
Bismuth-213 for Targeted Radionuclide Therapy: From Atom to Bedside

 , , and
, , and
Abstract
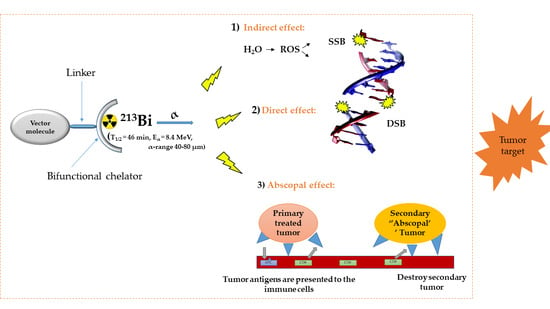
1. Introduction
2. Radionuclide Properties and Production of 225Ac and Its Daughter 213Bi
2.1. Decay Properties of 225Ac and 213Bi
2.2. Current Strategies for 225Ac Production
2.3. 225Ac/213Bi Radionuclide Generators
3. Fundamental Chemistry of Bi
4. Bifunctional Chelating Ligands for 213Bi
4.1. DTPA and DTPA-Derivatives
4.2. DOTA and DOTA-Derivatives
4.3. NETA and DEPA-Derivatives
5. General Considerations for Designing a 213Bi-radiopharmaceutical
6. Preclinical TAT Studies with 213Bi-labeled Probes
6.1. Antibodies
6.2. Antibody Fragments
6.3. Peptides
7. Clinical TAT Studies with 213Bi-labeled Radiopharmaceuticals
7.1. Locoregional Administration
7.1.1. Intravesical TRNT
7.1.2. Intracerebral Substance-P PRRT
7.1.3. Intralesional Melanoma TRNT
7.2. Systemic Administration
7.2.1. Acute Myeloid Leukemia TRNT
7.2.2. SSTR PRRT
7.2.3. 213Bi-PSMA
8. Future Perspectives of 213Bi-TAT
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vermeulen, K.; Vandamme, M.; Bormans, G.; Cleeren, F. Design and Challenges of Radiopharmaceuticals. Semin. Nucl. Med. 2019, 49, 339–356. [Google Scholar] [CrossRef] [PubMed]
- Kostelnik, T.I.; Orvig, C. Radioactive Main Group and Rare Earth Metals for Imaging and Therapy. Chem. Rev. 2019, 2, 902–956. [Google Scholar] [CrossRef] [PubMed]
- Price, E.W.; Orvig, C. Matching chelators to radiometals for radiopharmaceuticals. Chem. Soc. Rev. 2014, 43, 260–290. [Google Scholar] [CrossRef]
- Dekempeneer, Y.; Keyaerts, M.; Krasniqi, A.; Puttemans, J.; Muyldermans, S.; Lahoutte, T.; D’huyvetter, M.; Devoogdt, N. Targeted alpha therapy using short-lived alpha-particles and the promise of nanobodies as targeting vehicle. Expert Opin. Biol. Ther. 2016, 16, 1035–1047. [Google Scholar] [CrossRef]
- Dekempeneer, Y.; Caveliers, V.; Ooms, M.; Maertens, D.; Gysemans, M.; Lahoutte, T.; Xavier, C.; Lecocq, Q.; Maes, K.; Covens, P.; et al. The therapeutic efficacy of 213Bi-labeled sdAbs in a preclinical model of ovarian cancer. Mol. Pharm. 2020, 17, 3553–3566. [Google Scholar] [CrossRef]
- Heskamp, S.; Hernandez, R.; Molkenboer-Kuenen, J.D.M.; Essler, M.; Bruchertseifer, F.; Morgenstern, A.; Steenbergen, E.J.; Cai, W.; Seidl, C.; McBride, W.J.; et al. α-Versus β-Emitting radionuclides for pretargeted radioimmunotherapy of carcinoembryonic antigen-expressing human colon cancer xenografts. J. Nucl. Med. 2017, 58, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Bruchertseifer, F.; Kellerbauer, A.; Malmbeck, R.; Morgenstern, A. Targeted alpha therapy with bismuth-213 and actinium-225: Meeting future demand. J. Label. Compd. Rad. 2019, 62, 794–802. [Google Scholar] [CrossRef]
- Beyls, C.; Haustermans, K.; Deroose, C.M.; Pans, S.; Vanbeckevoort, D.; Verslype, C.; Dekervel, J. Could Autoimmune Disease Contribute to the Abscopal Effect in Metastatic Hepatocellular Carcinoma? Hepatology 2020, 72, 1152–1154. [Google Scholar] [CrossRef] [PubMed]
- Gill, M.R.; Vallis, K.A. Transition metal compounds as cancer radiosensitizers. Chem. Soc. Rev. 2019, 48, 540–557. [Google Scholar] [CrossRef]
- Lacoeuille, F.; Arlicot, N.; Faivre-Chauvet, A. Targeted alpha and beta radiotherapy: An overview of radiopharmaceutical and clinical aspects. Med. Nucl. 2018, 42, 32–44. [Google Scholar] [CrossRef]
- Turner, J.H. Recent advances in theranostics and challenges for the future. Br. J. Radiol. 2018, 91, 20170969. [Google Scholar] [CrossRef]
- Qaim, S.M. Theranostic radionuclides: Recent advances in production methodologies. J. Radioanal. Nucl. Chem. 2019, 322, 1257–1266. [Google Scholar] [CrossRef]
- Morgenstern, A.; Apostolidis, C.; Kratochwil, C.; Sathekge, M.; Krolicki, L.; Bruchertseifer, F. An Overview of Targeted Alpha Therapy with 225Actinium and 213Bismuth. Curr. Radiopharm. 2018, 11, 200–208. [Google Scholar] [CrossRef]
- Sgouros, G.; Roeske, J.C.; McDevitt, M.R.; Palm, S.; Allen, B.J.; Fisher, D.R.; Brill, A.B.; Song, H.; Howell, R.W.; Akabani, G.; et al. MIRD pamphlet No. 22 (Abridged): Radiobiology and dosimetry of α-particle emitters for targeted radionuclide therapy. J. Nucl. Med. 2010, 51, 311–328. [Google Scholar] [CrossRef]
- Jiao, R.; Allen, K.J.H.; Malo, M.E.; Helal, M.; Jiang, Z.; Smart, K.; Buhl, S.V.; Rickles, D.; Bryan, R.A.; Dadachova, E. Evaluation of novel highly specific antibodies to cancer testis antigen Centrin-1 for radioimmunoimaging and radioimmunotherapy of pancreatic cancer. Cancer Med. 2019, 8, 5289–5300. [Google Scholar] [CrossRef]
- Feuerecker, B.; Tauber, R.; Knorr, K.; Heck, M.; Beheshti, A.; Seidl, C.; Bruchertseifer, F.; Pickhard, A.; Gafita, A.; Kratochwil, C.; et al. Activity and Adverse Events of Actinium-225-PSMA-617 in Advanced Metastatic Castration-resistant Prostate Cancer After Failure of Lutetium-177-PSMA. Eur. Urol. Suppl. 2020, 79, 343–350. [Google Scholar] [CrossRef]
- Zacherl, M.J.; Gildehaus, F.J.; Mittlmeier, L.; Boening, G.; Gosewisch, A.; Wenter, V.; Schmidt-Hegemann, N.-S.; Belka, C.; Kretschmer, A.; Casuscelli, J.; et al. First clinical results for PSMA targeted alpha therapy using 225Ac-PSMA-I&T in advanced mCRPC patients. J. Nucl. Med. 2020. [Google Scholar] [CrossRef]
- Kratochwil, C.; Bruchertseifer, F.; Rathke, H.; Bronzel, M.; Apostolidis, C.; Weichert, W.; Haberkorn, U.; Giesel, F.L.; Morgenstern, A. Targeted a-therapy of metastatic castration-resistant prostate cancer with 225Ac-PSMA-617: Dosimetry estimate and empiric dose finding. J. Nucl. Med. 2017, 58, 1624–1631. [Google Scholar] [CrossRef]
- Kratochwil, C.; Giesel, F.L.; Bruchertseifer, F.; Mier, W.; Apostolidis, C.; Boll, R.; Murphy, K.; Haberkorn, U.; Morgenstern, A. 213Bi-DOTATOC receptor-targeted alpha-radionuclide therapy induces remission in neuroendocrine tumours refractory to beta radiation: A first-in-human experience. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 2106–2119. [Google Scholar] [CrossRef]
- Boden, S.; Vints, K.; Cagno, S.; Maertens, D.; Van Hecke, K.; Cardinaels, T. Thorium-229 quantified in historical Thorium-228 capsules. Appl. Radiat. Isot. 2017, 120, 40–44. [Google Scholar] [CrossRef]
- Apostolidis, C.; Molinet, R.; Rasmussen, G.; Morgenstern, A. Production of Ac-225 from Th-229 for targeted α therapy. Anal. Chem. 2005, 77, 6288–6291. [Google Scholar] [CrossRef]
- Apostolidis, C.; Molinet, R.; McGinley, J.; Abbas, K.; Möllenbeck, J.; Morgenstern, A. Cyclotron production of Ac-225 for targeted alpha therapy. Appl. Radiat. Isot. 2005, 62, 383–387. [Google Scholar] [CrossRef]
- Morgenstern, A.; Abbas, K.; Bruchertseifer, F.; Apostolidis, C. Production of Alpha Emitters for Targeted Alpha Therapy. Curr. Radiopharm. 2008, 1, 135–143. [Google Scholar] [CrossRef]
- Nesteruk, K.P.; Ramseyer, L.; Carzaniga, T.S.; Braccini, S. Measurement of the Beam Energy Distribution of a Medical Cyclotron with a Multi-Leaf Faraday Cup. Instruments 2019, 3, 4. [Google Scholar] [CrossRef]
- Engle, J.W.; Mashnik, S.G.; Weidner, J.W.; Wolfsberg, L.E.; Fassbender, M.E.; Jackman, K.; Couture, A.; Bitteker, L.J.; Ullmann, J.L.; Gulley, M.S.; et al. Cross sections from proton irradiation of thorium at 800 MeV. Phys. Rev. C 2013, 88, 014604. [Google Scholar] [CrossRef]
- Weidner, J.W.; Mashnik, S.G.; John, K.D.; Hemez, F.; Ballard, B.; Bach, H.; Birnbaum, E.R.; Bitteker, L.J.; Couture, A.; Dry, D.; et al. Proton-induced cross sections relevant to production of 225Ac and 223Ra in natural thorium targets below 200MeV. Appl. Radiat. Isot. 2012, 70, 2602–2607. [Google Scholar] [CrossRef]
- Melville, G.; Meriarty, H.; Metcalfe, P.; Knittel, T.; Allen, B.J. Production of Ac-225 for cancer therapy by photon-induced transmutation of Ra-226. Appl. Radiat. Isot. 2007, 65, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.K.H.; Ramogida, C.F.; Schaffer, P.; Radchenko, V. Development of 225Ac Radiopharmaceuticals: TRIUMF Perspectives and Experiences. Curr. Radiopharm. 2018, 11, 156–172. [Google Scholar] [CrossRef]
- Morgenstern, A.; Bruchertseifer, F.; Apostolidis, C. Bismuth-213 and Actinium-225—Generator Performance and Evolving Therapeutic Applications of Two Generator-Derived Alpha-Emitting Radioisotopes. Curr. Radiopharm. 2012, 5, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; McDevitt, M.R.; Finn, R.D.; Scheinberg, D.A. Breakthrough of 225Ac and its radionuclide daughters from an 225Ac/213Bi generator: Development of new methods, quantitative characterization, and implications for clinical use. Appl. Radiat. Isot. 2001, 55, 667–678. [Google Scholar] [CrossRef]
- Sinenko, I.L.; Kalmykova, T.P.; Likhosherstova, D.V.; Egorova, B.V.; Zubenko, A.D.; Vasiliev, A.N.; Vasiliev, A.N.; Ermolaev, S.V.; Lapshina, E.V.; Ostapenko, V.S.; et al. 213Bi production and complexation with new picolinate containing ligands. J. Radioanal. Nucl. Chem. 2019, 321, 531–540. [Google Scholar] [CrossRef]
- Morgenstern, A.; Apostolidis, C.; Bruchertseifer, F. Supply and Clinical Application of Actinium-. Semin. Nucl. Med. 2020, 50, 119–123. [Google Scholar] [CrossRef] [PubMed]
- McDevitt, M.R.; Finn, R.D.; Sgouros, G.; Ma, D.; Scheinberg, D.A. An 225Ac/213Bi generator system for therapeutic clinical applications: Construction and operation. Appl. Radiat. Isot. 1999, 50, 895–904. [Google Scholar] [CrossRef]
- Mehring, M. From molecules to bismuth oxide-based materials: Potential homo-and heterometallic precursors and model compounds. Coord. Chem. Rev. 2007, 251, 974–1006. [Google Scholar] [CrossRef]
- Briand, G.G.; Burford, N. Bismuth Compounds and Preparations with Biological or Medicinal Relevance. Chem. Rev. 1999, 99, 2601–2658. [Google Scholar] [PubMed]
- Ananthakrishnan, S.V. The electronic theory of valency. Curr. Sci. 1946, 15, 33–35. [Google Scholar]
- Pyykkö, P. Relativistic Effects in Structural Chemistry. Chem. Rev. 1988, 88, 563–594. [Google Scholar] [CrossRef]
- Ershov, B.G.; Akinshin, M.A.; Gordeev, A.V.; Sukhov, N.L. A pulse radiolysis study of the chloride complexes of Bi(II) and Bi(IV). Int. J. Radiat. Appl. Instrum. Part C Radiat. Phys. Chem. 1986, 27, 91–92. [Google Scholar] [CrossRef]
- Tooth, B.; Etschmann, B.; Pokrovski, G.S.; Testemale, D.; Hazemann, J.L.; Grundler, P.V.; Brugger, J. Bismuth speciation in hydrothermal fluids: An X-ray absorption spectroscopy and solubility study. Geochim. Cosmochim. Acta 2013, 101, 156–172. [Google Scholar] [CrossRef]
- Näslund, J.; Persson, I.; Sandström, M. Solvation of the bismuth(III) ion by water, dimethyl sulfoxide, N,N’-dimethylpropyleneurea, and N,N-dimethylthioformamide. An EXAFS, large-angle X-ray scattering, and crystallographic structural study. Inorg. Chem. 2000, 39, 4012–4021. [Google Scholar] [CrossRef]
- Sun, H.; Li, H.; Sadler, P.J. The biological and medicinal chemistry of bismuth. Chem. Ber. 1997, 130, 669–681. [Google Scholar] [CrossRef]
- Tooth, B. The Hydrothermal Chemistry of Bismuth and the Liquid Bismuth Collector Model. Ph.D. Thesis, University of Adelaide, Adelaide, Australia, 2013. [Google Scholar]
- Pearson, R.G. Hard and soft acids and bases, HSAB, part I: Fundamental principles. J. Chem. Ed. 1968, 45, 581–587. [Google Scholar] [CrossRef]
- Li, M.X.; Zhang, L.Z.; Yang, M.; Niu, J.Y.; Zhou, J. Synthesis, crystal structures, in vitro biological evaluation of zinc(II) and bismuth(III) complexes of 2-acetylpyrazine N(4)-phenylthiosemicarbazone. Bioorganic Med. Chem. Lett. 2012, 22, 2418–2423. [Google Scholar] [CrossRef]
- Ferraz, K.S.O.; Silva, N.F.; Da Silva, J.G.; De Miranda, L.F.; Romeiro, C.F.D.; Souza-Fagundes, E.M.; Mendes, I.C.; Beraldo, H. Investigation on the pharmacological profile of 2,6-diacetylpyridine bis(benzoylhydrazone) derivatives and their antimony(III) and bismuth(III) complexes. Eur. J. Med. Chem. 2012, 53, 98–106. [Google Scholar] [CrossRef]
- Sadler, P.J.; Li, H.; Sun, H. Coordination chemistry of metals in medicine: Target sites for bismuth. Coord. Chem. Rev. 1999, 185–186, 689–709. [Google Scholar] [CrossRef]
- Hancock, R.D.; Baloyi, J.; Mashishi, J. The Affinity of Bismuth (rii) for Nitrogen-donor Ligands. J. Chem. Soc. Dalt. Trans. 1993, 5, 2895–2899. [Google Scholar] [CrossRef]
- Guo, Z.; Sadler, P.J. Metals in Medicine: Metal-based drugs. Angew. Chem. Int. Ed. 1999, 38, 1512–1531. [Google Scholar] [CrossRef]
- Dorso, L.; Bigot-Corbel, E.; Abadie, J.; Diab, M.; Gouard, S.; Bruchertseifer, F.; Morgenstern, A.; Maurel, C.; Chérel, M.; Davodeau, F. Long-term toxicity of 213Bi-labelled BSA in mice. PLoS ONE 2016, 11, e0151330. [Google Scholar] [CrossRef]
- Garmestani, K.; Yao, Z.; Zhang, M.; Wong, K.; Park, C.W.; Pastan, I.; Carrasquillo, J.A.; Brechbiel, M.W. Synthesis and evaluation of a macrocyclic bifunctional chelating agent for use with bismuth radionuclides. Nucl. Med. Biol. 2001, 28, 409–418. [Google Scholar] [CrossRef]
- Bomanji, J.B.; Papathanasiou, N.D. 111In-DTPA0-octreotide (Octreoscan), 131I-MIBG and other agents for radionuclide therapy of NETs. Eur. J. Nucl. Med. Mol. Imaging 2012, 39 (Suppl. S1), S112–S125. [Google Scholar] [CrossRef] [PubMed]
- Rizzieri, D. Zevalin® (ibritumomab tiuxetan): After more than a decade of treatment experience, what have we learned? Crit. Rev. Oncol. Hematol. 2016, 105, 5–17. [Google Scholar] [CrossRef]
- Montavon, G.; Le Du, A.; Champion, J.; Rabung, T.; Morgenstern, A. DTPA complexation of bismuth in human blood serum. Dalt. Trans. 2012, 41, 8615–8623. [Google Scholar] [CrossRef]
- Brechbiel, M.W.; Gansow, O.A. Synthesis of C-functionalized trans-cyclohexyldiethylenetriaminepenta-acetic acids for labelling of monoclonal antibodies with the bismuth-212 α-particle emitter. J. Chem. Soc. Perkin Trans. 1 1992, 1173–1178. [Google Scholar] [CrossRef]
- Milenic, D.E.; Roselli, M.; Mirzadeh, S.; Pippin, C.G.; Gansow, O.A.; Colcher, D.; Brechbiel, M.W.; Schlom, J. In Vivo evaluation of bismuth-labeled monoclonal antibody comparing DTPA-derived bifunctional chelates. Cancer Biother. Radio. 2001, 16, 133–146. [Google Scholar] [CrossRef]
- Chan, H.S.; de Blois, E.; Morgenstern, A.; Bruchertseifer, F.; de Jong, M.; Breeman, W.; Konijnenberg, M. In Vitro comparison of 213Bi- and 177Lu-radiation for peptide receptor radionuclide therapy. PLoS ONE 2017, 12, e0181473. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.S.; Konijnenberg, M.W.; Daniels, T.; Nysus, M.; Makvandi, M.; de Blois, E.; Breeman, W.A.; Atcher, R.W.; de Jong, M.; Norenberg, J.P. Improved safety and efficacy of 213Bi-DOTATATE-targeted alpha therapy of somatostatin receptor-expressing neuroendocrine tumors in mice pre-treated with l-lysine. EJNMMI Res. 2016, 6, 83. [Google Scholar] [CrossRef] [PubMed]
- Csajbók, É.; Baranyai, Z.; Bányai, I.; Brücher, E.; Király, R.; Müller-Fahrnow, A.; Platzek, J.; Radüchel, B.; Schäfer, M. Equilibrium, 1 H and 13 C NMR Spectroscopy, and X-ray Diffraction Studies on the Complexes Bi(DOTA)-and Bi(DO3A-Bu). Inorg. Chem. 2003, 7, 2342–2349. [Google Scholar] [CrossRef] [PubMed]
- Šimeček, J.; Hermann, P.; Seidl, C.; Bruchertseifer, F.; Morgenstern, A.; Wester, H.J.; Notni, J. Efficient formation of inert Bi-213 chelates by tetraphosphorus acid analogues of DOTA: Towards improved alpha-therapeutics. EJNMMI Res. 2018, 8, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lima, L.M.P.; Beyler, M.; Delgado, R.; Platas-Iglesias, C.; Tripier, R. Investigating the Complexation of the Pb2+/Bi3+ Pair with Dipicolinate Cyclen Ligands. Inorg. Chem. 2015, 54, 7045–7057. [Google Scholar] [CrossRef]
- Lima, L.M.P.; Beyler, M.; Oukhatar, F.; Le Saec, P.; Faivre-Chauvet, A.; Platas-Iglesias, C.; Delgado, R.; Tripier, R. H2Me-do2pa: An attractive chelator with fast, stable and inert natBi3+ and 213Bi3+ complexation for potential α-radioimmunotherapy applications. Chem. Comm. 2014, 50, 12371–12374. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.S.; Song, H.A.; Birch, N.; Le, T.; Lim, S.; Ma, X. Efficient synthesis and evaluation of bimodal ligand NETA. Bioorg. Med. Chem. Lett. 2008, 18, 3436–3439. [Google Scholar] [CrossRef]
- Song, H.A.; Kang, C.S.; Baidoo, K.E.; Milenic, D.E.; Chen, Y.; Dai, A.; Brechbiel, M.W.; Chong, H.-S. Efficient Bifunctional Decadentate Ligand 3p-C-DEPA for Targeted α-Radioimmunotherapy Applications. Bioconjug. Chem. 2011, 22, 1128–1135. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.S.; Song, H.A.; Ma, X.; Milenic, D.E.; Brady, E.D.; Lim, S.; Lee, H.; Baidoo, K.; Cheng, D.; Brechbiel, M.W. Novel bimodal bifunctional ligands for radioimmunotherapy and targeted MRI. Bioconjug. Chem. 2008, 19, 1439–1447. [Google Scholar] [CrossRef][Green Version]
- Kang, C.S.; Song, H.A.; Milenic, D.E.; Baidoo, K.E.; Brechbiel, M.W.; Chong, H.S. Preclinical evaluation of NETA-based bifunctional ligand for radioimmunotherapy applications using 212Bi and 213Bi: Radiolabeling, serum stability, and biodistribution and tumor uptake studies. Nucl. Med. Biol. 2013, 40, 600–605. [Google Scholar] [CrossRef]
- Dadwal, M.; Kang, C.S.; Song, H.A.; Sun, X.; Dai, A.; Baidoo, K.E.; Brechbiel, M.W.; Chong, H.-S. Synthesis and evaluation of a bifunctional chelate for development of Bi(III)-labeled radioimmunoconjugates. Bioorg. Med. Chem. Lett. 2011, 21, 7513–7515. [Google Scholar] [CrossRef]
- Chong, H.S.; Lim, S.; Baidoo, K.E.; Milenic, D.E.; Ma, X.; Jia, F.; Song, H.A.; Brechbiel, M.W.; Lewis, M.R. Synthesis and biological evaluation of a novel decadentate ligand DEPA. Bioorg. Med. Chem. Lett. 2008, 18, 5792–5795. [Google Scholar] [CrossRef] [PubMed]
- Spreckelmeyer, S.; Ramogida, C.F.; Rousseau, J.; Arane, K.; Bratanovic, I.; Colpo, N.; Jermilova, U.; Dias, G.M.; Dude, I.; Jaraquemada-Peláez, M.G.; et al. p-NO2-Bn-H4neunpa and H4neunpa-Trastuzumab: Bifunctional Chelator for Radiometalpharmaceuticals and 111In Immuno-Single Photon Emission Computed Tomography Imaging. Bioconjug. Chem. 2017, 28, 2145–2159. [Google Scholar] [CrossRef]
- Milenic, D.E.; Brady, E.D.; Garmestani, K.; Albert, P.S.; Abdulla, A.; Brechbiel, M.W. Improved efficacy of α-particle-targeted radiation therapy: Dual targeting of human epidermal growth factor receptor-2 and tumor-associated glycoprotein 72. Cancer 2010, 116 (Suppl. S4), 1059–1066. [Google Scholar] [CrossRef]
- Jurcic, J. Alpha-Particle Therapy for Acute Myeloid Leukemia. J. Med Imaging Radiat. Sci. 2019, 50, S86–S87. [Google Scholar] [CrossRef]
- Rosenblat, T.L.; McDevitt, M.R.; Mulford, D.A.; Pandit-Taskar, N.; Divgi, C.R.; Panageas, K.S.; Heaney, M.L.; Chanel, S.; Morgenstern, A.; Sgouros, G.; et al. Sequential cytarabine and α-particle immunotherapy with bismuth-213-lintuzumab (HuM195) for acute myeloid leukemia. Clin. Cancer Res. 2010, 16, 5303–5311. [Google Scholar] [CrossRef]
- Jurcic, J.G.; Larson, S.M.; Sgouros, G.; McDevitt, M.R.; Finn, R.D.; Divgi, C.R.; Ballangrud, Å.M.; Hamacher, K.A.; Ma, D.; Humm, J.L.; et al. Targeted α particle immunotherapy for myeloid leukemia. Blood 2002, 100, 1233–1239. [Google Scholar] [CrossRef]
- Bethge, W.A.; Wilbur, D.S.; Storb, R.; Hamlin, D.K.; Santos, E.B.; Brechbiel, M.W.; Sandmaier, B.M. Radioimmunotherapy with bismuth-213 as conditioning for nonmyeloablative allogeneic hematopoietic cell transplantation in dogs: A dose deescalation study. Transplantation 2004, 78, 352–359. [Google Scholar] [CrossRef]
- Kneifel, S.; Cordier, D.; Good, S.; Ionescu, M.C.S.; Ghaffari, A.; Hofer, S.; Kretzschmar, M.; Tolnay, M.; Apostolidis, C.; Waser, B.; et al. Local targeting of malignant gliomas by the diffusible peptidic vector 1.,4,7,10-tetraazacyclododecane-1-glutaric acid-4,7,10-triacetic acid-substance P. Clin. Cancer Res. 2006, 12, 3843–3850. [Google Scholar] [CrossRef]
- Królicki, L.; Bruchertseifer, F.; Kunikowska, J.; Koziara, H.; Królicki, B.; Jakuciński, M.; Pawlak, D.; Apostolidis, C.; Mirzadeh, S.; Rola, R.; et al. Safety and efficacy of targeted alpha therapy with 213 Bi-DOTA-substance P in recurrent glioblastoma. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Cordier, D.; Forrer, F.; Bruchertseifer, F.; Morgenstern, A.; Apostolidis, C.; Good, S.; Müller-Brand, J.; Mäcke, H.; Reubi, J.C.; Merlo, A. Targeted alpha-radionuclide therapy of functionally critically located gliomas with 213Bi-DOTA-[Thi8,Met(O2)11]- substance P: A pilot trial. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 1335–1344. [Google Scholar] [CrossRef] [PubMed]
- Kratochwil, C.; Schmidt, K.; Afshar-Oromieh, A.; Bruchertseifer, F.; Rathke, H.; Morgenstern, A.; Haberkorn, U.; Giesel, F. Targeted alpha therapy of mCRPC: Dosimetry estimate of 213Bismuth-PSMA-617. Eur J. Nucl. Med. Mol. Imaging. 2018, 45, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Autenrieth, M.E.; Seidl, C.; Bruchertseifer, F.; Horn, T.; Kurtz, F.; Feuerecker, B.; D’Alessandria, C.; Pfob, C.; Nekolla, S.; Apostolidis, C.; et al. Treatment of carcinoma in situ of the urinary bladder with an alpha-emitter immunoconjugate targeting the epidermal growth factor receptor: A pilot study. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 1364–1371. [Google Scholar] [CrossRef]
- Hong, H.; Sun, J.; Cai, W. Radionuclide-Based Cancer Imaging Targeting the Carcinoembryonic Antigen. Biomark. Insights 2008, 3, 435–451. [Google Scholar] [CrossRef]
- Sathekge, M.; Knoesen, O.; Meckel, M.; Modiselle, M.; Vorster, M.; Marx, S. 213Bi-PSMA-617 targeted alpha-radionuclide therapy in metastatic castration-resistant prostate cancer. Eur. J. Nucl. Med. 2017, 44, 1099–1100. [Google Scholar] [CrossRef]
- Fazel, J.; Rötzer, S.; Seidl, C.; Feuerecker, B.; Autenrieth, M.; Weirich, G.; Bruchertseifer, F.; Morgenstern, A.; Senekowitsch-Schmidtke, R. Fractionated intravesical radioimmunotherapy with 213Bi-anti-EGFR-MAb is effective without toxic side-effects in a nude mouse model of advanced human bladder carcinoma. Cancer Biol. Ther. 2015, 16, 1526–1534. [Google Scholar] [CrossRef]
- Jiao, R.; Allen, K.J.H.; Malo, M.E.; Rickles, D.; Dadachova, E. Evaluating the combination of radioimmunotherapy and immunotherapy in a melanoma mouse model. Int. J. Mol. Sci. 2020, 21, 773. [Google Scholar] [CrossRef] [PubMed]
- Nosanchuk, J.D.; Jeyakumar, A.; Ray, A.; Revskaya, E.; Jiang, Z.; Bryan, R.A.; Allen, K.J.H.; Jiao, R.; Malo, M.E.; Gómez, B.L.; et al. Structure-function analysis and therapeutic efficacy of antibodies to fungal melanin for melanoma radioimmunotherapy. Nat. Sci. Rep. 2018, 8, 5466. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson-Lutz, A.; Bäck, T.; Aneheim, E.; Hultborn, R.; Palm, S.; Jacobsson, L.; Morgenstern, A.; Bruchertseifer, F.; Albertsson, P.; Lindegren, S. Therapeutic efficacy of α-radioimmunotherapy with different activity levels of the 213Bi-labeled monoclonal antibody MX35 in an ovarian cancer model. EJNMMI Res. 2017, 7, 38. [Google Scholar] [CrossRef] [PubMed]
- Revskaya, E.; Jiang, Z.; Morgenstern, A.; Bruchertseifer, F.; Sesay, M.; Walker, S.; Fuller, S.; Lebowitz, M.S.; Gravekamp, C.; Ghanbari, H.A.; et al. A Radiolabeled Fully Human Antibody to Human Aspartyl (Asparaginyl) β-Hydroxylase Is a Promising Agent for Imaging and Therapy of Metastatic Breast Cancer. Cancer Biother. Radiopharm. 2017, 32, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Derrien, A.; Gouard, S.; Maurel, C.; Gaugler, M.H.; Bruchertseifer, F.; Morgenstern, A.; Faivre-Chauvet, A.; Classe, J.M.; Chérel, M. Therapeutic efficacy of alpha-RIT using a 213Bi-anti-hCD138 antibody in a mouse model of ovarian peritoneal carcinomatosis. Front. Med. 2015, 2, 88. [Google Scholar] [CrossRef] [PubMed]
- Fichou, N.; Gouard, S.; Maurel, C.; Barbet, J.; Ferrer, L.; Morgenstern, A.; Bruchertseifer, F.; Faivre-Chauvet, A.; Bigot-Corbel, E.; Davodeau, F.; et al. Single-dose anti-CD138 radioimmunotherapy: Bismuth-213 is more efficient than lutetium-177 for treatment of multiple myeloma in a preclinical model. Front. Med. 2015, 2, 76. [Google Scholar] [CrossRef]
- Feuerecker, B.; Michalik, M.; Hundshammer, C.; Schwaiger, M.; Bruchertseifer, F.; Morgenstern, A.; Seidl, C. Assessment of 213Bi-anti-EGFR MAb treatment efficacy in malignant cancer cells with [1−13C]pyruvate and [18F]FDG. Nat. Sci. Rep. 2019, 9, 8294. [Google Scholar] [CrossRef] [PubMed]
- Ménager, J.; Gorin, J.B.; Maurel, C.; Drujont, L.; Gouard, S.; Louvet, C.; Chérel, M.; Faivre-Chauvet, A.; Morgenstern, A.; Bruchertseifer, F.; et al. Combining α-radioimmunotherapy and adoptive T cell therapy to potentiate tumor destruction. PLoS ONE 2015, 10, e0130249. [Google Scholar] [CrossRef]
- Teiluf, K.; Seidl, C.; Blechert, B.; Gaertner, F.C.; Gilbertz, K.P.; Fernandez, V.; Bassermann, F.; Endell, J.; Boxhammer, R.; Leclair, S.; et al. α-radioimmunotherapy with 213Bi-anti-CD38 immunoconjugates is effective in a mouse model of human multiple myeloma. Oncotarget 2015, 6, 4692–4703. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Hedayati, M.; Hobbs, R.F.; Shao, C.; Bruchertseifer, F.; Morgenstern, A.; DeWeese, T.L.; Sgouros, G. Targeting aberrant DNA double-strand break repair in triple-negative breast cancer with alpha-particle emitter radiolabeled anti-EGFR antibody. Mol. Cancer Ther. 2013, 12, 2043–2054. [Google Scholar] [CrossRef]
- Roscher, M.; Hormann, I.; Leib, O.; Marx, S.; Moreno, J.; Miltner, E.; Friesen, C. Targeted alpha-therapy using [Bi-213]anti-CD20 as novel treatment option for radio-and chemoresistant non-Hodgkin lymphoma cells. Oncotarget 2013, 4, 218–230. [Google Scholar] [CrossRef]
- Pagel, J.M.; Kenoyer, A.L.; Bäck, T.; Hamlin, D.K.; Wilbur, D.S.; Fisher, D.R.; Park, S.I.; Frayo, S.; Axtman, A.; Orgun, N.; et al. Anti-CD45 pretargeted radioimmunotherapy using bismuth-213: High rates of complete remission and long-term survival in a mouse myeloid leukemia xenograft model. Blood 2011, 118, 703–711. [Google Scholar] [CrossRef]
- Allen, B.J.; Abbas Rizvi, S.M.; Qu, C.F.; Smith, R.C. Targeted alpha therapy approach to the management of pancreatic cancer. Cancers 2011, 3, 1821–1843. [Google Scholar] [CrossRef] [PubMed]
- Park, S.I.; Shenoi, J.; Page, J.M.; Hamlin, D.K.; Wilbur, D.S.; Orgun, N.; Kenoyer, A.L.; Frayo, S.; Axtman, A.; Bäck, T.; et al. Conventional and pretargeted radioimmunotherapy using bismuth-213 to target and treat non-Hodgkin lymphomas expressing CD20: A preclinical model toward optimal consolidation therapy to eradicate minimal residual disease. Blood 2010, 116, 4231–4239. [Google Scholar] [CrossRef] [PubMed]
- Lingappa, M.; Song, H.; Thompson, S.; Bruchertseifer, F.; Morgenstern, A.; Sgouros, G. Immunoliposomal delivery of 213Bi for α-emitter targeting of metastatic breast cancer. Cancer Res. 2010, 70, 6815–6823. [Google Scholar] [CrossRef] [PubMed]
- Drecoll, E.; Gaertner, F.C.; Miederer, M.; Blechert, B.; Vallon, M.; Müller, J.M.; Alke, A.; Seidl, C.; Bruchertseifer, F.; Morgenstern, A.; et al. Treatment of peritoneal carcinomatosis by targeted delivery of the radio-labeled tumor homing peptide 213Bi-DTPA-[F3]2 into the nucleus of tumor cells. PLoS ONE 2009, 4, e5715. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Rizvi, S.M.A.; Ranson, M.; Allen, B.J. 213Bi-PAI2 conjugate selectively induces apoptosis in PC3 metastatic prostate cancer cell line and shows anti-cancer activity in a xenograft animal model. Br. J. Cancer 2002, 86, 1197–1203. [Google Scholar] [CrossRef]
- Verhoeven, M.; Seimbille, Y.; Dalm, S.U. Therapeutic applications of pretargeting. Pharmaceutics 2019, 11, 434. [Google Scholar] [CrossRef]
- Listek, M.; Hönow, A.; Gossen, M.; Hanack, K. A novel selection strategy for antibody producing hybridoma cells based on a new transgenic fusion cell line. Nat. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Holliger, P.; Hudson, P.J. Engineered antibody fragments and the rise of single domains. Nat. Biotechnol. 2005, 23, 1126–1136. [Google Scholar] [CrossRef]
- Adams, G.P.; Shaller, C.C.; Chappell, L.L.; Wu, C.; Horak, E.M.; Simmons, H.H.; Litwin, S.; Marks, J.D.; Weiner, L.M.; Brechbiel, M.W. Delivery of the α-emitting radioisotope bismuth-213 to solid tumors via single-chain Fv and diabody molecules. Nucl. Med. Biol. 2000, 27, 339–346. [Google Scholar] [CrossRef]
- Strosberg, J.; El-Haddad, G.; Wolin, E.; Hendifar, A.; Yao, J.; Chasen EM, B.; Kunz, P.L.; Kulke, M.H.; Jacene, H.; Bushnell, D.; et al. Phase 3 Trial of 177Lu-Dotatate for Midgut Neuroendocrine Tumors. N. Engl. J. Med. 2017, 2, 125–135. [Google Scholar] [CrossRef]
- Bidwell, G.L.; Raucher, D. Therapeutic peptides for cancer therapy. Part I—Peptide inhibitors of signal transduction cascades. Expert Opin. Drug Deliv. 2009, 6, 1033–1047. [Google Scholar] [CrossRef] [PubMed]
- McGregor, D.P. Discovering and improving novel peptide therapeutics. Curr. Opin. Pharmacol. 2008, 8, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Fjelde, A.; Id, M.; Saidi, A.; Torgue, J.; Heyerdahl, H.; Stallons, T.A.R.; Kolstad, A.; Dahle, J. Targeted alpha therapy for chronic lymphocytic leukaemia and non-Hodgkin’s lymphoma with the anti-CD37 radioimmunoconjugate 212Pb-NNV003. PLoS ONE 2020, 15, e023. [Google Scholar]
- Ebbers, S.C.; Braat, A.J.A.T.; Moelker, A.; Stokkel, M.P.M.; Lam, M.G.E.H.; Barentsz, M.W. Intra-arterial versus standard intravenous administration of lutetium-177-DOTA-octreotate in patients with NET liver metastases: Study protocol for a multicenter, randomized controlled trial (LUTIA trial). Trials 2020, 21, 141. [Google Scholar] [CrossRef]
- Graf, J.; Pape, U.F.; Jann, H.; Denecke, T.; Arsenic, R.; Brenner, W.; Pavel, M.; Prasad, V. Prognostic Significance of Somatostatin Receptor Heterogeneity in Progressive Neuroendocrine Tumor Treated with Lu-177 DOTATOC or Lu-177 DOTATATE. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 881–894. [Google Scholar] [CrossRef]
- Delpassand, E.S.; Samarghandi, A.; Zamanian, S.; Wolin, E.M.; Hamiditabar, M.; Espenan, G.D.; Erion, J.L.; O’Dorisio, T.M.; Kvols, L.K.; Simon, J.; et al. Peptide receptor radionuclide therapy with 177Lu-DOTATATE for patients with somatostatin receptor-expressing neuroendocrine tumors: The first US phase 2 experience. Pancreas 2014, 43, 518–525. [Google Scholar] [CrossRef]
- Allen, B.J.; Raja, C.; Rizvi, S.; Li, Y.; Tsui, W.; Graham, P.; Thompson, J.F.; Reisfeld, R.A.; Kearsley, J.; Morgenstern, A.; et al. Intralesional targeted alpha therapy for metastatic melanoma. Cancer Biol. Ther. 2005, 4, 1318–1324. [Google Scholar] [CrossRef]
- Finn, L.E.; Levy, M.; Orozco, J.J.; Park, J.H.; Atallah, E.; Craig, M.; Perl, A.E.; Scheinberg, D.A.; Cicic, D.; Bergonio, G.R.; et al. A Phase 2 Study of Actinium-225 (225Ac)-Lintuzumab in Older Patients with Previously Untreated Acute Myeloid Leukemia (AML) Unfit for Intensive Chemotherapy. Blood 2017, 130 (Suppl. S1), 2638. [Google Scholar]
- Jurcic, J.G.; Ravandi, F.; Pagel, J.M.; Park, J.H.; Smith, B.D.; Douer, D.; Levy, M.Y.; Estey, E.; Kantarjian, H.M.; Earle, D.; et al. Phase I trial of α-particle therapy with actinium-225 (225Ac)-lintuzumab (anti-CD33) and low-dose cytarabine (LDAC) in older patients with untreated acute myeloid leukemia (AML. J. Clin. Oncol. 2015, 33 (Suppl. S15), 7050. [Google Scholar] [CrossRef]
- Juzeniene, A.; Stenberg, V.Y.; Bruland, Ø.S.; Larsen, R.H. Preclinical and Clinical Status of PSMA-Targeted Alpha Therapy for Metastatic Castration-Resistant Prostate Cancer. Cancers 2021, 13, 779. [Google Scholar] [CrossRef]
- Allen, B.J.; Singla, A.A.; Rizvi, S.M.A.; Graham, P.; Bruchertseifer, F.; Apostolidis, C.; Morgenstern, A. Analysis of patient survival in a Phase i trial of systemic targeted α?-therapy for metastatic melanoma. Immunotherapy 2011, 3, 1041–1050. [Google Scholar] [CrossRef] [PubMed]
- Raja, C.; Graham, P.; Abbas Rizvi, S.M.; Song, E.; Goldsmith, H.; Thompson, J.; Bosserhoff, A.; Morgenstern, A.; Apostolidis, C.; Kearsley, J.; et al. Interim analysis of toxicity and response in phase 1 trial of systemic targeted alpha therapy for metastatic melanoma. Cancer Biol. Ther. 2007, 6, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Krolicki, L.; Bruchertseifer, F.; Kunikowska, J.; Koziara, H.; Królicki, B.; Jakuciński, M.; Pawlak, D.; Apostolidis, C.; Mirzadeh, S.; Rola, R.; et al. Prolonged survival in secondary glioblastoma following local injection of targeted alpha therapy with 213Bi-substance P analogue. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 1636–1644. [Google Scholar] [CrossRef]
- De Kruijff, R.; Wolterbeek, H.; Denkova, A. A Critical Review of Alpha Radionuclide Therapy—How to Deal with Recoiling Daughters? Pharmaceuticals 2015, 8, 321–336. [Google Scholar] [PubMed]
- Jansen, K.; Heirbaut, L.; Cheng, J.D.; Joossens, J.; Ryabtsova, O.; Cos, P.; Maes, L.; Lambeir, A.M.; De Meester, I.; Augustyns, K.; et al. Selective inhibitors of fibroblast activation protein (FAP) with a (4-quinolinoyl)-glycyl-2-cyanopyrrolidine scaffold. ACS Med. Chem. Lett. 2013, 4, 491–496. [Google Scholar] [CrossRef]
- Giesel, F.L.; Kratochwil, C.; Lindner, T.; Marschalek, M.M.; Loktev, A.; Lehnert, W.; Debus, J.; Jäger, D.; Flechsig, P.; Altmann, A.; et al. 68Ga-FAPI PET/CT: Biodistribution and preliminary dosimetry estimate of 2 DOTA-containing FAP-targeting agents in patients with various cancers. J. Nucl. Med. 2019, 60, 386–392. [Google Scholar] [CrossRef]
- Lindner, T.; Loktev, A.; Altmann, A.; Giesel, F.; Kratochwil, C.; Debus, J.; Jäger, D.; Mier, W.; Haberkorn, U. Development of quinoline-based theranostic ligands for the targeting of fibroblast activation protein. J. Nucl. Med. 2018, 59, 1415–1422. [Google Scholar] [CrossRef]
- Loktev, A.; Lindner, T.; Mier, W.; Debus, J.; Altmann, A.; Jäger, D.; Giesel, F.; Kratochwil, C.; Barthe, P.; Roumestand, C.; et al. A tumor-imaging method targeting cancer-associated fibroblasts. J. Nucl. Med. 2018, 59, 1423–1429. [Google Scholar] [CrossRef]
- Jaggi, J.S.; Seshan, S.V.; McDevitt, M.R.; Sgouros, G.; Hyjek, E.; Scheinberg, D.A. Mitigation of radiation nephropathy after internal α-particle irradiation of kidneys. Int. J. Radiat. Oncol. Biol. Phys. 2006, 64, 1503–1512. [Google Scholar] [CrossRef]
- Chatal, J.F.; Davodeau, F.; Cherel, M.; Barbet, J. Different ways to improve the clinical effectiveness of radioimmunotherapy in solid tumors. J. Cancer Res. Ther. 2009, 5 (Suppl. S1), S36–S40. [Google Scholar] [CrossRef] [PubMed]
- Song, E.Y.; Abbas Rizvi, S.M.; Qu, C.F.; Raja, C.; Brechbiel, M.W.; Morgenstern, A.; Apostolidis, C.; Allen, B.J. Pharmacokinetics and toxicity of 213Bi-labeled PAI2 in preclinical targeted alpha therapy for cancer. Cancer Biol. Ther. 2007, 6, 898–904. [Google Scholar] [CrossRef] [PubMed]
- Bendre, S.; Zhang, Z.; Kuo, H.-T.; Rousseau, J.; Zhang, C.; Merkens, H.; Roxin, Á.; Bénard, F.; Lin, K.-S. Evaluation of Met-Val-Lys as a Renal Brush Border Enzyme-Cleavable Linker to Reduce Kidney Uptake of 68Ga-Labeled DOTA-Conjugated Peptides and Peptidomimetics. Molecules 2020, 25, 3854. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, C.; Uehara, T.; Kanazawa, N.; Wada, S.; Suzuki, H.; Arano, Y. Preferential Cleavage of a Tripeptide Linkage by Enzymes on Renal Brush Border Membrane To Reduce Renal Radioactivity Levels of Radiolabeled Antibody Fragments. J. Med. Chem. 2018, 61, 5257–5268. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Coordination Number | Geometry | Example |
|---|---|---|
| 3 | Pyramidal | Bi(SAr)3 |
| 4 | Trigonal bipyramidal | [Bi{OP(NMe2)3}2][Fe(CO)2(η5–Cp)5F2][PF6] |
| 5 | Square-based pyramidal | Na2[Bi(SC6F5)5)](THF)4 |
| 5 | Trigonal antiprism | {Bi(NO3)bis[1-azepanyl-4-(2-thieniyl)-2,3-diazapenta-1-3 diene- 1 -thlolato-N3,S]} |
| 6 | Octahedral | [Bi6O4(OH4)]6+ |
| 7 | Trigonal Dodecahedron | {Bi(NO3)bis[1-azepanyl-4-(2-pyridyl)-2,3-diazapenta-1,3 diene-1-thiolato-N’,N3,S]} |
| 8 | Bicapped trigonal prism | [Bi(nta)(H2O)2] |
| 9 | Tricapped trigonal prism | [Bi(H2O)9](CF3SO3)3 |
| 9 | Monocapped square antiprism | (guanidinium)2[Bi(dtpa)]·4H2O |
| Metal Ion | Ligand | Coordinating Nuclei | Geometry | LogKML b | pM c |
|---|---|---|---|---|---|
| Bi3+ | DOTA | N4O4 | Square antiprism | 30.3 | 27.0 |
| Bi3+ | Me-DO2PA | N6O2 | Square antiprism | 34.2 | 28.6 |
| Bi3+ | DTPA | N3O5 | Square antiprism | 33.9–35.2 | - |
| Bi3+ | CHX-DTPA | N3O5 | Square antiprism | 34.9–35.6 | - |
| Bi3+ | NETA | N4O4 | Square antiprism | - | - |
| Bi3+ | DEPA | N4O5/N5O4 a | Distorted dodecahedron | - | - |
| Bi3+ | H4neunpa | N5O4 | Distorted dodecahedron | 28.8 | - |
| Bioconjugate | Key Findings | Cancer Type | Reference |
|---|---|---|---|
| 213Bi-anti-EGFR-mAb | The animals survived for an average of 131.8 d after fractionated treatment with 0.46 MBq 213Bi-anti-EGFR-mAb, with 30% remaining for more than 300 days. Even after treatment with 3.7 MBq of 213Bi-anti-EGFR-mAb, no toxic side effects on normal urothelium were observed. | Human bladder carcinoma (local instillation of 213Bi-anti-EGFR-mAb) | [81] |
| 213Bi-69-11 antibody | Antibody 69-11 localized significantly in pancreatic ductal adenocarcinoma cancer (PDAC) xenografts in mice in vivo and ex vivo. TAT of PDAC xenografts with 213Bi-69-11 was effective, safe, and CETN1-specific. | Pancreatic cancer | [15] |
| 213Bi-h8C3 antibody | Treatments with anti-PD-1 antibody alone had a modest impact on tumor size, while the combination therapy with 213Bi-h8C3 resulted in a substantial slowing of tumor development and improved animal survival. | Melanoma | [82] |
| 213Bi-8C3 or 213Bi-6D2 antibody | Antibody binding to melanin was shown to be dependent on both charge and hydrophobic interactions, and in vivo evidence supports the development of 8C3 IgG as a radioimmunotherapy reagent for metastatic melanoma. | Melanoma | [83] |
| 213Bi-DOTATATE | A 10% cell survival of CA20948 was reached at doses of 3 Gy with 213Bi-DOTATATE, a factor six lower than the 18 Gy found for 177Lu-DOTATATE and below the 5 Gy after 137Cs external exposure. | Pancreatic cancer | [56] |
| 213Bi-IMP288-mAb | 213Bi-IMP288 cleared from the bloodstream rapidly; blood levels were 0.44 ± 0.28% ID/g 30 min after injection. Except for the kidneys, where uptake was 1.8 ± 1.1% ID/g 30 min after injection, uptake in normal tissues was poor. | Colon cancer | [6] |
| 213Bi-MX35-mAb | The tumor-free fraction in animals given 3 MBq/mL of 213Bi-MX35 was 0.55, while it was 0.78 in animals given 9 MBq/mL of 213Bi-MX35. The tumor-free fraction in the control group treated with unlabeled MX35 was 0.15. There was no significant drop in white blood cell counts or weight loss. | Ovarian cancer | [84] |
| 213Bi-DTPA-PAN-622-mAb | A pilot therapy study with 213Bi-DTPA-PAN-622 demonstrated a significant effect on the primary tumor. | Breast cancer | [85] |
| 213Bi-Anti-hCD138 Antibody | TAT of 7.4 MBq and 11.1 MBq significantly improved survival (p = 0.0303 and p = 0.0070, respectively), whereas HIPEC and HIPEC + TAT treatments did not significantly ameliorate survival as compared with the control group. | Ovarian cancer | [86] |
| 213Bi-DOTA-9E7.4-mAb | TAT with 3.7 MBq of 213Bi-labeled 9E7.4 anti-CD138 mAb increased median survival to 80 days compared with 37 days in the untreated control group and resulted in effected cure in 45% of the animals. | Multiple myeloma (MM) | [87] |
| 213Bi-anti-EGFR-mAb | Treatment with 213Bi-anti-EGFR-mAb resulted in an effective induction of cell death in EJ28Luc and LN18 cells. | Bladder carcinoma | [88] |
| 213Bi-CHX-A’’-DTPA-anti-CD138-mAb | The combined treatment resulted in significant tumor growth suppression and improved survival in the animals. | MM | [89] |
| 213Bi-DTPA-anti-CD38-MAb | Treatment with 213Bi-anti-CD38-mAb suppressed tumor growth in myeloma xenografts by inducing apoptosis in tumor tissue and significantly extended survival relative to controls. | MM | [90] |
| 213Bi-DTPA-Cetuximab | 213Bi-cetuximab was found to be significantly more effective in the BRCA-1-mutated triple negative breast cancer (TNBC) cell line HCC1937 than BRCA-1-competent TNBC cell MDA-MB-231. siRNA knockdown of BRCA-1 or DNA-dependent protein kinase, catalytic subunit (DNA-PKcs), a key gene in non-homologous end-joining DSB repair pathway, also sensitized TNBC cells to 213Bi-cetuximab. | Breast cancer | [91] |
| 213Bi-DTPA-anti-CD20-mAb | In CD20-expressing sensitive as well as chemoresistant, beta-radiation resistant, and gamma-radiation resistant NHL cells, 213Bi-anti-CD20 induced apoptosis; activated caspase-3, caspase-2, and caspase-9; and cleaved PARP. | Non-Hodgkin lymphoma | [92] |
| 213Bi-DOTA-biotin | Treated with anti-CD45 Ab-SA conjugate followed by 29.6 MBq of 213Bi- or 90Y-DOTA-biotin, 80% and 20% of mice survived leukemia-free for more than 100 days with limited toxicity, respectively. | Myeloid leukemia | [93] |
| 213Bi-DTPA-C595-mAb and 213Bi-DTPA-PAI2-mAb | After 16 weeks, systemic injections of 213Bi-conjugate at doses of 111, 222, and 333 MBq/kg induced significant tumor growth delay in a dose-dependent manner, compared with the non-specific control at 333 MBq/kg. | Pancreatic cancer | [94] |
| 213Bi-DOTA-biotin | Mice injected with anti-CD20 PTRNT or 22.2 MBq 213Bi-DOTA-biotin had significantly slower tumor growth than controls (mean tumor volume 0.01 ± 0.02 vs. 203.38 ± 83.03 mm3 after 19 days, respectively). | Non-Hodgkin lymphoma | [95] |
| 213Bi-CHX-A”-DTPA-7.16.4-mAb | In the same animal model, 213Bi radiolabeled immunoliposomes were successful in treating early-stage micrometastases, with median survival times comparable with those obtained with antibody-mediated 213Bi delivery. | Breast cancer | [96] |
| 213Bi-CHX-A”-DTPA-HuCC49ΔCH2 | The median survival time after treatment with 213Bi-HuCC49ΔCH2 was 45 days, which was equivalent to the median survival time after treatment with 213Bi-trastuzumab. | Colon carcinoma | [69] |
| 213Bi (213Bi-DTPA-[F3]2) | Except for the kidneys, where 213Bi-DTPA-[F3]2 was present due to renal excretion, 213Bi-DTPA-[F3]2 accumulated significantly in tumors, but only low activities were found in control organs. | Peritoneal carcinomatosis | [97] |
| 213Bi-DTPA-2Rs15d sdAb | Median survival significantly increased when 213Bi-DTPA-2Rs15d was given alone or in combination with trastuzumab. | Ovarian cancer | [5] |
| 213Bi-DTPA-PAI2-mAb | At 2 days and 2 weeks after cell inoculation, no lymphatic cancer spread was observed in the 222 MBq/kg 213Bi-DTPA-PAI2-mAb treated class. | Prostate cancer | [98] |
| Cancer Type | Radioligand | Patients | Reference |
|---|---|---|---|
| Leukemia | 213Bi-anti-CD33-mAb (SA) | 49 | [72] |
| Melanoma | 213Bi-anti-MCSP-mAb (SA) | 54 | [110,114,115] |
| Glioma | 213Bi-Substance P (SA) | 68 | [74,75,76,116] |
| Bladder cancer | 213Bi-anti-EGFR-mAb (LR) | 12 | [78] |
| Neuroendocrine tumor | 213Bi-DOTATOC (SA) | 25 | [19] |
| mCRPCa | 213Bi-PSMA-617 | 1 | [80] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahenkorah, S.; Cassells, I.; Deroose, C.M.; Cardinaels, T.; Burgoyne, A.R.; Bormans, G.; Ooms, M.; Cleeren, F. Bismuth-213 for Targeted Radionuclide Therapy: From Atom to Bedside. Pharmaceutics 2021, 13, 599. https://doi.org/10.3390/pharmaceutics13050599
Ahenkorah S, Cassells I, Deroose CM, Cardinaels T, Burgoyne AR, Bormans G, Ooms M, Cleeren F. Bismuth-213 for Targeted Radionuclide Therapy: From Atom to Bedside. Pharmaceutics. 2021; 13(5):599. https://doi.org/10.3390/pharmaceutics13050599
Chicago/Turabian StyleAhenkorah, Stephen, Irwin Cassells, Christophe M. Deroose, Thomas Cardinaels, Andrew R. Burgoyne, Guy Bormans, Maarten Ooms, and Frederik Cleeren. 2021. "Bismuth-213 for Targeted Radionuclide Therapy: From Atom to Bedside" Pharmaceutics 13, no. 5: 599. https://doi.org/10.3390/pharmaceutics13050599
APA StyleAhenkorah, S., Cassells, I., Deroose, C. M., Cardinaels, T., Burgoyne, A. R., Bormans, G., Ooms, M., & Cleeren, F. (2021). Bismuth-213 for Targeted Radionuclide Therapy: From Atom to Bedside. Pharmaceutics, 13(5), 599. https://doi.org/10.3390/pharmaceutics13050599