
Mutual Effects of Hydrogen Bonding and Polymer Hydrophobicity on Ibuprofen Crystal Inhibition in Solid Dispersions with Poly(N-vinyl pyrrolidone) and Poly(2-oxazolines)



Abstract
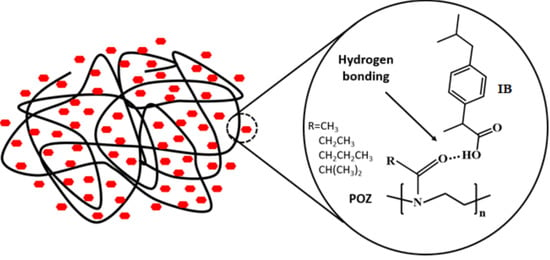
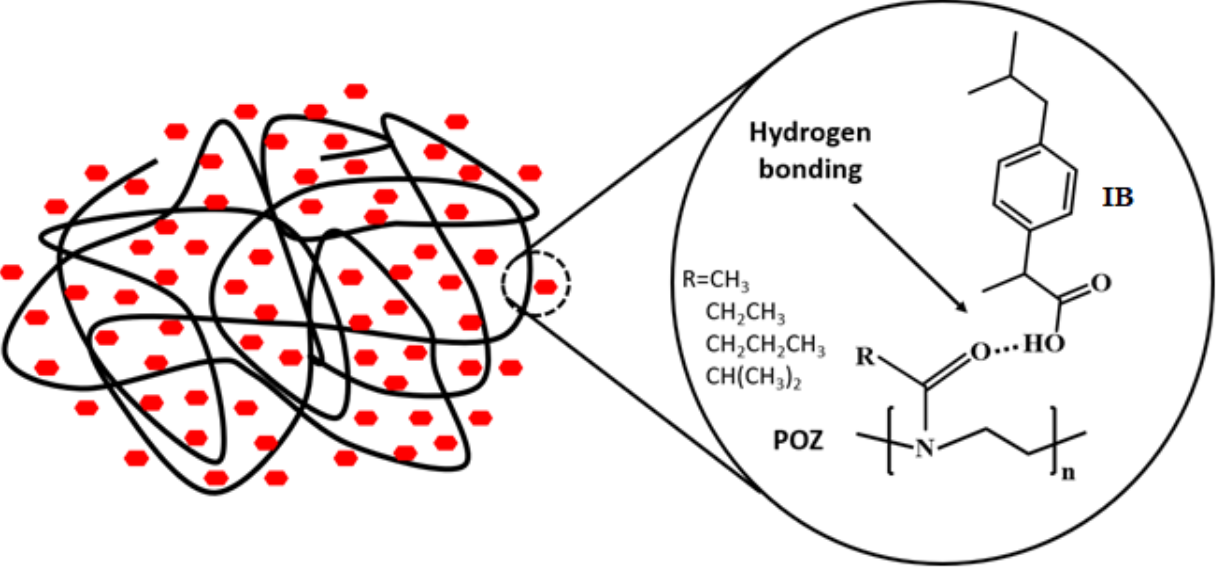
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Polymer–IB Solid Dispersions
2.3. Characterization of Solid Dispersions
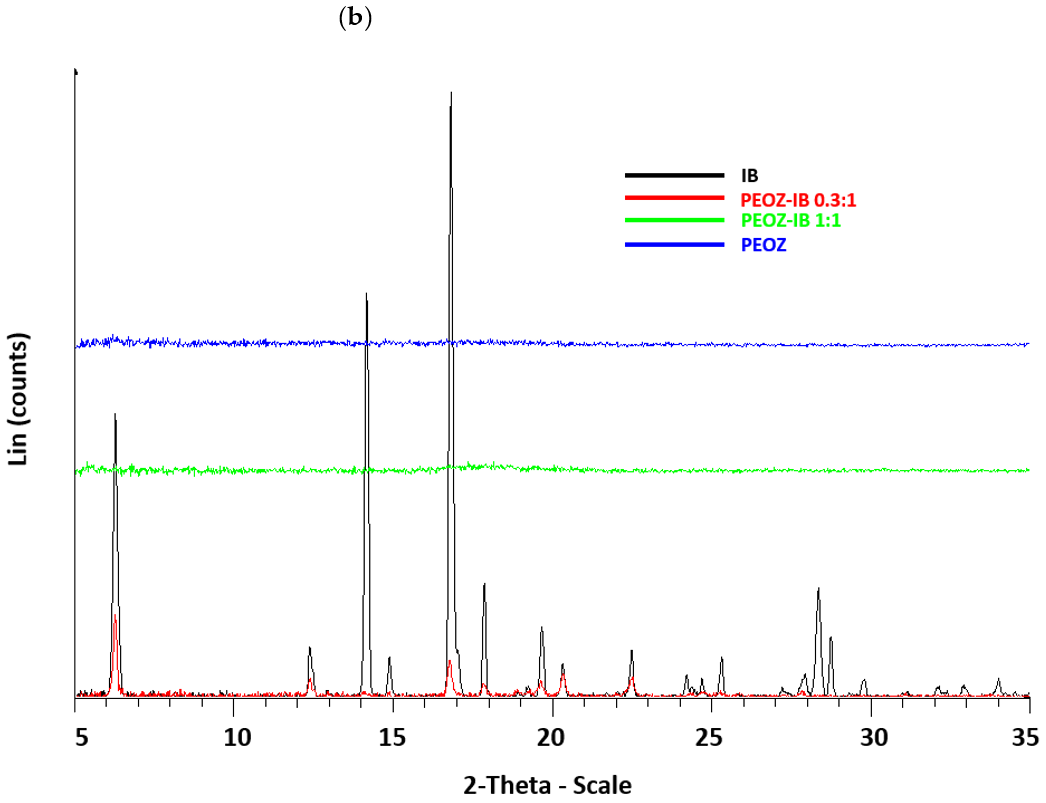
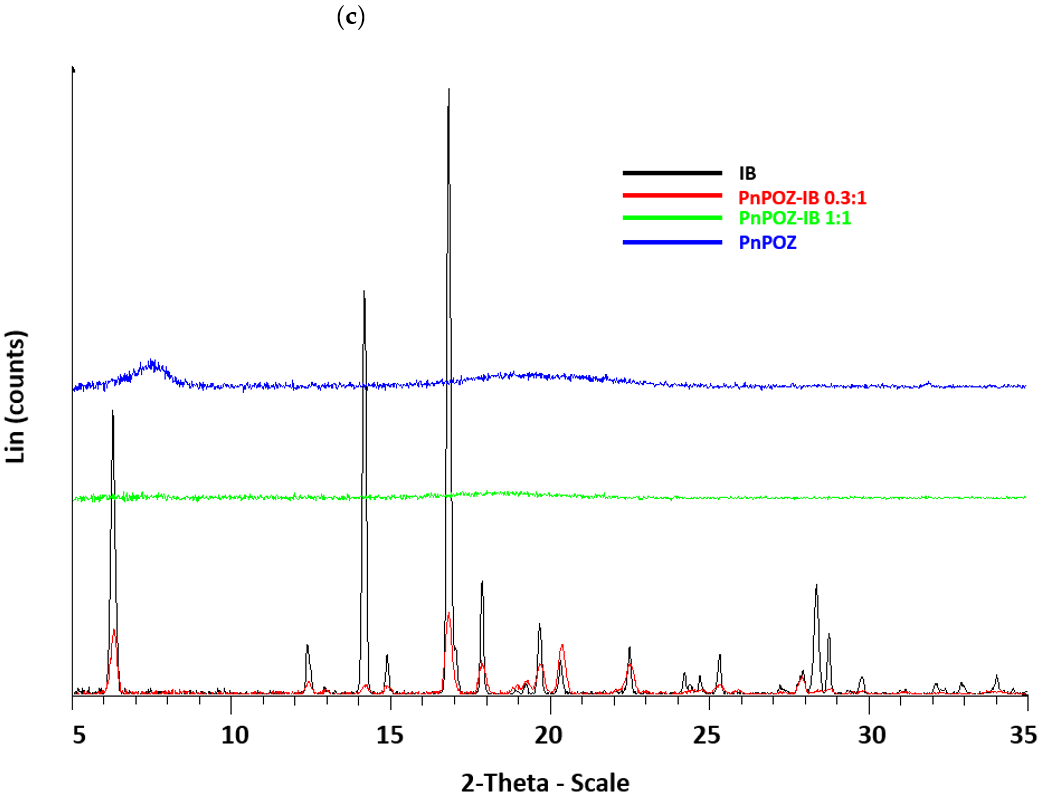
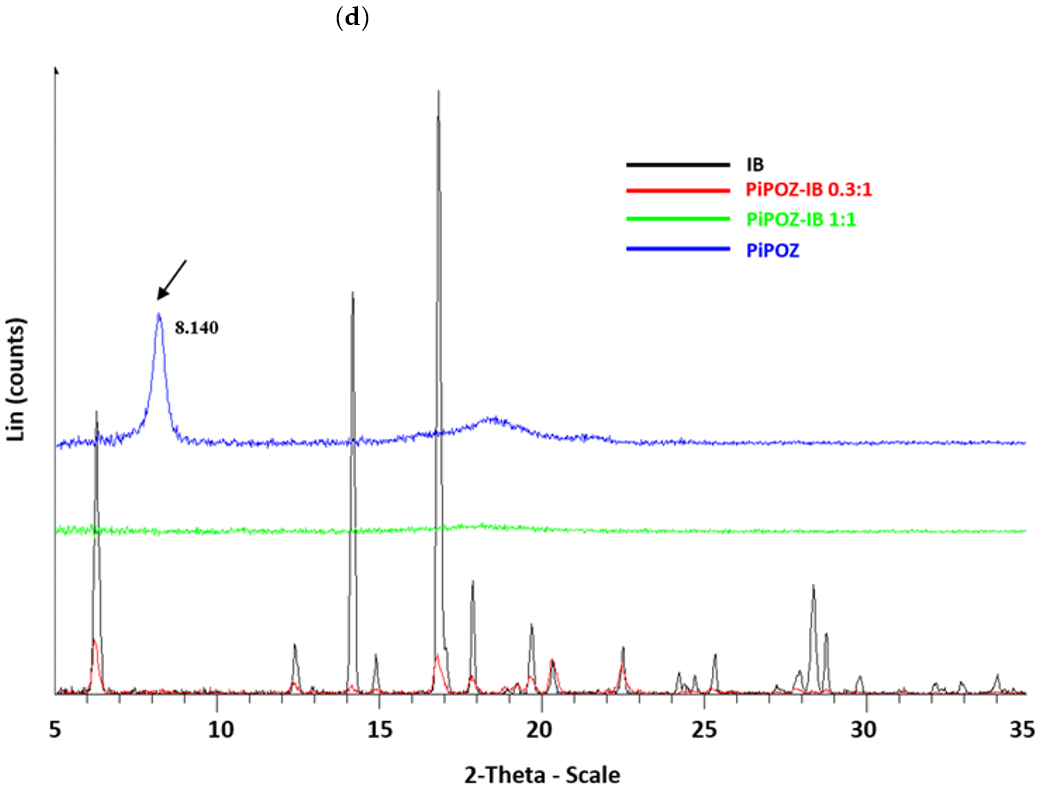
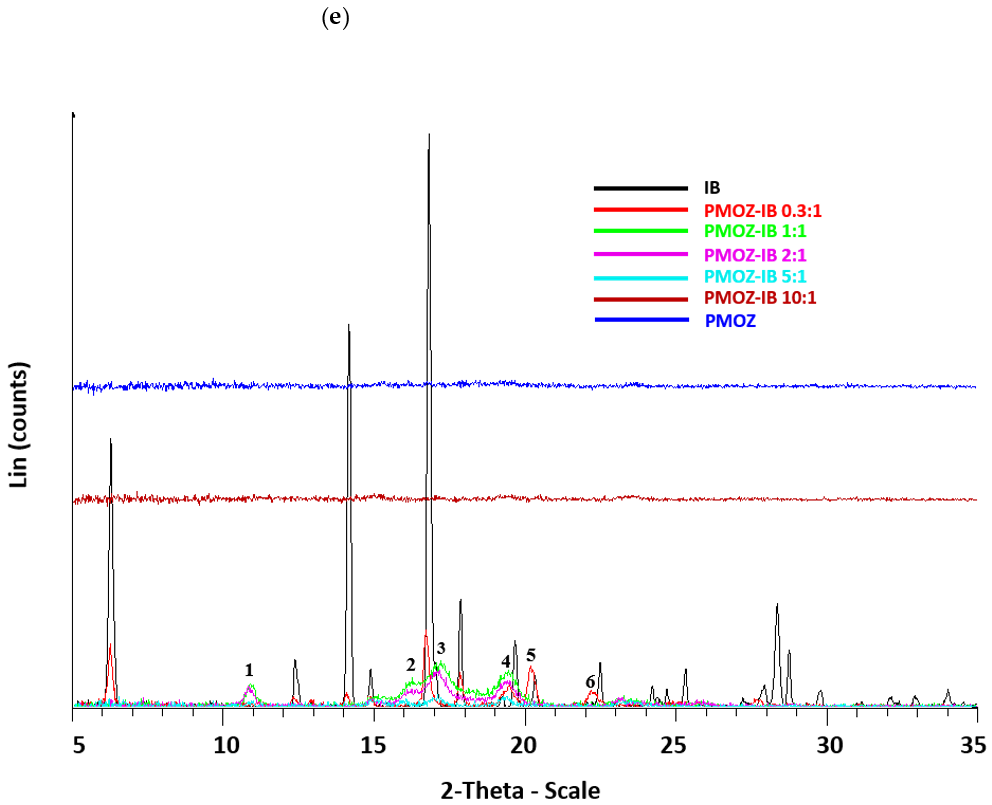
2.3.1. Powder X-ray Diffractometry (PXRD)
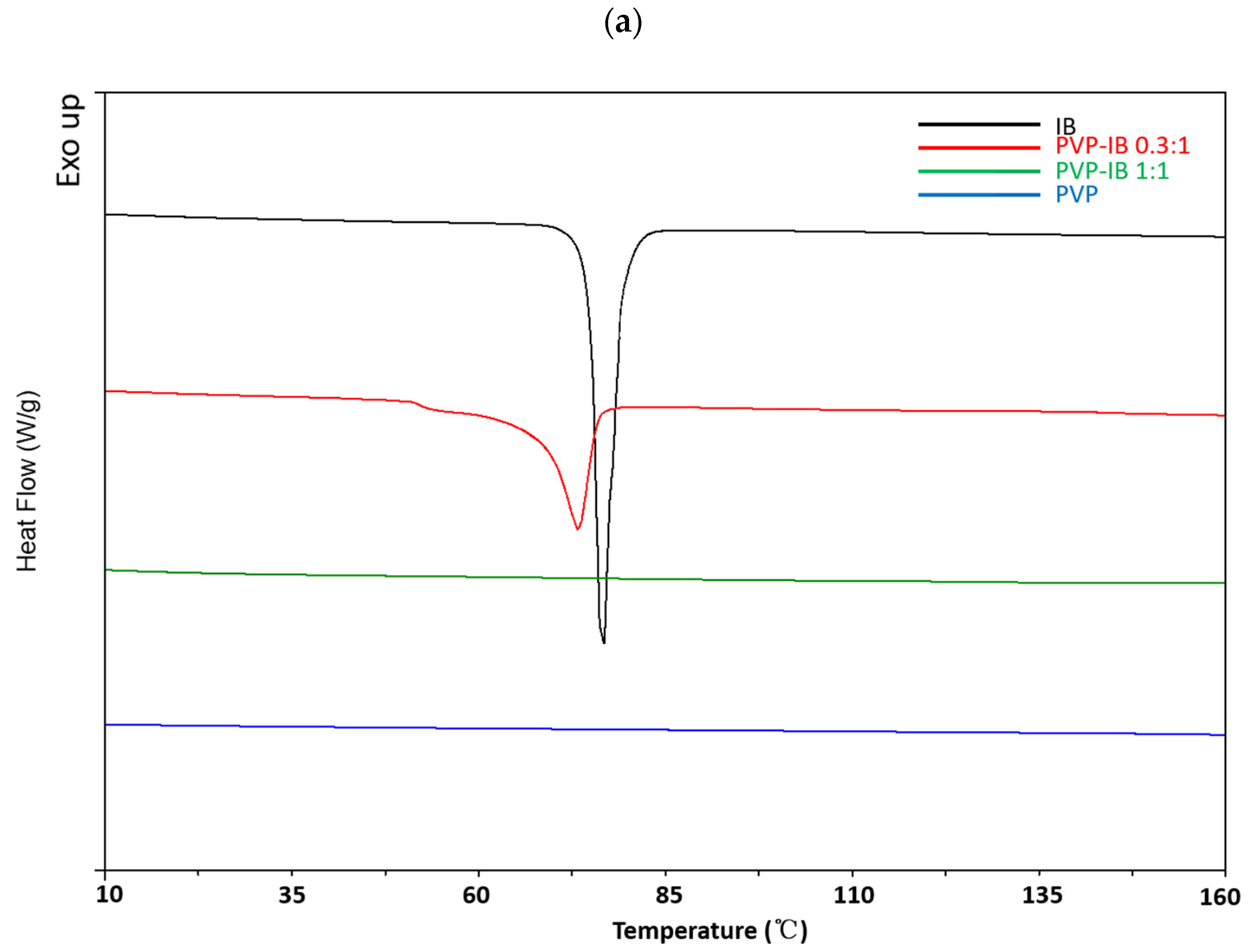
2.3.2. Differential Scanning Calorimetry (DSC)
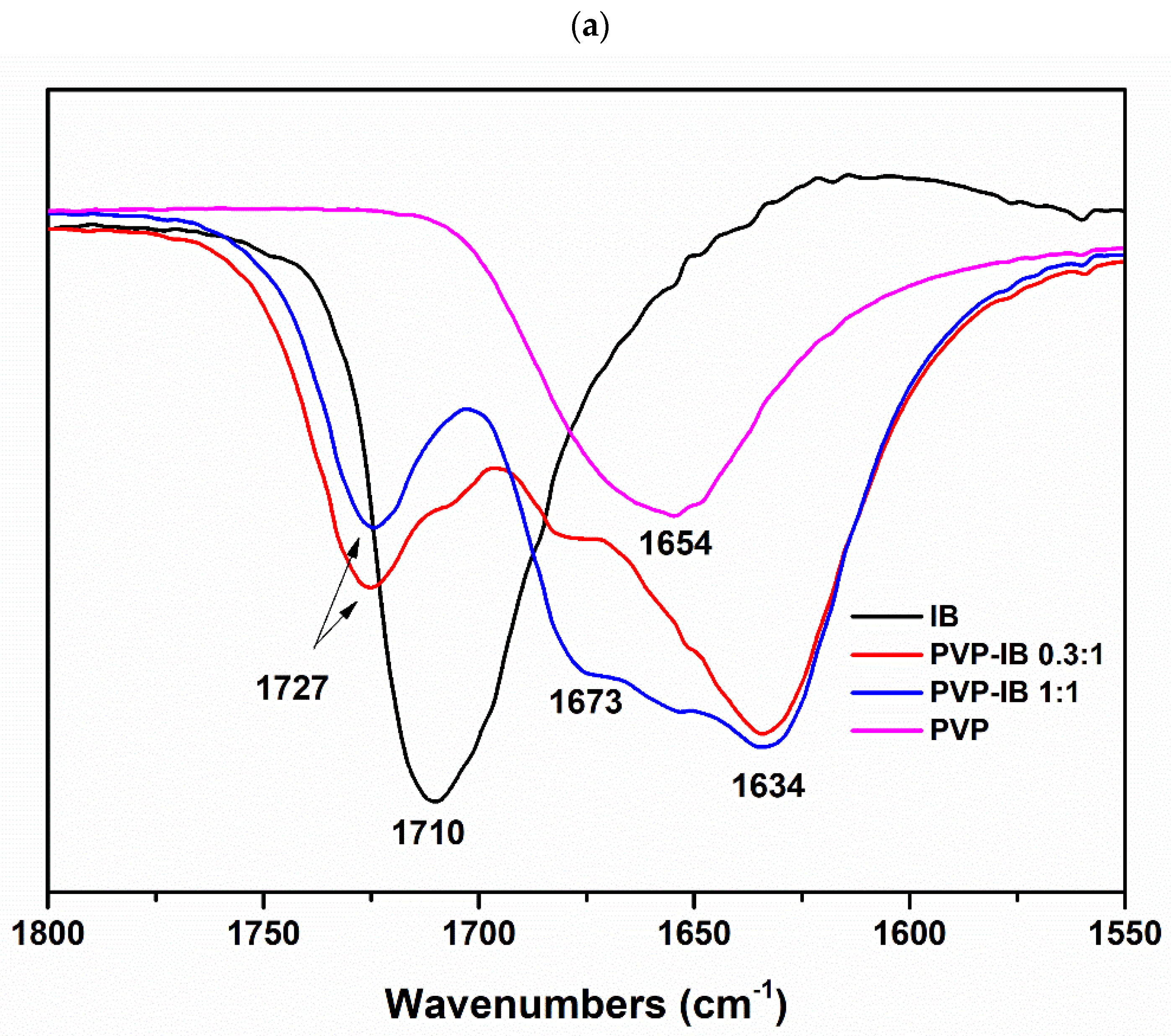
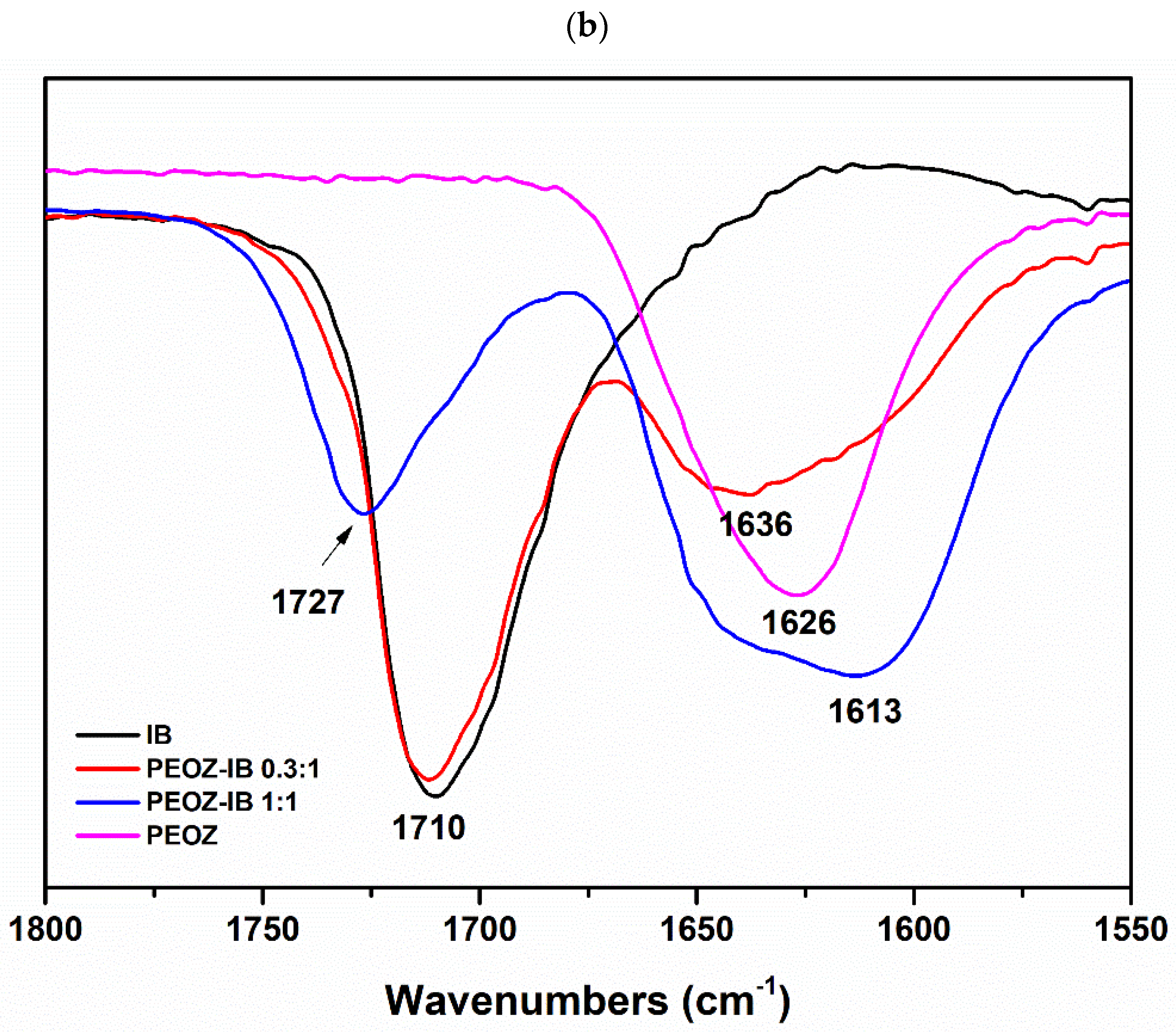
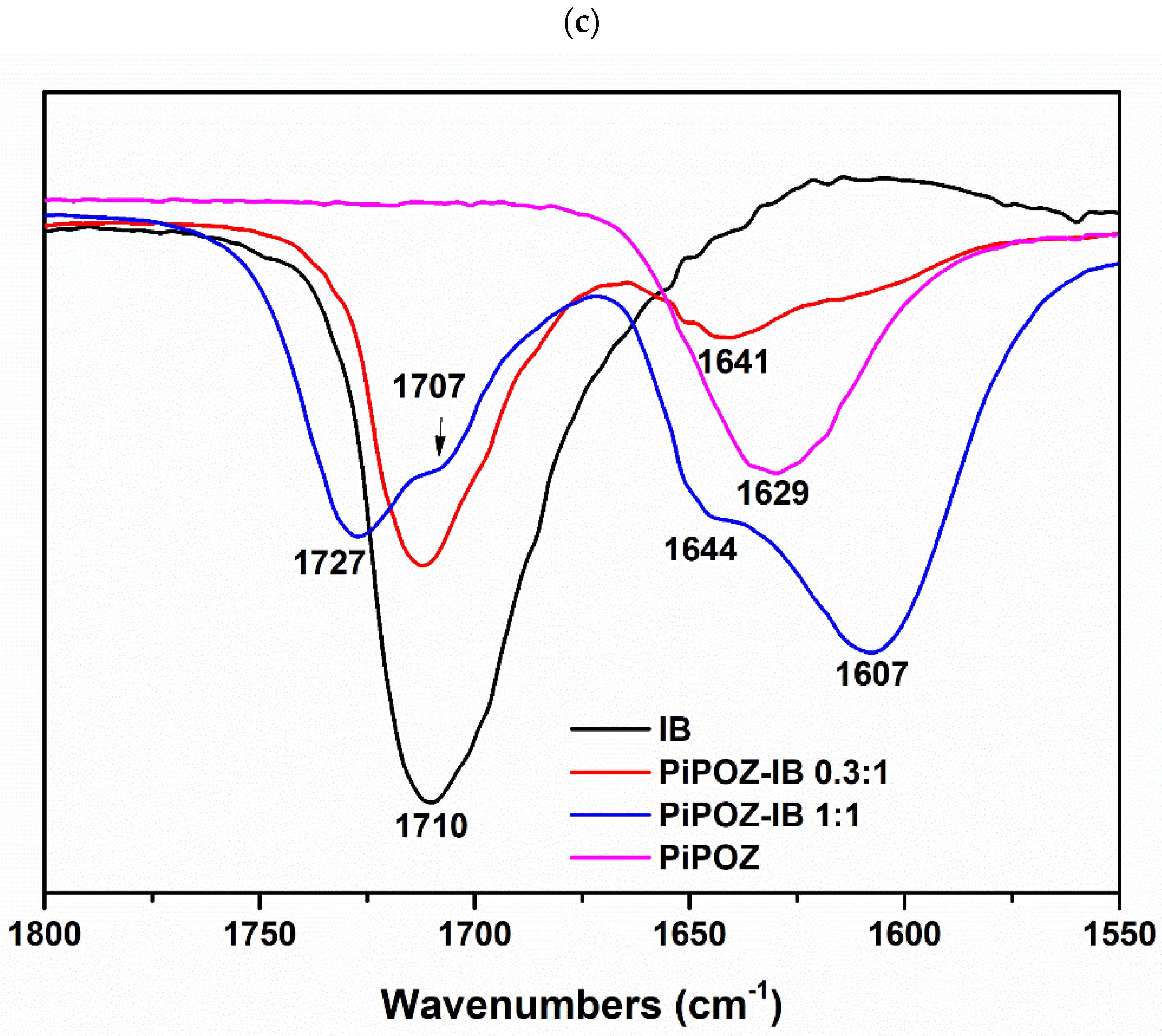
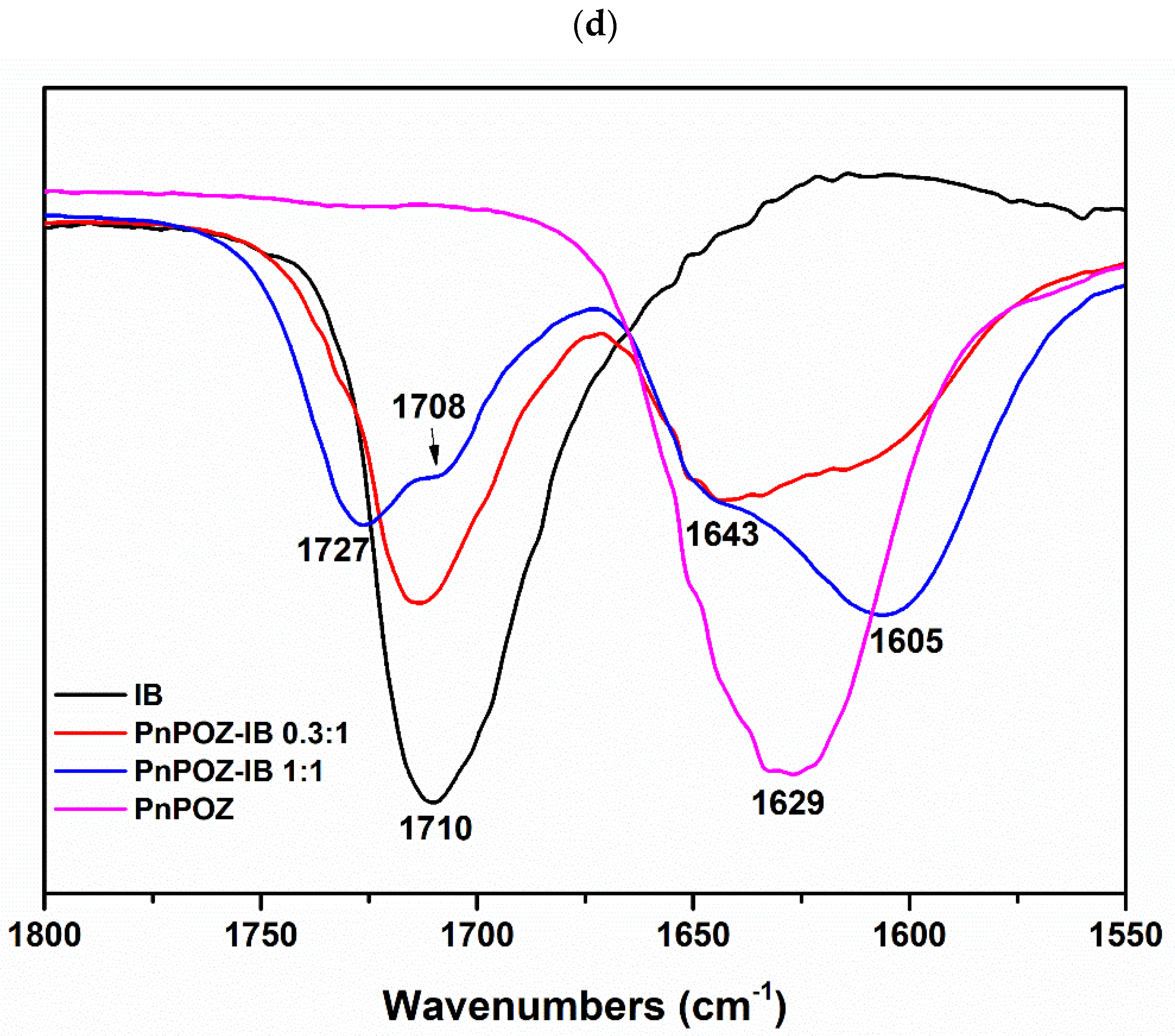
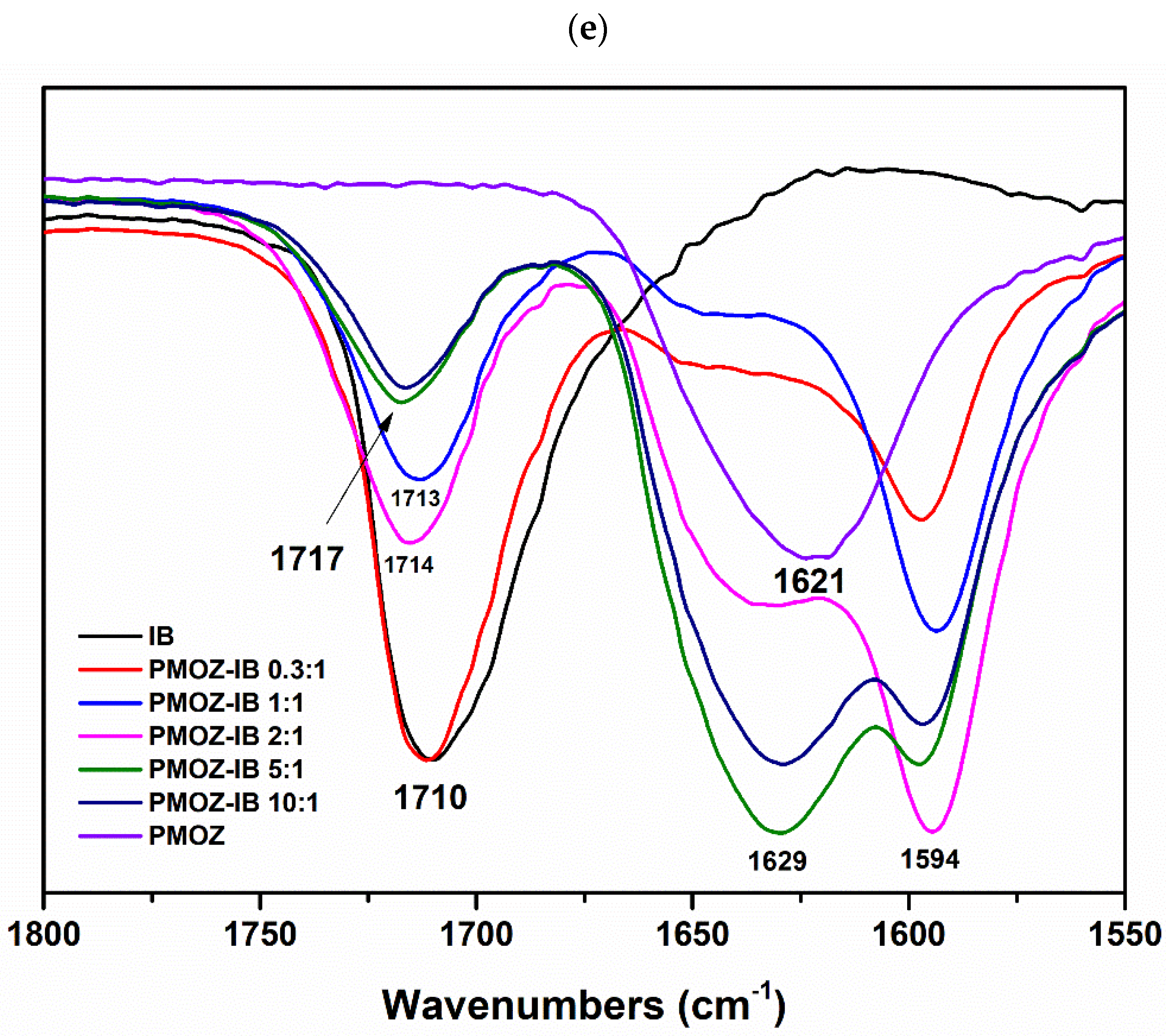
2.3.3. Fourier Transform Infrared (FTIR) Spectroscopy
2.4. In Vitro Dissolution Studies
2.5. Statistical Analysis
3. Results and Discussion
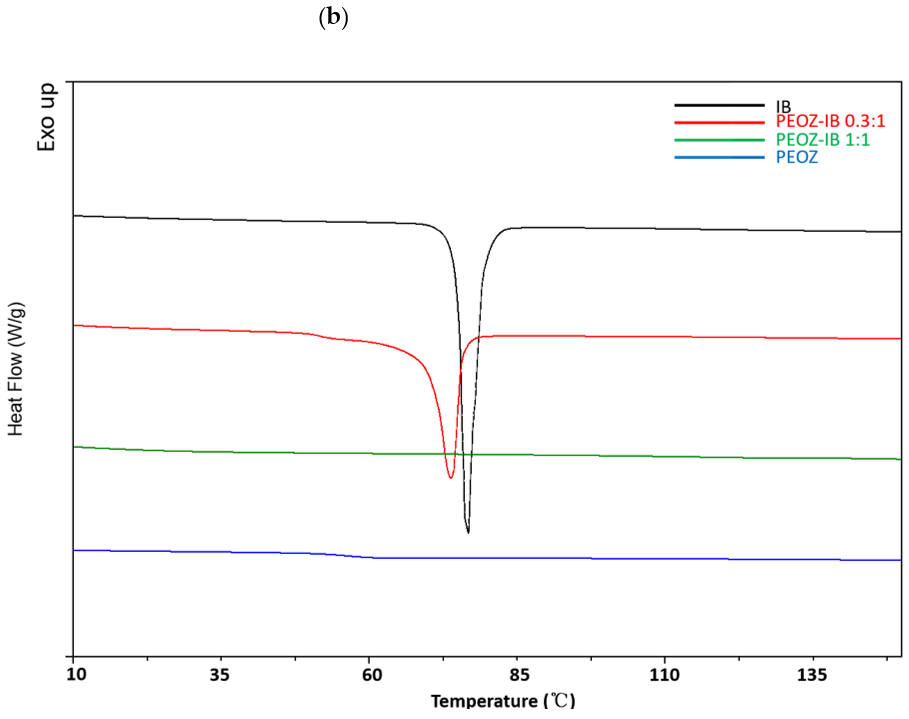
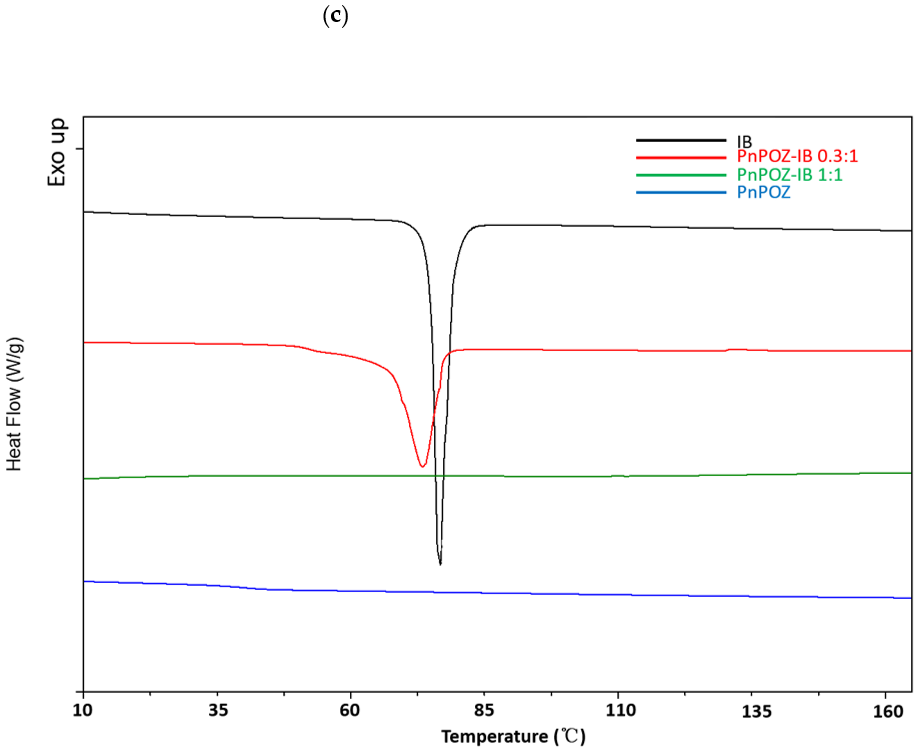
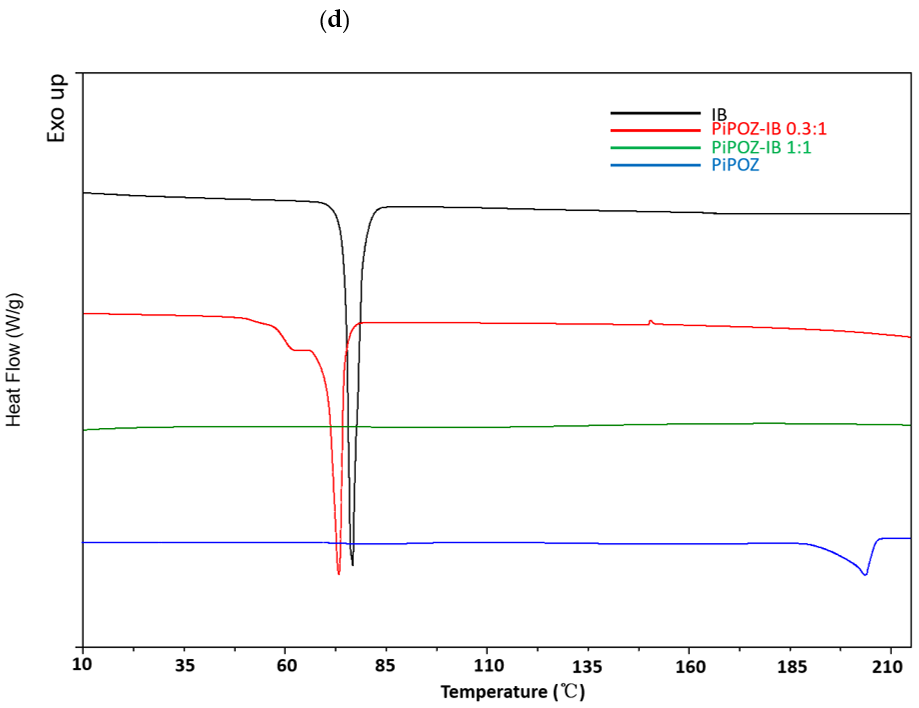
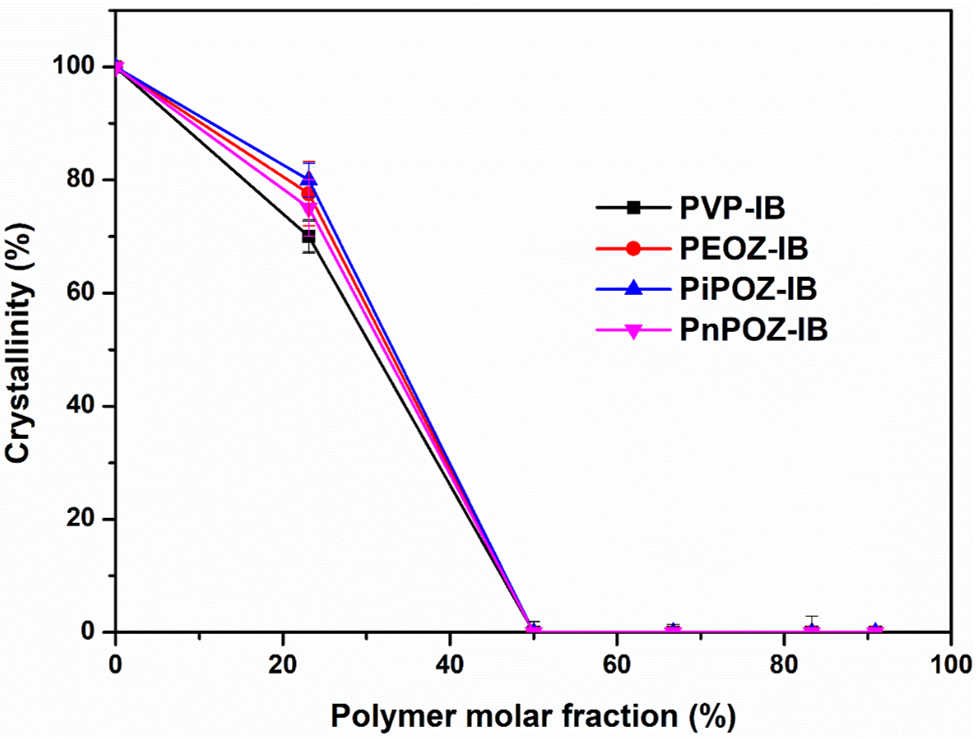
3.1. Preparation and Characterization of Solid Dispersions
3.2. Theoretical Evaluation of Drug-Polymer Miscibility
3.2.1. Solubility Parameters
3.2.2. Flory–Huggins Interaction Parameter
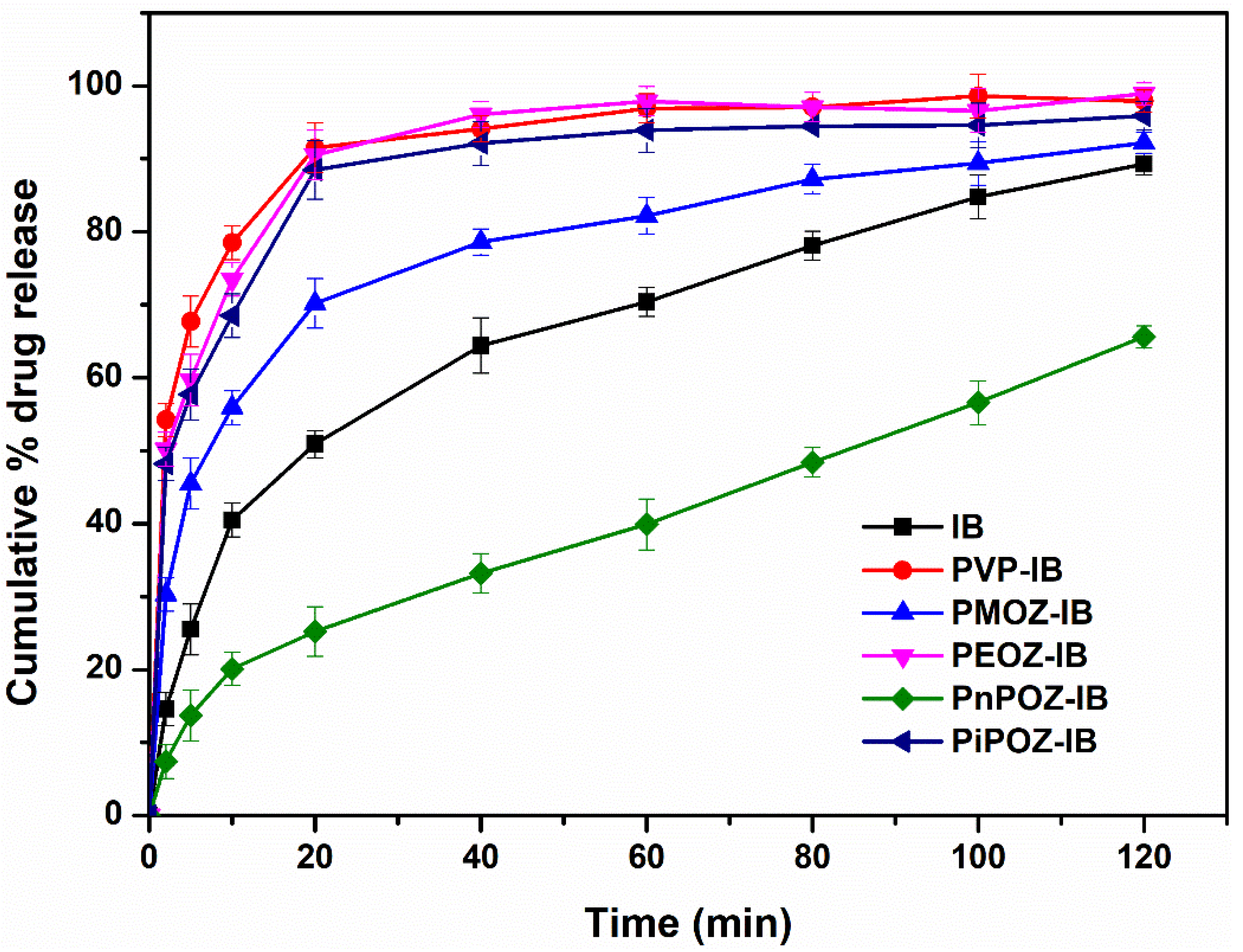
3.3. In Vitro Dissolution Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baghel, S.; Cathcart, H.; O’Reilly, N.J. Polymeric amorphous solid dispersions: A review of amorphization, crystallization, stabilization, solid-state characterization, and aqueous solubilization of biopharmaceutical classification system class ii drugs. J. Pharm. Sci. 2016, 105, 2527–2544. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.T.; Lee, J.Y.; Lee, M.Y.; Song, C.K.; Choi, J.H.; Kim, D.D. Solid dispersions as a drug delivery system. J. Pharm. Investig. 2011, 41, 125–142. [Google Scholar] [CrossRef]
- Tekade, A.R.; Yadav, J.N. A review on solid dispersion and carriers used therein for solubility enhancement of poorly water soluble drugs. Adv. Pharm. Bull. 2020, 10, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Biswal, S.; Sahoo, J.; Murthy, P.N. Physicochemical properties of solid dispersions of gliclazide in polyvinylpyrrolidone k90. AAPS Pharm. Sci. Tech 2009, 10, 329–334. [Google Scholar] [CrossRef]
- Franco, P.; De Marco, I. The use of poly(N-vinyl pyrrolidone) in the delivery of drugs: A review. Polymers 2020, 12, 1114. [Google Scholar] [CrossRef]
- Rumondor, A.C.; Ivanisevic, I.; Bates, S.; Alonzo, D.E.; Taylor, L.S. Evaluation of drug-polymer miscibility in amorphous solid dispersion systems. Pharm. Res. 2009, 26, 2523–2534. [Google Scholar] [CrossRef]
- Indulkar, A.S.; Lou, X.; Zhang, G.G.Z.; Taylor, L.S. Insights into the dissolution mechanism of ritonavir-copovidone amorphous solid dispersions: Importance of congruent release for enhanced performance. Mol. Pharm. 2019, 16, 1327–1339. [Google Scholar] [CrossRef]
- Panini, P.; Rampazzo, M.; Singh, A.; Vanhoutte, F.; Van den Mooter, G. Myth or truth: The glass forming ability class iii drugs will always form single-phase homogenous amorphous solid dispersion formulations. Pharmaceutics 2019, 11, 529. [Google Scholar] [CrossRef]
- Que, C.; Lou, X.; Zemlyanov, D.Y.; Mo, H.; Indulkar, A.S.; Gao, Y.; Zhang, G.G.Z.; Taylor, L.S. Insights into the dissolution behavior of ledipasvir-copovidone amorphous solid dispersions: Role of drug loading and intermolecular interactions. Mol. Pharm. 2019, 16, 5054–5067. [Google Scholar] [CrossRef]
- Biswal, S.; Sahoo, J.; Murthy, P.N.; Giradkar, R.P.; Avari, J.G. Enhancement of dissolution rate of gliclazide using solid dispersions with polyethylene glycol 6000. AAPS Pharm. Sci. Tech 2008, 9, 563–570. [Google Scholar] [CrossRef]
- Urbanetz, N.A. Stabilization of solid dispersions of nimodipine and polyethylene glycol 2000. Eur. J. Pharm. Sci. 2006, 28, 67–76. [Google Scholar] [CrossRef]
- Ghosh, I.; Snyder, J.; Vippagunta, R.; Alvine, M.; Vakil, R.; Tong, W.Q.; Vippagunta, S. Comparison of hpmc based polymers performance as carriers for manufacture of solid dispersions using the melt extruder. Int. J. Pharm. 2011, 419, 12–19. [Google Scholar] [CrossRef]
- Li, B.; Harich, K.; Wegiel, L.; Taylor, L.S.; Edgar, K.J. Stability and solubility enhancement of ellagic acid in cellulose ester solid dispersions. Carbohydr. Polym. 2013, 92, 1443–1450. [Google Scholar] [CrossRef]
- Ali, W.; Williams, A.C.; Rawlinson, C.F. Stochiometrically governed molecular interactions in drug: Poloxamer solid dispersions. Int. J. Pharm. 2010, 391, 162–168. [Google Scholar] [CrossRef]
- Newa, M.; Bhandari, K.H.; Oh, D.H.; Kim, Y.R.; Sung, J.H.; Kim, J.O.; Woo, J.S.; Choi, H.G.; Yong, C.S. Enhanced dissolution of ibuprofen using solid dispersion with poloxamer 407. Arch. Pharm. Res. 2008, 31, 1497–1507. [Google Scholar] [CrossRef]
- Ilevbare, G.A.; Liu, H.; Edgar, K.J.; Taylor, L.S. Understanding polymer properties important for crystal growth inhibition—impact of chemically diverse polymers on solution crystal growth of ritonavir. Cryst. Growth Des. 2012, 12, 3133–3143. [Google Scholar] [CrossRef]
- Li, Y.; Pang, H.; Guo, Z.; Lin, L.; Dong, Y.; Li, G.; Lu, M.; Wu, C. Interactions between drugs and polymers influencing hot melt extrusion. J. Pharm. Pharmacol. 2014, 66, 148–166. [Google Scholar] [CrossRef]
- Tran, T.T.D.; Tran, P.H.L. Molecular interactions in solid dispersions of poorly water-soluble drugs. Pharmaceutics 2020, 12, 745. [Google Scholar] [CrossRef]
- Niemczyk, A.I.; Williams, A.C.; Rawlinson-Malone, C.F.; Hayes, W.; Greenland, B.W.; Chappell, D.; Khutoryanskaya, O.; Timmins, P. Novel polyvinylpyrrolidones to improve delivery of poorly water-soluble drugs: From design to synthesis and evaluation. Mol. Pharm. 2012, 9, 2237–2247. [Google Scholar] [CrossRef]
- Rawlinson, C.F.; Williams, A.C.; Timmins, P.; Grimsey, I. Polymer-mediated disruption of drug crystallinity. Int. J. Pharm. 2007, 336, 42–48. [Google Scholar] [CrossRef]
- Van Nguyen, H.; Baek, N.; Lee, B.J. Enhanced gastric stability of esomeprazole by molecular interaction and modulation of microenvironmental ph with alkalizers in solid dispersion. Int. J. Pharm. 2017, 523, 189–202. [Google Scholar] [CrossRef]
- Ozeki, T.; Yuasa, H.; Kanaya, Y. Application of the solid dispersion method to the controlled release of medicine. Ix. Difference in the release of flurbiprofen from solid dispersions with poly(ethylene oxide) and hydroxypropylcellulose and the interact. Int. J. Pharm. 1997, 155, 209–217. [Google Scholar] [CrossRef]
- Huang, J.; Wigent, R.J.; Schwartz, J.B. Drug-polymer interaction and its significance on the physical stability of nifedipine amorphous dispersion in microparticles of an ammonio methacrylate copolymer and ethylcellulose binary blend. J. Pharm. Sci. 2008, 97, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Rasenack, N.; Hartenhauer, H.; Müller, B.W. Microcrystals for dissolution rate enhancement of poorly water-soluble drugs. Int. J. Pharm. 2003, 254, 137–145. [Google Scholar] [CrossRef]
- Zimmermann, A.; Millqvist-Fureby, A.; Elema, M.R.; Hansen, T.; Mullertz, A.; Hovgaard, L. Adsorption of pharmaceutical excipients onto microcrystals of siramesine hydrochloride: Effects on physicochemical properties. Eur. J. Pharm. Biopharm. 2009, 71, 109–116. [Google Scholar] [CrossRef]
- Fael, H.; Rafols, C.; Demirel, A.L. Poly(2-ethyl-2-oxazoline) as an alternative to poly(vinylpyrrolidone) in solid dispersions for solubility and dissolution rate enhancement of drugs. J. Pharm. Sci. 2018, 107, 2428–2438. [Google Scholar] [CrossRef] [PubMed]
- Boel, E.; Smeets, A.; Vergaelen, M.; De la Rosa, V.R.; Hoogenboom, R.; Van den Mooter, G. Comparative study of the potential of poly(2-ethyl-2-oxazoline) as carrier in the formulation of amorphous solid dispersions of poorly soluble drugs. Eur. J. Pharm. Biopharm. 2019, 144, 79–90. [Google Scholar] [CrossRef]
- Everaerts, M.; Tigrine, A.; de la Rosa, V.R.; Hoogenboom, R.; Adriaensens, P.; Clasen, C.; Van den Mooter, G. Unravelling the miscibility of poly(2-oxazoline)s: A novel polymer class for the formulation of amorphous solid dispersions. Molecules 2020, 25, 3587. [Google Scholar] [CrossRef]
- Shan, X.; Williams, A.C.; Khutoryanskiy, V.V. Polymer structure and property effects on solid dispersions with haloperidol: Poly(n-vinyl pyrrolidone) and poly(2-oxazolines) studies. Int. J. Pharm. 2020, 590, 119884. [Google Scholar] [CrossRef]
- Greenhalgh, D.J.; Williams, A.C.; Timmins, P.; York, P. Solubility parameters as predictors of miscibility in solid dispersions. J. Pharm. Sci. 1999, 88, 1182–1190. [Google Scholar] [CrossRef] [PubMed]
- Dudognon, E.; Danede, F.; Descamps, M.; Correia, N.T. Evidence for a new crystalline phase of racemic ibuprofen. Pharm. Res. 2008, 25, 2853–2858. [Google Scholar] [CrossRef]
- Rostkowska, H.; Nowak, M.J.; Lapinski, L.; Adamowicz, L. Ir spectral and theoretical characterization of intramolecular hydrogen bonds closing five-membered rings. Phys. Chem. Chem. Phys. 2001, 3, 3012–3017. [Google Scholar] [CrossRef]
- Rumondor, A.C.; Wikstrom, H.; Van Eerdenbrugh, B.; Taylor, L.S. Understanding the tendency of amorphous solid dispersions to undergo amorphous-amorphous phase separation in the presence of absorbed moisture. AAPS Pharm. Sci. Tech 2011, 12, 1209–1219. [Google Scholar] [CrossRef]
- Teberekidis, V.I.; Sigalas, M.P. Theoretical study of hydrogen bond interactions of felodipine with polyvinylpyrrolidone and polyethyleneglycol. J. Mol. Struct. 2007, 803, 29–38. [Google Scholar] [CrossRef]
- Ramukutty, S.; Ramachandran, E. Growth, spectral and thermal studies of ibuprofen crystals. Cryst. Res. Technol. 2012, 47, 31–38. [Google Scholar] [CrossRef]
- Bogdanova, S.; Pajeva, I.; Nikolova, P.; Tsakovska, I.; Müller, B. Interactions of poly(vinylpyrrolidone) with ibuprofen and naproxen: Experimental and modeling studies. Pharm. Res. 2005, 22, 806–815. [Google Scholar] [CrossRef]
- Iannuccelli, V.; Coppi, G.; Leo, E.; Fontana, F.; Bernabei, M.T. Pvp solid dispersions for the controlled release of furosemide from a floating multiple-unit system. Drug Dev. Ind. Pharm. 2000, 26, 595–603. [Google Scholar] [CrossRef]
- Chadha, R.; Kapoor, V.K.; Kumar, A. Analytical techniques used to characterize drug-polyvinylpyrrolidone systems in solid and liquid states—An overview. J. Sci. Ind. Res. 2006, 65, 459–469. [Google Scholar]
- Hosono, T.; Tsuchiya, S.; Matsumaru, H. Model of interaction of ajmaline with polyvinylpyrrolidone. J. Pharm. Sci. 1980, 69, 824–826. [Google Scholar] [CrossRef]
- Sekizaki, H.; Danjo, K.; Eguchi, H.; Yonezawa, Y.; Sunada, H.; Otsuka, A. Solid-state interaction of ibuprofen with polyvinylpyrrolidone. Chem. Pharm. Bull. 1995, 43, 988–993. [Google Scholar] [CrossRef]
- Williams, A.C.; Timmins, P.; Lu, M.; Forbes, R.T. Disorder and dissolution enhancement: Deposition of ibuprofen on to insoluble polymers. Eur. J. Pharm. Sci. 2005, 26, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, S.; Tsakiridou, G.; Ditzinger, F.; Koehl, N.J.; Price, D.J.; Ilie, A.R.; Kalantzi, L.; Kimpe, K.; Holm, R.; Nair, A.; et al. Application of the solubility parameter concept to assist with oral delivery of poorly water-soluble drugs—A PEARRL review. J. Pharm. Pharmacol. 2019, 71, 441–463. [Google Scholar] [CrossRef] [PubMed]
- Krevelen, D.W.V. Properties of Polymers; Elsevier: Amsterdam, The Netherlands, 1990; pp. 189–221. [Google Scholar]
- Chokshi, R.J.; Sandhu, H.K.; Iyer, R.M.; Shah, N.H.; Malick, A.W.; Zia, H. Characterization of physico-mechanical properties of indomethacin and polymers to assess their suitability for hot-melt extrusion processes as a means to manufacture solid dispersion/solution. J. Pharm. Sci. 2005, 94, 2463–2474. [Google Scholar] [CrossRef] [PubMed]
- Fedors, R.F. A method for estimating both the solubility parameters and molar volumes of liquids. Polym. Eng. Sci. 1974, 14, 147–154. [Google Scholar] [CrossRef]
- Kitak, T.; Dumicic, A.; Planinsek, O.; Sibanc, R.; Srcic, S. Determination of solubility parameters of ibuprofen and ibuprofen lysinate. Molecules 2015, 20, 21549–21568. [Google Scholar] [CrossRef]
- Barton, A.F.M. Solubility parameters. Chem. Rev. 1975, 75, 731–753. [Google Scholar] [CrossRef]
- Flory, P.J. Principles of Polymer Chemistry; Cornell University Press: Ithaca, NY, USA, 1953. [Google Scholar]
- Huang, Y.; Dai, W.G. Fundamental aspects of solid dispersion technology for poorly soluble drugs. Acta Pharm. Sin. B 2014, 4, 18–25. [Google Scholar] [CrossRef]
- Lin, D.; Huang, Y. A thermal analysis method to predict the complete phase diagram of drug-polymer solid dispersions. Int. J. Pharm. 2010, 399, 109–115. [Google Scholar] [CrossRef]
- Moseson, D.E.; Taylor, L.S. The application of temperature-composition phase diagrams for hot melt extrusion processing of amorphous solid dispersions to prevent residual crystallinity. Int. J. Pharm. 2018, 553, 454–466. [Google Scholar] [CrossRef]
- Kong, C.Y.; Sugiura, K.; Funazukuri, T.; Miyake, K.; Okajima, I.; Badhulika, S.; Sako, T. The retention factors and partial molar volumes of ibuprofen at infinite dilution in supercritical carbon dioxide at t = (308.15, 313.15, 323.15, 333.15, 343.15 and 353.15) k. J. Mol. Liq. 2019, 296, 111849. [Google Scholar] [CrossRef]
- Bouten, P.; Lava, K.; van Hest, J.; Hoogenboom, R. Thermal properties of methyl ester-containing poly(2-oxazoline)s. Polymers 2015, 7, 1998–2008. [Google Scholar] [CrossRef]
- Park, J.S.; Kataoka, K. Comprehensive and accurate control of thermosensitivity of poly(2-alkyl-2-oxazoline)s via well-defined gradient or random copolymerization. Macromolecules 2007, 40, 3599–3609. [Google Scholar] [CrossRef]
- Han, Y.R.; Lee, P.I. Effect of extent of supersaturation on the evolution of kinetic solubility profiles. Mol. Pharm. 2017, 14, 206–220. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer–Drug | Wavenumbers (cm−1) | |||
|---|---|---|---|---|
| 0.3:1 mol | Red Shift | 1:1 mol | Red Shift | |
| PVP-IB | 1727 | 17 | 1727 | 17 |
| PMOZ-IB | 1710 | 0 | 1713 | 3 |
| PEOZ-IB | 1711 | 1 | 1727 | 17 |
| PnPOZ-IB | 1713 | 3 | 1727 | 17 |
| PiPOZ-IB | 1712 | 2 | 1727 | 17 |
| Drug and Polymers | Solubility Parameters (δ) (MPa1/2) | Group Classification | |
|---|---|---|---|
| Van Krevelen Method | Δδ | ||
| IB | 19.4 | ||
| PVP | 26.3 | 6.9 | Miscible |
| PMOZ | 27.0 | 7.6 | Not miscible |
| PEOZ | 24.5 | 5.1 | Miscible |
| PnPOZ | 22.9 | 3.5 | Miscible |
| PiPOZ | 22.5 | 3.1 | Miscible |
| Polymer–Drug | VPolymer Repeat Unit a (cm3/mol) | V Polymer b (cm3/mol) | VDrug c (cm3/mol) | M d | Tm (°C) | χ |
|---|---|---|---|---|---|---|
| PVP-IB | 80.0 | 40,000 | 195.5 | 204.60 | 73.27 | −3.71 |
| PEOZ-IB | 74.1 | 37,050 | 189.51 | 73.80 | −3.85 | |
| PnPOZ-IB | 90.2 | 45,100 | 230.69 | 73.50 | −3.32 | |
| PiPOZ-IB | 90.5 | 45,250 | 231.46 | 73.34 | −3.52 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shan, X.; Moghul, M.A.; Williams, A.C.; Khutoryanskiy, V.V. Mutual Effects of Hydrogen Bonding and Polymer Hydrophobicity on Ibuprofen Crystal Inhibition in Solid Dispersions with Poly(N-vinyl pyrrolidone) and Poly(2-oxazolines). Pharmaceutics 2021, 13, 659. https://doi.org/10.3390/pharmaceutics13050659
Shan X, Moghul MA, Williams AC, Khutoryanskiy VV. Mutual Effects of Hydrogen Bonding and Polymer Hydrophobicity on Ibuprofen Crystal Inhibition in Solid Dispersions with Poly(N-vinyl pyrrolidone) and Poly(2-oxazolines). Pharmaceutics. 2021; 13(5):659. https://doi.org/10.3390/pharmaceutics13050659
Chicago/Turabian StyleShan, Xiaoning, Maryam A. Moghul, Adrian C. Williams, and Vitaliy V. Khutoryanskiy. 2021. "Mutual Effects of Hydrogen Bonding and Polymer Hydrophobicity on Ibuprofen Crystal Inhibition in Solid Dispersions with Poly(N-vinyl pyrrolidone) and Poly(2-oxazolines)" Pharmaceutics 13, no. 5: 659. https://doi.org/10.3390/pharmaceutics13050659
APA StyleShan, X., Moghul, M. A., Williams, A. C., & Khutoryanskiy, V. V. (2021). Mutual Effects of Hydrogen Bonding and Polymer Hydrophobicity on Ibuprofen Crystal Inhibition in Solid Dispersions with Poly(N-vinyl pyrrolidone) and Poly(2-oxazolines). Pharmaceutics, 13(5), 659. https://doi.org/10.3390/pharmaceutics13050659