diego, H.; Rades, T.; Lobmann, K. Amorphization within the tablet: Using microwave irradiation to form a glass solution in situ. Int. J. Pharm. 2017, 519, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Hempel, N.J.; Knopp, M.M.; Berthelsen, R.; Zeitler, J.A.; Lobmann, K. The influence of drug and polymer particle size on the in situ amorphization using microwave irradiation. Eur. J. Pharm. Biopharm. 2020, 149, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Qiang, W.; Lobmann, K.; McCoy, C.P.; Andrews, G.P.; Zhao, M. Microwave-induced in situ amorphization: A new strategy for tackling the stability issue of amorphous solid dispersions. Pharmaceutics 2020, 12, 655. [Google Scholar] [CrossRef] [PubMed]
- Hempel, N.J.; Knopp, M.M.; Berthelsen, R.; Lobmann, K. Convection-induced vs. microwave radiation-induced in situ drug amorphization. Molecules 2020, 25, 1068. [Google Scholar] [CrossRef]
- Hempel, N.J.; Morsch, F.; Knopp, M.M.; Berthelsen, R.; Lobmann, K. The use of glycerol as an enabling excipient for microwave-induced in situ drug amorphization. J. Pharm. Sci. 2021, 110, 155–163. [Google Scholar] [CrossRef]
- Hempel, N.J.; Dao, T.; Knopp, M.M.; Berthelsen, R.; Lobmann, K. The influence of temperature and viscosity of polyethylene glycol on the rate of microwave-induced in situ amorphization of celecoxib. Molecules 2020, 26, 110. [Google Scholar] [CrossRef]
- Demuth, B.; Nagy, Z.K.; Balogh, A.; Vigh, T.; Marosi, G.; Verreck, G.; Van Assche, I.; Brewster, M.E. Downstream processing of polymer-based amorphous solid dispersions to generate tablet formulations. Int. J. Pharm. 2015, 486, 268–286. [Google Scholar] [CrossRef]
- Van den Mooter, G. The use of amorphous solid dispersions: A formulation strategy to overcome poor solubility and dissolution rate. Drug Discov. Today Technol. 2012, 9, e71–e174. [Google Scholar] [CrossRef]
- Baghel, S.; Cathcart, H.; O’Reilly, N.J. Polymeric amorphous solid dispersions: A review of amorphization, crystallization, stabilization, solid-state characterization, and aqueous solubilization of biopharmaceutical classification system class II drugs. J. Pharm. Sci. 2016, 105, 2527–2544. [Google Scholar] [CrossRef]
- Doreth, M.; Löbmann, K.; Grohganz, H.; Holm, R.; Lopez de Diego, H.; Rades, T.; Priemel, P.A. Glass solution formation in water—In situ amorphization of naproxen and ibuprofen with Eudragit® E PO. J. Drug Deliv. Sci. Technol. 2016, 34, 32–40. [Google Scholar] [CrossRef]
- Petry, I.; Lobmann, K.; Grohganz, H.; Rades, T.; Leopold, C.S. In situ co-amorphisation of arginine with indomethacin or furosemide during immersion in an acidic medium—A proof of concept study. Eur. J. Pharm. Biopharm. 2018, 133, 151–160. [Google Scholar] [CrossRef]
- Holm, T.P.; Knopp, M.M.; Löbmann, K.; Berthelsen, R. Microwave induced in situ amorphisation facilitated by crystalline hydrates. Eur. J. Pharm. Sci. 2021. [Google Scholar] [CrossRef]
- Hempel, N.-J.; Merkl, P.; Asad, S.; Knopp, M.M.; Berthelsen, R.; Bergström, C.A.S.; Teleki, A.; Sotiriou, G.A.; Löbmann, K. Utilizing laser activation of photothermal plasmonic nanoparticles to induce on-demand drug amorphization inside a tablet. Mol. Pharm. 2021. [Google Scholar] [CrossRef]
- Zhang, X.; Rajagopalan, K.; Lei, H.; Ruan, R.; Sharma, B.K. An overview of a novel concept in biomass pyrolysis: Microwave irradiation. Sustain. Energy Fuels 2017, 1, 1664–1699. [Google Scholar] [CrossRef]
- Grant, E.; Halstead, B.J. Dielectric parameters relevant to microwave dielectric heating. Chem. Soc. Rev. 1998, 27, 213–224. [Google Scholar]
- Heng, P.W.; Loh, Z.H.; Liew, C.V.; Lee, C.C. Dielectric properties of pharmaceutical materials relevant to microwave processing: Effects of field frequency, material density, and moisture content. J. Pharm. Sci. 2010, 99, 941–957. [Google Scholar] [CrossRef]
- McLoughlin, C.M.; McMinn, W.A.M.; Magee, T.R.A. Physical and dielectric properties of pharmaceutical powders. Powder Technol. 2003, 134, 40–51. [Google Scholar] [CrossRef]
- Koizumi, N.; Hanai, T. Dielectric Properties of Polyethylene Glycols: Dielectric Relaxation in Solid State (Special Issue on Polymer Chemistry, I). Bull. Instit. Chem. Res. 1964, 42, 115–127. [Google Scholar]
- McDuffie, G.E.; Quinn, R.G.; Litovitz, T.A. Dielectric properties of glycerol—Water mixtures. J. Chem. Phys. 1962, 37, 239–242. [Google Scholar] [CrossRef]
- Edinger, M.; Knopp, M.M.; Kerdoncuff, H.; Rantanen, J.; Rades, T.; Lobmann, K. Quantification of microwave-induced amorphization of celecoxib in PVP tablets using transmission Raman spectroscopy. Eur. J. Pharm. Sci. 2018, 117, 62–67. [Google Scholar] [CrossRef]
- Doreth, M.; Lobmann, K.; Priemel, P.; Grohganz, H.; Taylor, R.; Holm, R.; Lopez de Diego, H.; Rades, T. Influence of PVP molecular weight on the microwave assisted in situ amorphization of indomethacin. Eur. J. Pharm. Biopharm. 2018, 122, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Noyes, A.A.; Whitney, W.R. The rate of solution of solid substances in their own solutions. J. Am. Chem. Soc. 2002, 19, 930–934. [Google Scholar] [CrossRef]
- Rask, M.B.; Knopp, M.M.; Olesen, N.E.; Holm, R.; Rades, T. Influence of PVP/VA copolymer composition on drug–polymer solubility. Eur. J. Pharm. Sci. 2016, 85, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Parker, T.G. Dielectric properties of polymers, II. In Polymer Science; Elsevier: Amsterdam, The Netherlands, 1972; pp. 1297–1327. [Google Scholar]
- Oksanen, C.A.; Zografi, G. Molecular mobility in mixtures of absorbed water and solid poly(vinylpyrrolidone). Pharm. Res. 1993, 10, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Barmpalexis, P.; Karagianni, A.; Kachrimanis, K. Molecular simulations for amorphous drug formulation: Polymeric matrix properties relevant to hot-melt extrusion. Eur. J. Pharm. Sci. 2018, 119, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Rask, M.B.; Knopp, M.M.; Olesen, N.E.; Holm, R.; Rades, T. Comparison of two DSC-based methods to predict drug-polymer solubility. Int. J. Pharm. 2018, 540, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Knopp, M.M.; Olesen, N.E.; Holm, P.; Langguth, P.; Holm, R.; Rades, T. Influence of polymer molecular weight on drug-polymer solubility: A comparison between experimentally determined solubility in pvp and prediction derived from solubility in monomer. J. Pharm. Sci. 2015, 104, 2905–2912. [Google Scholar] [CrossRef]
- Gordon, M.; Taylor, J.S. Ideal copolymers and the second-order transitions of synthetic rubbers. I. Non-crystalline copolymers. J. Appl. Chem. 1952, 2, 493–500. [Google Scholar] [CrossRef]
- Roudaut, G.; Simatos, D.; Champion, D.; Contreras-Lopez, E.; Le Meste, M. Molecular mobility around the glass transition temperature: A mini review. Innov. Food Sci. Emerg. Technol. 2004, 5, 127–134. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).