
Development and Bio-Predictive Evaluation of Biopharmaceutical Properties of Sustained-Release Tablets with a Novel GPR40 Agonist for a First-in-Human Clinical Trial

 , , , , , ,
, , , , , , 
Abstract
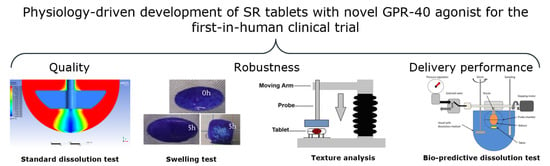
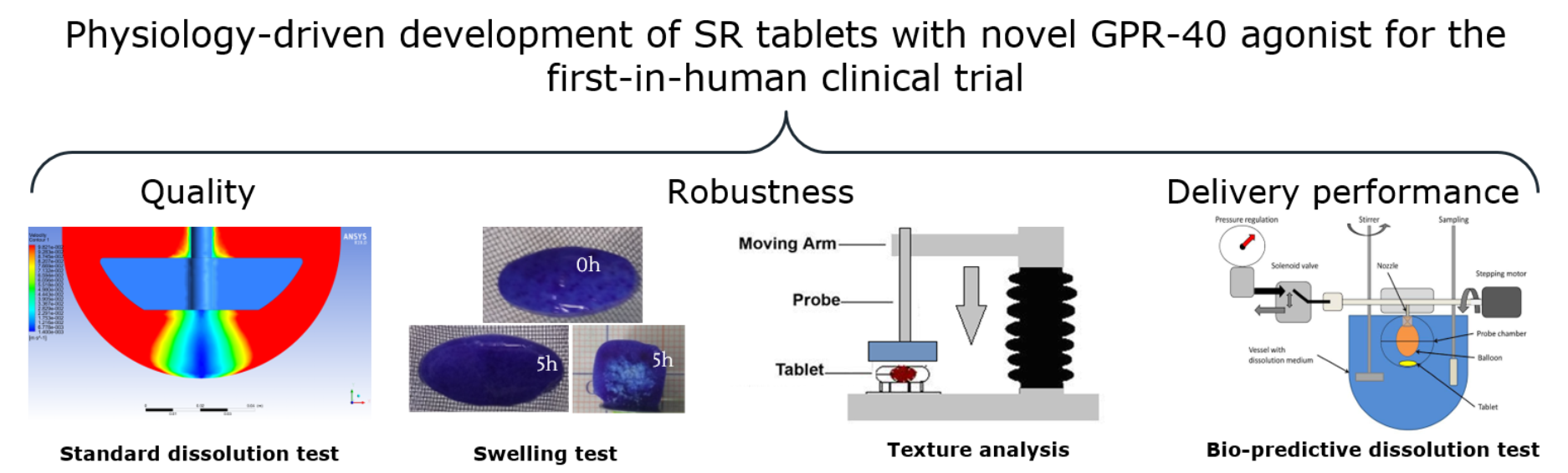
1. Introduction
2. Materials and Methods
2.1. Basic Characteristics of the Formulations
2.2. Analytical Methods and Dissolution Tests Setup
2.2.1. Standard Dissolution Testing
Test Conditions
Determination of the API in Standard Dissolution Media
2.2.2. Prototype Performance Evaluation
Water Sorption and Swelling Kinetics Analysis
Texture Kinetics
2.2.3. Biorelevant Dissolution Tests
Biorelevant Test Protocols
Media
Determination of the API in the Media
2.3. Experiment Workflow
3. Results
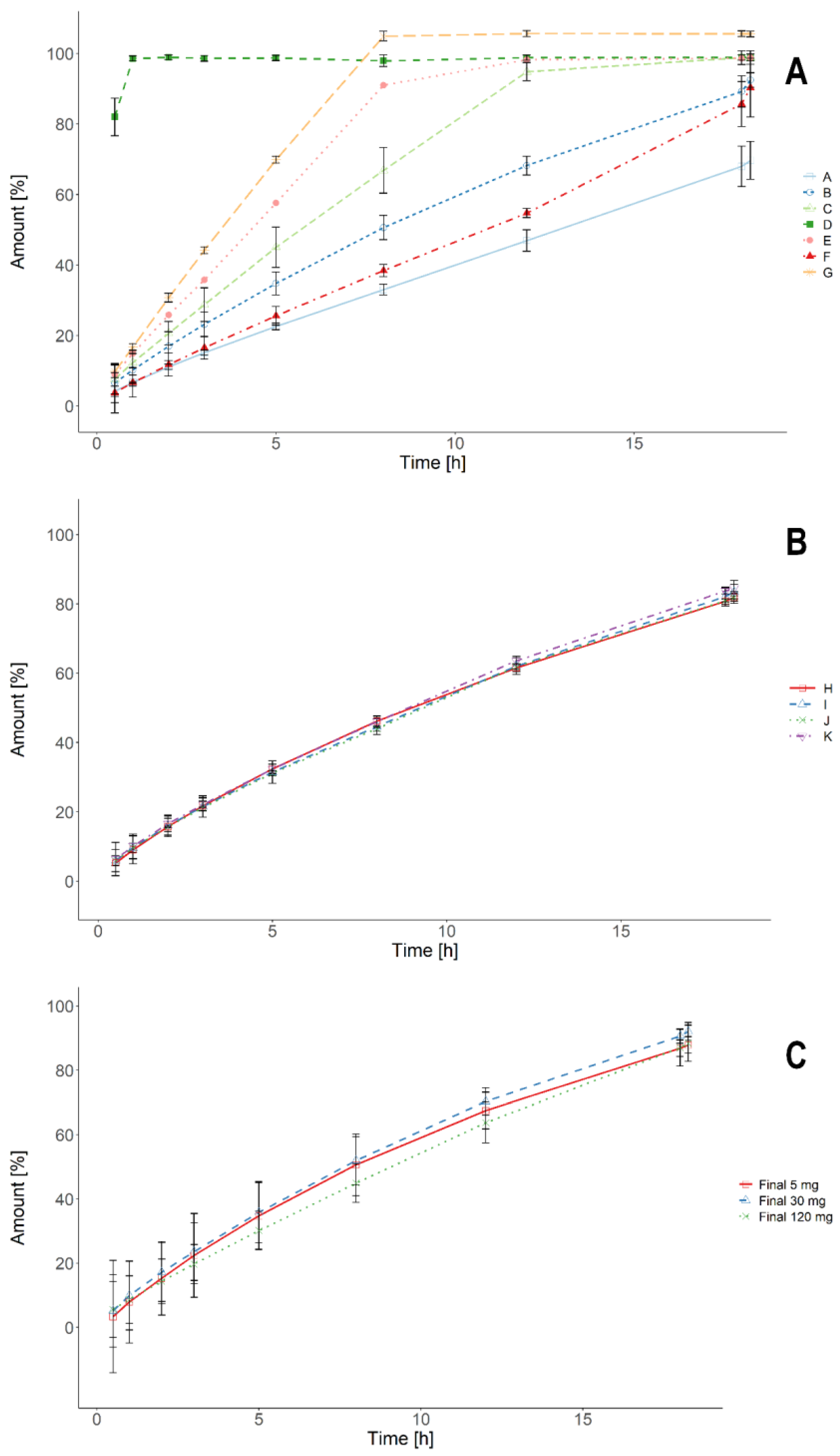
3.1. Standard USP Dissolution Tests
3.2. Preformulation Analysis
3.2.1. Water Sorption Analysis
3.2.2. Swelling Kinetics
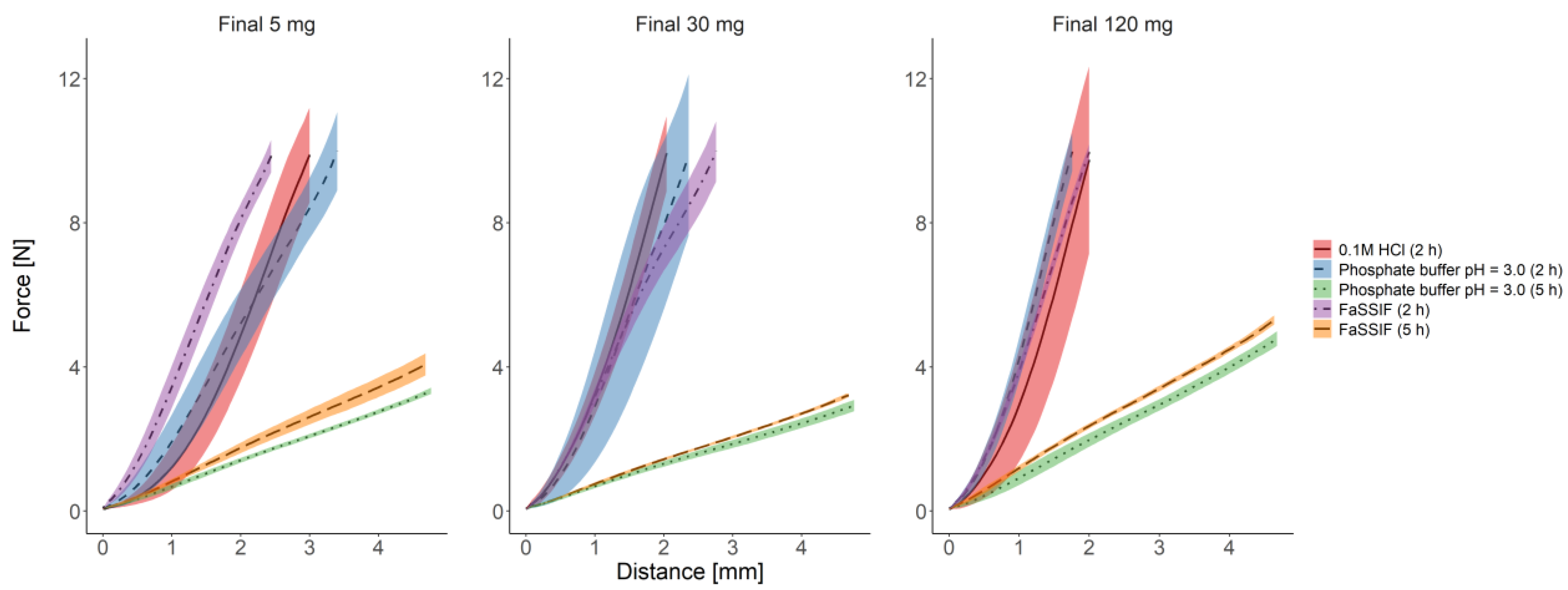
3.2.3. Texture Analysis
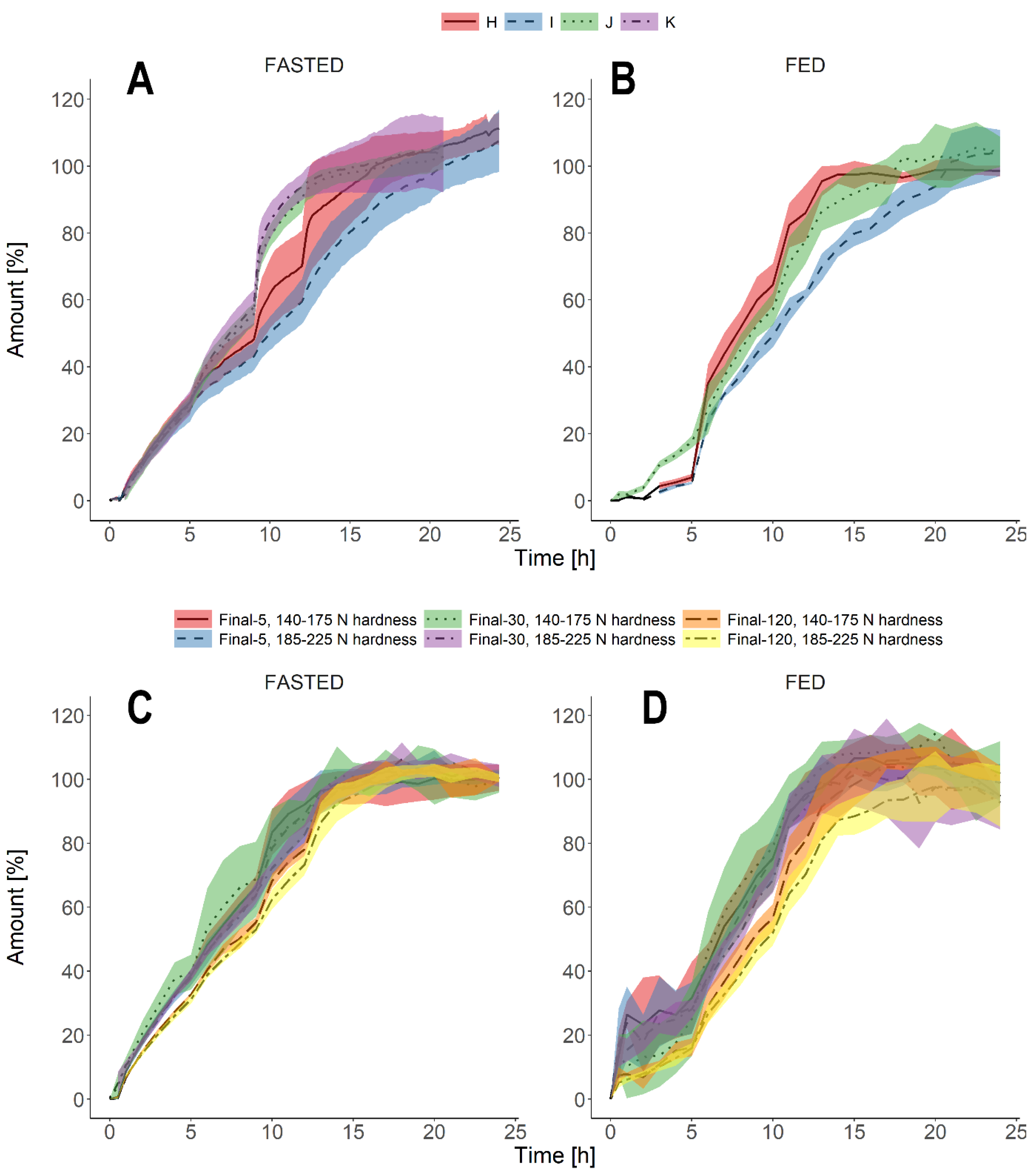
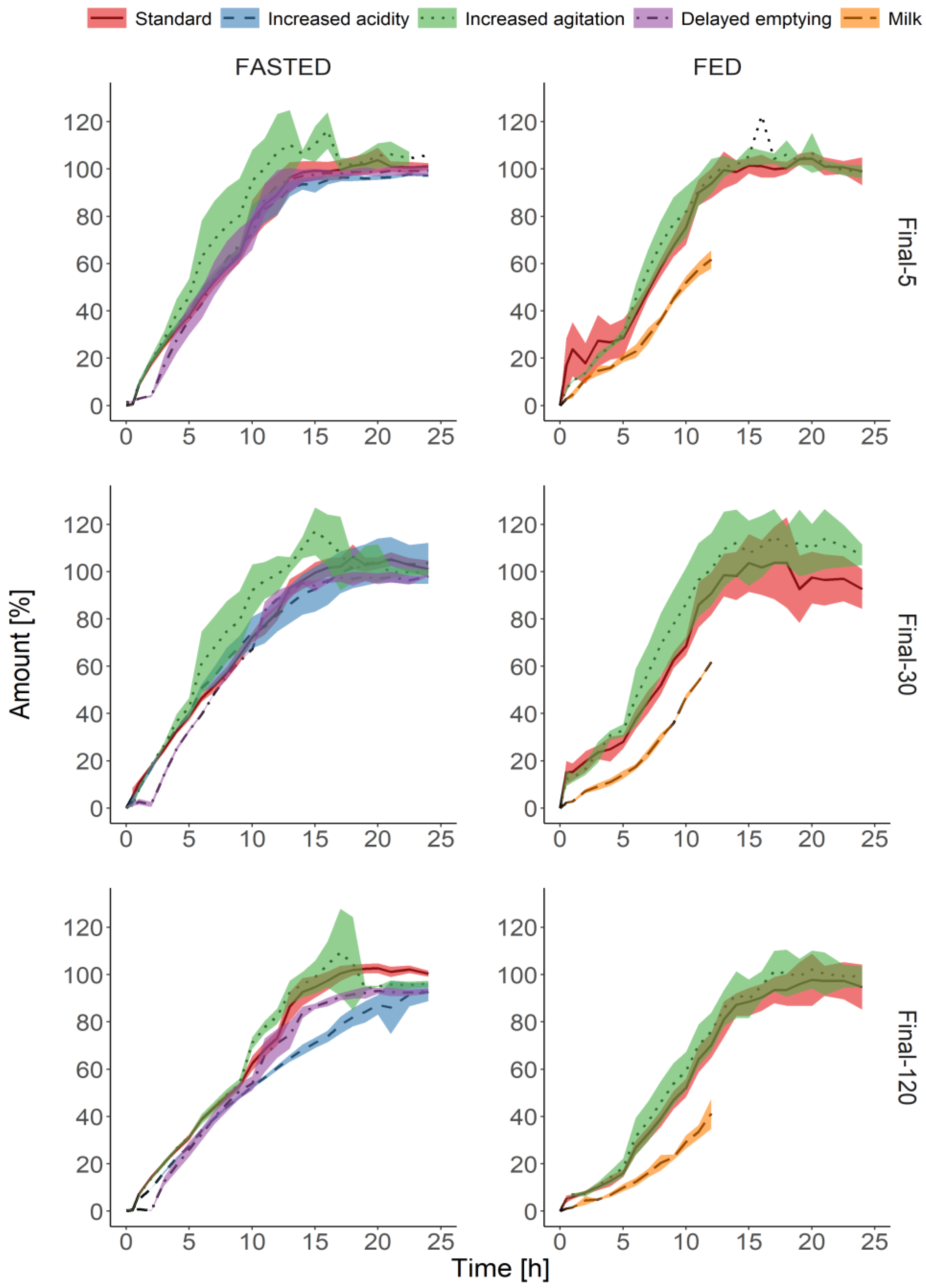
3.3. Biorelevant Dissolution Tests
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Levy, J.; Cobas, R.A.; Gomes, M.B. Assessment of efficacy and tolerability of once- daily extended release metformin in patients with type 2 diabetes mellitus. Diabetol. Metab. Syndr. 2010, 2, 16. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, L.A.; Morris, A.D.; Pearson, E.R. Adherence in patients transferred from immediate release metformin to a sustained release formulation: A population-based study. DiabetesObes. Metab. 2009, 11, 338–342. [Google Scholar] [CrossRef]
- Lu, C.; Chang, C.; Chuang, L.; Wang, C.Y.; Jiang, Y.D.; Wu, H.P. Double-blind, randomized, multicentre study of the efficacy and safety of gliclazide-modified release in the treatment of Chinese type 2 diabetic patients. Diabetes Obes. Metab. 2006, 8, 184–191. [Google Scholar] [CrossRef]
- Wang, L.; Sun, X.; Du, L.; Yuan, Q.; Li, H.; Tian, H.; Li, Y. Effects and patient compliance of sustained-release versus immediate-release glipizides in patients with type 2 diabetes mellitus: A systematic review and meta-analysis. J. Evid. Based Med. 2011, 4, 232–241. [Google Scholar] [CrossRef]
- Van Dijk, F.; Teekamp, N.; Beljaars, L.; Post, E.; Zuidema, J.; Steendam, R.; Kim, Y.O.; Frijlink, H.W.; Schuppan, D.; Poelstra, K.; et al. Pharmacokinetics of a sustained release formulation of PDGF β -receptor directed carrier proteins to target the fi brotic liver. J. Control. Release 2018, 269, 258–265. [Google Scholar] [CrossRef]
- Trofimiuk, M.; Wasilewska, K.; Winnicka, K. How to Modify Drug Release in Paediatric Dosage Forms? Novel Technologies and Modern Approaches with Regard to Children’s Population. Int. J. Mol. Sci. 2019, 20, 3200. [Google Scholar] [CrossRef] [PubMed]
- Keraliya, R.A.; Patel, C.; Patel, P.; Keraliya, V.; Soni, T.G.; Patel, R.C.; Patel, M.M. Osmotic Drug Delivery System as a Part of Modified Release Dosage Form. Int. Sch. Res. Not. 2012, 2012, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Malaterre, V.; Ogorka, J.; Loggia, N.; Gurny, R. Oral osmotically driven systems: 30 Years of development and clinical use. Eur. J. Pharm. Biopharm. 2009, 73, 311–323. [Google Scholar] [CrossRef]
- Maderuelo, C.; Zarzuelo, A.; Lanao, J.M. Critical factors in the release of drugs from sustained release hydrophilic matrices. J. Control. Release 2011, 154, 2–19. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Kao, W.J. Drug release kinetics and transport mechanisms of non-degradable and degradable polymeric delivery systems. Expert Opin. Drug Deliv. 2010, 7, 429–444. [Google Scholar] [CrossRef]
- Garbacz, G.; Wedemeyer, R.S.; Nagel, S.; Giessmann, T.; Mönnikes, H.; Wilson, C.G.; Siegmund, W.; Weitschies, W. Irregular absorption profiles observed from diclofenac extended release tablets can be predicted using a dissolution test apparatus that mimics in vivo physical stresses. Eur. J. Pharm. Biopharm. 2008, 70, 421–428. [Google Scholar] [CrossRef]
- Garbacz, G.; Rappen, G.; Koziolek, M.; Weitschies, W. Dissolution of mesalazine modified release tablets under standard and bio-relevant test conditions. J. Pharm. Pharmacol. 2014, 67, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Schneider, F.; Beeck, R.; Hoppe, M.; Koziolek, M.; Weitschies, W. In vitro simulation of realistic gastric pressure profiles. Eur. J. Pharm. Sci. 2017, 107, 71–77. [Google Scholar] [CrossRef]
- Reynolds, T.D.; Mitchell, S.A.; Balwinski, K.M. Investigation of the Effect of Tablet Surface Area / Volume on Drug Release from Hydroxypropylmethylcellulose Controlled-Release Matrix Tablets. Drug Dev. Ind. Pharm. 2002, 28, 457–466. [Google Scholar] [CrossRef]
- Quality Guidelines: ICH. Available online: https://www.ich.org/products/guidelines/quality/article/quality-guidelines.html (accessed on 20 September 2019).
- Garbacz, G.; Klein, S.; Weitschies, W. Review A biorelevant dissolution stress test device -- background and experiences. Expert Opin. Drug Deliv. 2010, 7, 1251–1261. [Google Scholar] [CrossRef]
- Bland, J.M.; Altman, D.G. Statistical methods for assessing agreement between two methods of clinical measurement. Int. J. Nurs. Stud. 2010, 47, 931–936. [Google Scholar] [CrossRef]
- Colombo, P.; Bettini, R.; Santi, P.; Peppas, N.A. Swellable matrices for controlled drug delivery: Gel-layer behaviour, mechanisms and optimal performance. Pharm. Sci. Technol. Today 2000, 3, 198–204. [Google Scholar] [CrossRef]
- European Medicines Agency, Strategies to Identify and Mitigate Risks for First-in-Human Early Clinical Trials with Investigational Medicinal Products. 2018. Available online: https://www.ema.europa.eu/en/strategies-identify-mitigate-risks-first-human-early-clinical-trials-investigational-medicinal (accessed on 7 March 2020).
- Shah, S.M.; Jain, A.S.; Kaushik, R.; Nagarsenker, M.S.; Nerurkar, M.J. Preclinical Formulations: Insight, Strategies, and Practical Considerations. Aaps Pharmscitech 2014, 15, 1307–1323. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Swift, B.; Mamelok, R.; Pine, S.; Sinclair, J.; Attar, M. Design and Conduct Considerations for First-in-Human Trials. Clin. Transl. Sci. 2019, 12, 6–19. [Google Scholar] [CrossRef]
- Strovel, J.; Sittampalam, S.; Coussens, N.P.; Hughes, M.; Inglese, J.; Kurtz, A.; Andalibi, A.; Patton, L.; Austin, C.; Baltezor, M.; et al. Early Drug Discovery and Development Guidelines: For Academic Researchers, Collaborators, and Start-up Companies. In Assay Guidance Manual; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2004. [Google Scholar]
- Ku, M.S.; Dulin, W. A biopharmaceutical classification-based Right- First-Time formulation approach to reduce human pharmacokinetic variability and project cycle time from First-In-Human to clinical Proof-Of-Concept. Pharm. Dev. Technol. 2012, 17, 285–302. [Google Scholar] [CrossRef]
- Koziolek, M.; Grimm, M.; Garbacz, G.; Kühn, J.P.; Weitschies, W. Intragastric Volume Changes after Intake of a High-Caloric, High-Fat Standard Breakfast in Healthy Human Subjects Investigated by MRI. Mol. Pharm. 2014, 11, 1632–1639. [Google Scholar] [CrossRef]
- Schneider, F.; Grimm, M.; Koziolek, M.; Modeß, C.; Dokter, A.; Roustom, T.; Siegmund, W.; Weitschies, W. Resolving the physiological conditions in bioavailability and bioequivalence studies: Comparison of fasted and fed state. Eur. J. Pharm. Biopharm. 2016, 108, 214–219. [Google Scholar] [CrossRef]
- Cassilly, D.; Kantor, S.; Knight, L.C.; Maurer, A.H.; Fisher, R.S.; Semler, J.; Parkman, H.P. Gastric emptying of a non-digestible solid: Assessment with simultaneous SmartPill pH and pressure capsule antroduodenal manometry, gastric emptying scintigraphy. Neurogastroenterol. Motil. 2008, 20, 311–319. [Google Scholar] [CrossRef]
- Garbacz, G.; Kołodziej, B.; Koziolek, M.; Weitschies, W.; Klein, S. An Automated System for Monitoring and Regulating the pH of Bicarbonate Buffers. Aaps Pharmscitech 2013, 14, 517–522. [Google Scholar] [CrossRef]
- Haznar-Garbacz, D.; Kaminska, E.; Zakowiecki, D.; Lachmann, M.; Kaminski, K.; Garbacz, G.; Dorożyński, P.; Kulinowski, P. Melts of Octaacetyl Sucrose as Oral-Modified Release Dosage Forms for Delivery of Poorly Soluble Compound in Stable Amorphous Form. Aaps Pharmscitech 2018, 19, 951–960. [Google Scholar] [CrossRef]
- Milanowski, B.; Hejduk, A.; Bawiec, M.A.; Jakubowska, E.; Urbańska, A.; Wiśniewska, A.; Garbacz, G.; Lulek, J. Biorelevant In Vitro Release Testing and In Vivo Study of Extended-Release Niacin Hydrophilic Matrix Tablets. Aaps Pharmscitech 2020, 21, 83. [Google Scholar] [CrossRef]
- Drechsler, M.; Garbacz, G.; Thomann, R.; Schubert, R. Development and evaluation of chitosan and chitosan/Kollicoat® Smartseal 30 D film-coated tablets for colon targeting. Eur. J. Pharm. Biopharm. 2014, 88, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Danielak, D.; Milanowski, B.; Wentowski, K.; Nogowska, M.; Kątny, M.; Rogowski, P.; Konwicki, Ł.; Puk, E.; Pieczuro, J.; Bawiec, M.; et al. Physiologically Based Dissolution Testing in a Drug Development Process—A Case Study of a Successful Application in a Bioequivalence Study of Trazodone ER Formulations Under Fed Conditions. Aaps Pharmscitech 2020, 21, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.J.; Hsu, P.I.; Chen, C.H.; Hsiao, M.; Chang, W.C.; Tseng, H.H.; Lin, K.H.; Chuah, S.K.; Chen, H.C. Gastric juice acidity in upper gastrointestinal diseases. World J. Gastroenterol. 2010, 16, 5496–5501. [Google Scholar] [CrossRef] [PubMed]
- Kuo, B.; McCallum, R.W.; Koch, K.L.; Sitrin, M.D.; Wo, J.M.; Chey, W.D.; Hasler, W.L.; Lackner, J.M.; Katz, L.A.; Semler, J.R.; et al. Comparison of gastric emptying of a nondigestible capsule to a radio-labelled meal in healthy and gastroparetic subjects. Aliment. Pharmacol. Ther. 2008, 27, 186–196. [Google Scholar] [CrossRef]
- Hasler, W.L. The use of SmartPill for gastric monitoring. Expert Rev. Gastroenterol. Hepatol. 2014, 8, 587–600. [Google Scholar] [CrossRef]
- Diakidou, A.; Vertzoni, M.; Abrahamsson, B.; Dressman, J.; Reppas, C. Simulation of gastric lipolysis and prediction of felodipine release from a matrix tablet in the fed stomach. Eur. J. Pharm. Sci. 2009, 37, 133–140. [Google Scholar] [CrossRef]
- Jantratid, E.; Janssen, N.; Reppas, C.; Dressman, J.B. Dissolution Media Simulating Conditions in the Proximal Human Gastrointestinal Tract: An Update. Pharm. Res. 2008, 25, 1663–1676. [Google Scholar] [CrossRef] [PubMed]
- Jantratid, E.; Dressman, J. Biorelevant Dissolution Media Simulating the Proximal Human Gastrointestinal Tract: An Update. Dissolution Technol. 2009, 16, 21–25. [Google Scholar] [CrossRef]
- Parojčić, J.; Vasiljević, D.; Ibrić, S.; Djurić, Z. Tablet disintegration and drug dissolution in viscous media: Paracetamol IR tablets. Int. J. Pharm. 2008, 355, 93–99. [Google Scholar] [CrossRef]
- Williams, H.D.; Nott, K.P.; Barrett, D.A.; Ward, R.; Hardy, I.J.; Melia, C.D. Drug Release from HPMC Matrices in Milk and Fat-Rich Emulsions. J. Pharm. Sci. 2011, 100, 4823–4835. [Google Scholar] [CrossRef] [PubMed]
- Abrahamsson, B.; Albery, T.; Eriksson, A.; Gustafsson, I.; Sjöberg, M. Food effects on tablet disintegration. Eur. J. Pharm. Sci. 2004, 22, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Galia, E.; Nicolaides, E.; Hörter, D.; Löbenberg, R.; Reppas, C.; Dressman, J.B. Evaluation of various dissolution media for predicting in vivo performance of class I and II drugs. Pharm. Res. 1998, 15, 698–705. [Google Scholar] [CrossRef]
- Macheras, P.E.; Koupparis, M.A.; Antimisiaris, S.G. Drug binding and solubility in milk. Pharm. Res. 1990, 7, 537–541. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Program | Intragastric Conditions | Gastric Emptying | Intestinal Passage | Ileocaecal Passage | Colonic Passage | |
|---|---|---|---|---|---|---|
| Simulated fasted conditions | ||||||
| Program 1 Regular gastric emptying | Time points Pressure Mech. agitation | 0–0.5 h None No agitation | 0.5 h 3 × 300 mbar 60 s × 100 rpm | 1, 2 and 3 h 1 × 150 mbar 30 s × 50 rpm | 5 h 2 × 200 mbar 30 s × 50 rpm | 9, 12, 16 and 20 h 2 × 200 mbar 30 s × 50 rpm |
| Medium | USP SGF sine pepsin, pH = 1.8 | 50 mM potassium phosphate buffer pH = 6.8 + 2.24 g/L FaSSIF/FeSSIF/FaSSGF | ||||
| Program 2 Acidified gastric environment | Time points Pressure Mech. agitation | 0–0.5 h None No agitation | 0.5 h 3 × 300 mbar 60 s × 100 rpm | 1, 2 and 3 h 1 × 150 mbar 30 s × 50 rpm | 5 h 2 × 200 mbar 30 s × 50 rpm | 9, 12, 16 and 20 h 2 × 200 mbar 30 s × 50 rpm |
| Medium | USP SGF sine pepsin, pH = 1.2 | 50 mM potassium phosphate buffer pH = 6.8 + 2.24 g/L FaSSIF/FeSSIF/FaSSGF | ||||
| Program 3 Increased mechanical agitation | Time points Pressure Mech. agitation | 0–0.5 h None No agitation | 0.5 h 3 × 350 mbar 80 s × 100 rpm | 1, 2 and 3 h 1 × 300 mbar 30 s × 50 rpm | 5 h 2 × 300 mbar 30 s × 50 rpm | 9, 12, 16 and 20 h 2 × 300 mbar 30 s × 50 rpm |
| Medium | USP SGF sine pepsin, pH = 1.8 | 50 mM potassium phosphate buffer pH = 6.8 + 2.24 g/L FaSSIF/FeSSIF/FaSSGF | ||||
| Program 4 Delayed gastric emptying | Time points Pressure Mech. agitation | 0–2 h None No agitation | 2 h 3 × 300 mbar 60 s × 100 rpm | 2.5, 3.5, and 4.5 h 1 × 150 mbar 30 s × 50 rpm | 6.5 h 2 × 200 mbar 30 s × 50 rpm | 11.5, 13.5, 17.5, and 21.5 h 2 × 200 mbar 30 s × 50 rpm |
| Medium | USP SGF sine pepsin, pH = 1.8 | 50 mM potassium phosphate buffer pH = 6.8 + 2.24 g/L FaSSIF/FeSSIF/FaSSGF | ||||
| Simulated fed conditions | ||||||
| Program 5 Regular gastric emptying | Time points Pressure Mech. agitation | 2, 3, and 4 h 2 × 200 mbar 30 s × 50 rpm | 5 h 3 × 300 mbar 60 s × 100 rpm | 6, 7, and 8 h 2 × 150 mbar 30 s × 50 rpm | 10 h 2 × 200 mbar 60 s × 50 rpm | 12, 16, and 20 h 3 × 200 mbar 30 s × 50 rpm |
| Medium | 50 mM potassium phosphate buffer 0–0.5 h: pH = 4.5; 0.5–1 h: pH = 3.5; 1–2 h: pH = 3.0; 2–5 h: pH = 2.0 | Addition of 40 mL FaSSIF/FeSSIF/FaSSGF dissolved in 50 mM phosphate buffer up to the final concentration of 11.2 g/L, and adjustment of pH to 6.5 | ||||
| Program 6 Regular gastric emptying in milk with addition of digestive enzymes | Time points Pressure Mech. agitation | 2, 3, and 4 h 2 × 200 mbar 30 s × 50 rpm | 5 h 3 × 300 mbar 60 s × 100 rpm | 6, 7, and 8 h 2 × 150 mbar 30 s × 50 rpm | 10 h 2 × 200 mbar 60 s × 50 rpm | 12, 16, and 20 h 3 × 200 mbar 30 s × 50 rpm |
| Medium | Whole milk (3.5% fat) 0–0.5 h: pH = 4.5; 0.5–1 h: pH = 3.5; 1–2 h: pH = 3.0; 2–5 h: pH = 2.0 Addition of 1 mL pepsin solution after 0, 0.5, 1 and 2 h up to the final concentration of 2.0 g/L | Addition of 40 mL of aqueous solution containing FaSSIF/FeSSIF/FaSSGF and pancreatin. Final concentration of FaSSIF/FeSSIF/FaSSGF was 11.2 g/L, and final concentration of pancreatin was 4.57 g/L. Adjustment of pH to 6.5 | ||||
| Program 7 Increased gastric mechanical agitation | Time points Pressure Mech. agitation | 2, 3, and 4 h 4 × 300 mbar 30 s × 50 rpm | 5 h 4 × 350 mbar 80 s × 100 rpm | 6, 7, and 8 h 2 × 150 mbar 30 s × 50 rpm | 10 h 2 × 200 mbar 60 s × 50 rpm | 12, 16, and 20 h 3 × 200 mbar 30 s × 50 rpm |
| Medium | 50 mM potassium phosphate buffer 0–0.5 h: pH = 4.5; 0.5–1 h: pH = 3.5; 1–2 h: pH = 3.0; 2–5 h: pH = 2.0 | Addition of 40 mL of FaSSIF/FeSSIF/FaSSGF dissolved in 50 mM phosphate buffer up to the final concentration of 11.2 g/L, and adjustment of pH to 6.5 | ||||
| Medium | Formulation | ||||
|---|---|---|---|---|---|
| J | Final-5 | Final-30 | Final-120 | ||
| Water sorption [%] (n = 3) | |||||
| 0.1% HCl (2 h) | 97.26 ± 3.82 | 94.91 ± 2.44 | 95.58 ± 1.24 | 95.80 ± 0.59 | |
| PP pH = 3.0 + 0.1% Tween (5 h) | 205.79 ± 4.03 | 169.49 ± 4.55 | 179.44 ± 2.23 | 173.19 ± 3.58 | |
| FaSSIF (5 h) | 199.19 ± 2.48 | 168.79 ± 0.62 | 161.59 ± 4.18 | 154.93 ± 0.16 | |
| Swelling kinetics [%] (n = 3) | |||||
| 0.1% HCl (2 h) | Height | 184.33 ± 17.42 | 168.55 ± 5.30 | 179.94 ± 14.20 | 174.07 ± 16.38 |
| Width | 152.60 ± 16.07 | 125.24 ± 5.82 | 133.06 ± 13.37 | 118.93 ± 2.95 | |
| Dry area | 26.68 ± 2.15 | 50.66 ± 3.25 | 50.50 ± 6.26 | 60.12 ± 4.61 | |
| PP pH = 3.0 + 0.1% Tween (5 h) | Height | 204.44 ± 8.27 | 178.92 ± 17.64 | 214.64 ± 9.84 | 188.21 ± 13.48 |
| Width | 145.01 ± 3.81 | 117.51 ± 15.78 | 127.09 ± 5.83 | 119.43 ± 10.72 | |
| Dry area | 33.50 ± 2.10 | 38.91 ± 7.18 | 43.14 ± 10.71 | 37.27 ± 10.41 | |
| FaSSIF (5 h) | Height | 190.14 ± 2.89 | 179.43 ± 6.72 | 191.85 ± 11.31 | 179.43 ± 8.07 |
| Width | 131.55 ± 3.45 | 117.98 ± 5.68 | 118.35 ± 14.94 | 118.42 ± 13.03 | |
| Dry area | 26.65 ± 2.34 | 45.27 ± 9.36 | 42.84 ± 2.81 | 38.53 ± 2.27 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Juszczyk, E.; Kisło, K.; Żero, P.; Tratkiewicz, E.; Wieczorek, M.; Paszkowska, J.; Banach, G.; Wiater, M.; Hoc, D.; Garbacz, G.; et al. Development and Bio-Predictive Evaluation of Biopharmaceutical Properties of Sustained-Release Tablets with a Novel GPR40 Agonist for a First-in-Human Clinical Trial. Pharmaceutics 2021, 13, 804. https://doi.org/10.3390/pharmaceutics13060804
Juszczyk E, Kisło K, Żero P, Tratkiewicz E, Wieczorek M, Paszkowska J, Banach G, Wiater M, Hoc D, Garbacz G, et al. Development and Bio-Predictive Evaluation of Biopharmaceutical Properties of Sustained-Release Tablets with a Novel GPR40 Agonist for a First-in-Human Clinical Trial. Pharmaceutics. 2021; 13(6):804. https://doi.org/10.3390/pharmaceutics13060804
Chicago/Turabian StyleJuszczyk, Ewelina, Kamil Kisło, Paweł Żero, Ewa Tratkiewicz, Maciej Wieczorek, Jadwiga Paszkowska, Grzegorz Banach, Marcela Wiater, Dagmara Hoc, Grzegorz Garbacz, and et al. 2021. "Development and Bio-Predictive Evaluation of Biopharmaceutical Properties of Sustained-Release Tablets with a Novel GPR40 Agonist for a First-in-Human Clinical Trial" Pharmaceutics 13, no. 6: 804. https://doi.org/10.3390/pharmaceutics13060804
APA StyleJuszczyk, E., Kisło, K., Żero, P., Tratkiewicz, E., Wieczorek, M., Paszkowska, J., Banach, G., Wiater, M., Hoc, D., Garbacz, G., Sczodrok, J., & Danielak, D. (2021). Development and Bio-Predictive Evaluation of Biopharmaceutical Properties of Sustained-Release Tablets with a Novel GPR40 Agonist for a First-in-Human Clinical Trial. Pharmaceutics, 13(6), 804. https://doi.org/10.3390/pharmaceutics13060804