4.1. Study Design and Rationale for Interventions
The design of the present study and its interventions (study arms) was based on a mechanistic model describing drug release from ASDs and subsequent absorption in the gastrointestinal tract. This conceptual model consists of three different phases (
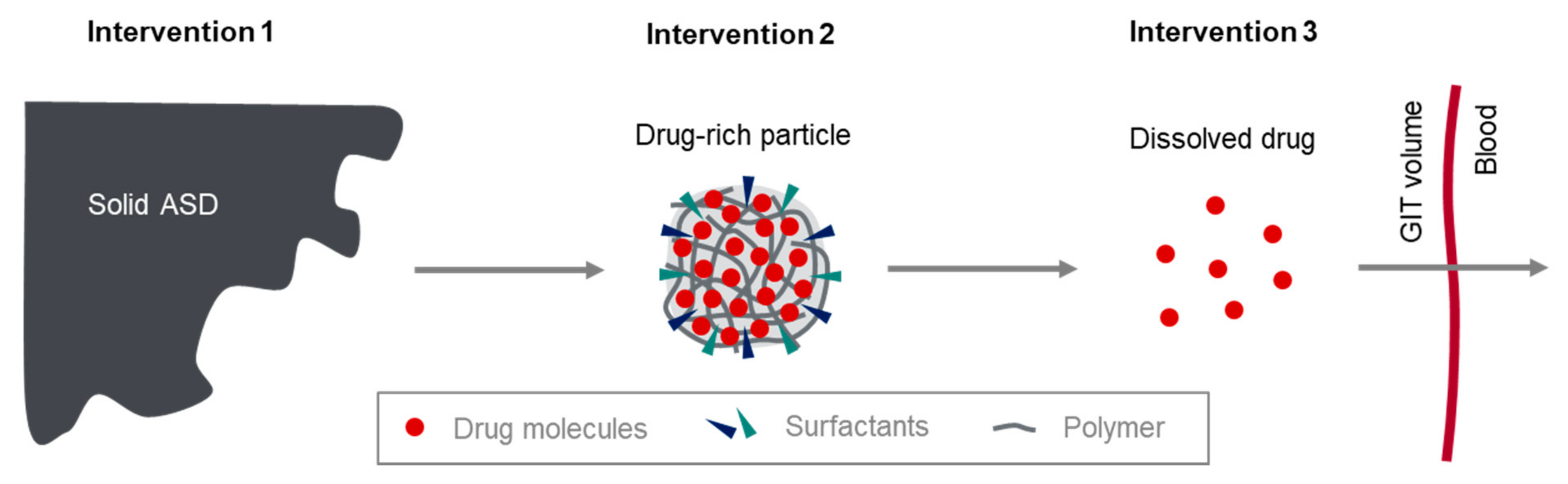
Figure 4), which can be summarized as follows:
The dissolution from solid ASD into the dissolved state of the ASD, showing drug-rich particles;
Drug liberation from the dissolved state (phase 1) of the ASD to molecularly dissolved drug;
Absorption of the molecularly dissolved drug in the gastrointestinal tract.
In the present study, three different drug formulations (study intervention 1–3) were tested, allowing for the investigation of the transition between the three conceptual stages outlined in
Figure 4 in humans. Intervention 3 was used to study intestinal absorption of the molecularly dissolved drug, intervention 2 represents the drug in form of drug-rich particles, and intervention 1 is the solid ASD.
It should be noted that the formation of the drug-rich particles is concentration-dependent and that the solubility of efavirenz in water is limited. Therefore, it was not possible to administer the same doses in all interventions. The volume of the drinking solution was maximized to 500 mL to maximize the dose of efavirenz that could be delivered in a solution (3 mg). This was necessary to guarantee sufficient plasma levels for concentration measurement. The dose of efavirenz delivered as ASD (50 mg) was increased high enough to guarantee the formation of a drug-rich particle but as close as possible to the comparative dose of 3 mg, even though literature data suggest that efavirenz has linear pharmacokinetics over an extended dose range [
19,
29]. Furthermore, saturation effects leading to nonlinear pharmacokinetics are unlikely at low doses. Linearity in the dose range of this study is supported by the almost identical pharmacokinetic profile of intervention 2 (dissolved ASD of efavirenz 50 mg) and intervention 3 (efavirenz solution 3 mg). Since no intravenous formulation of efavirenz is available [
27], as is the case for many poorly soluble drugs, the low dose oral solution seems to be the best comparator for study formulation effects. As the therapeutic dose of efavirenz is 600 mg daily, the doses used in the present study (3 and 50 mg) can be considered as a micro-dosing approach. Since a possible increase in efavirenz plasma concentrations compared to the marketed formulations could not be excluded in advance, the micro-dosing approach mitigates the risk of non-tolerable plasma levels in subjects.
It is exceedingly difficult to identify the reasons for reported inefficiencies of ASDs in animal or clinical studies. Our approach made it possible to investigate the mechanisms of increased bioavailability from ASDs in humans with a comparably simple and safe clinical study. The conceptual phases of our mechanistic model can be understood as a series of critical steps covering drug release from the formulation and drug absorption. To the best of our knowledge, this is the first study using such an approach to identify mechanisms limiting or enhancing the performance of ASDs.
The comparison of the novel efavirenz ASD formulation to existing formulations was not the primary aim of this study and was therefore not implemented as an additional study arm within the cross-over design. However, a comparison of our study results with existing data of a marketed formulation of efavirenz 50 mg gives insights into the performance of the novel formulation compared to a benchmark product.
Efavirenz has been used frequently for the research on ASDs [
30,
31,
32] since it is classified by some authors as a BCS class II drug [
33], i.e., with poor solubility but high permeability. Due to the lack of an intravenous formulation, no human data on the absolute bioavailability of efavirenz [
27] are available. Data in animals might hint toward a poor absolute bioavailability (16% in rats and 42% in monkeys [
19]); however, even for the more reliable data for bioavailability from monkeys [
34], a direct translation of animal data to humans is questionable. Various authors state the absolute bioavailability in humans to be 40–45%, but without referencing original data in humans. However, looking at the obtained results, it seems that efavirenz might be subject to a ceiling effect. Indeed, all interventions as well as the marked formulation show comparable
AUC0–t. It is therefore tempting to speculate that the absolute bioavailability of efavirenz might be higher than observed in preclinical studies, making it challenging to increase the bioavailability further. This is also suggested by the rapid absorption (high
ka), indicating that the absorption of efavirenz might be complete. On the other hand, some authors proposed that efavirenz is less permeable than commonly described in the literature [
35], which could reduce the sensitivity to detect formulation effects on the pharmacokinetics of efavirenz (
tmax and
ka values). However, taking into account the linear pharmacokinetics and the distinctly different values for
ka observed in the different interventions in this study, it is suggested that permeability does not limit the bioavailability (
AUC0–t) of efavirenz. Regarding the metabolism of efavirenz, it should be noted that in the present study only plasma concentrations of the parent drug were determined, as drug elimination was not a focus of this study. Furthermore, the metabolites (mainly 8-hydroxy-efavirenz through CYP2B6 but also 7-hydroxy-efavirenz through CYP2A6 [
36]) are not relevant pharmacologically [
37].
The sample size in this study was determined based on experience with previous pharmacokinetic studies. Here, the low within-subject CV of 6.9% (
AUC0–24 h ratio between cross-over interventions) indicates that the sample size of 16 subjects is large enough to differentiate between interventions [
18]. Furthermore, to compare the existing data of a marketed formulation to our study data, an equal sample size maximizes the ANOVA robustness. For the chosen sample size, normality violations were observed in some cases, which, despite the robustness of variance analysis regarding these violations, represent a limitation in the statistical analysis. For further details, refer to
Appendix D. The wash-out period of at least 14 days was chosen to keep the inclusion time of study subjects short. In some subjects, baseline corrections were necessary due to the long half-life of efavirenz. Quantitatively, the corrections were negligible and should therefore not have influenced our interpretations of the results.
4.2. Effects of ASD on the Bioavailability of Efavirenz in Humans
Looking at the pharmacokinetic analysis, it is striking that intervention 2 (dissolved ASD of efavirenz 50 mg) behaved almost identically to intervention 3 (solution of efavirenz 3 mg). This indicates that even though intervention 2 consisted of a supersaturated aqueous solution containing drug-rich particles as shown earlier by our group [
21], it pharmacokinetically behaved like a solution. Under the assumption of passive absorption of molecularly dissolved efavirenz, it therefore can be deduced that the dissolved drug concentration in the intestine (the site of absorption) of intervention 2 was proportionally higher than in intervention 3 (by a factor of 50/3 = 16.7). The drug concentration in intervention 2 (50 mg/500 mL) exceeded the aqueous solubility of efavirenz (approximately 10 mg/L [
35]) by far, even when taking into account the possibility of larger volumes of gastrointestinal fluids being present before administration. We could therefore show for the first time in humans, that drug-rich particles resulting from ASDs are an efficient oral drug-delivery system with a rapid and complete transformation of the drug into the systemic circulation.
Regarding the equilibria of the different states of the drug (molecularly dissolved, amorphous liquid phase separation, and crystalline), the solution-like behavior indicates the absence of any hindrance to drug absorption caused by the delivery system, even if part of the drug is expected to be in the drug-rich particles initially. Based on our review [
28], these results support the hypothesis that (1) crystallization of drug could be prevented in the investigated formulation, (2) there was no permanent (irreversible) solubilization of drug into micelles which would prevent drug absorption, and (3) the polymeric particles did not reduce the intestinal concentration of molecularly dissolved drug in the chosen study setting. These are important prerequisites for an ASD to function as a bioavailability-increasing drug-delivery system. Drug delivery in the form of drug-rich particles seems to facilitate a fast and efficient drug absorption and to reduce the previously modeled late efavirenz absorption from standard formulations in distant intestinal parts [
38].
The question arises to which degree bile salts might influence the ASDs performance in the present study. Subjects have fasted, therefore, the aqueous drinking solution is expected to trigger only a partial gall bladder emptying [
39]. However, it was reported that bile salts significantly increase the solubility of efavirenz from 10 to 194 mg/L in fasted simulated intestinal fluid containing bile salts (FaSSIF) [
40]. Further, no food effect on bioavailability at therapeutic doses was observed at a dose for 100 mg [
40], indicating that bioavailability could be high at low doses, which would explain the comparable
AUC0–t for the different interventions as well as for the marketed formulation. For the rationale of the choice of the dose regimen used in this study, refer to
Section 4.1. Based on these results, dose-escalation studies would be necessary to shed more light on the factors influencing the bioavailability of efavirenz in humans.
As intervention 2 (dissolved ASD of efavirenz 50 mg) showed a complete and fast absorption, the delayed absorption in intervention 1 (ASD of efavirenz 50 mg) is most likely caused by the dissolution of the solid ASD to drug-rich particles. This process was slightly faster and more efficient than dissolution from the marketed tablet (refer to
Section 4.4). This is a positive finding in the context of improving human bioavailability, showing that the ASD formulation approach can result in an efficient dissolution process. Regarding dissolution mechanisms from ASDs into drug-rich particles in humans, detailed conclusions are not possible based on this study.
4.4. ASD Formulation
The presented data from the novel ASD formulation utilized in the present study were compared to an existing marketed formulation [
18]. Based on the obtained
tmax and
Cmax values, the dissolution process of the ASD formulation was not different from the marketed formulation based on the statistics performed.
Based on
AUC0–t values (as an indicator for bioavailability), the ASD formulation of efavirenz performed as well as the marketed formulation. It is worthwhile to note, that the marketed tablet formulation we used for our comparisons contains solubility enhancers (sodium lauryl sulfate) [
41] and therefore has a higher bioavailability than the marked solution in triglycerides [
19]. Overall, the novel ASD formulation used in this study compared favorably to the marketed product regarding pharmacokinetic performance. Furthermore, this can be regarded as a positive example for the use of the surfactants sucrose palmitate and polysorbate 80 in an ASD formulation in humans. Further comparative investigations would be necessary to elucidate the isolated effects of the surfactants and if their effect would align with observations made in preclinical development [
21]. A limitation of this comparison is that a high to complete bioavailability of efavirenz at the doses used in this study cannot be excluded.
A limitation of this comparison is the non-crossover design (different subject groups) and a relatively small sample size. Furthermore, the clinical procedure for the administration of the marked formulation was slightly different from the one in this study: For the marketed formulation, subjects ingested the tablet with only 240 mL of water compared to 500 mL of buffer in our study.
In an attempt to shed light on the dissolution behavior of the formulation in vivo, a PBPK model was fitted to the experimentally measured PK profiles in order to extract simulated in vivo dissolution curves. Their discussion including limitations are shown in
Supplementary Material S1, PBPK Modeling: 4 Discussion.
From the pharmaceutics point of view, an unaltered implementation of this novel formulation as a therapeutic product would be challenging due to the low drug load of 22% (w/w), resulting in a high pill burden. A dose-escalation study would be necessary to investigate the possible increase in bioavailability, and therefore reduction of dose, at therapeutic doses.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



