
Peptide-Functionalized Dendrimer Nanocarriers for Targeted Microdystrophin Gene Delivery

 , ,
, , 
Abstract
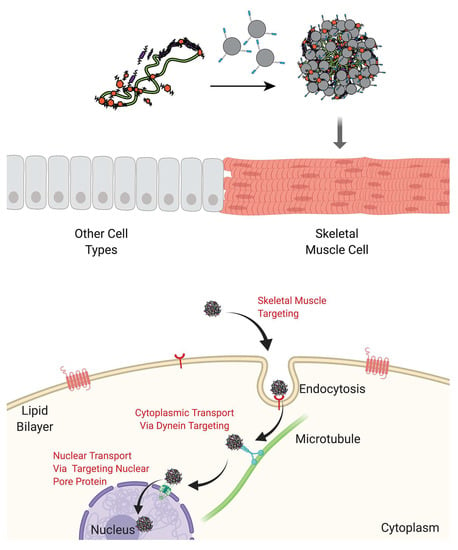
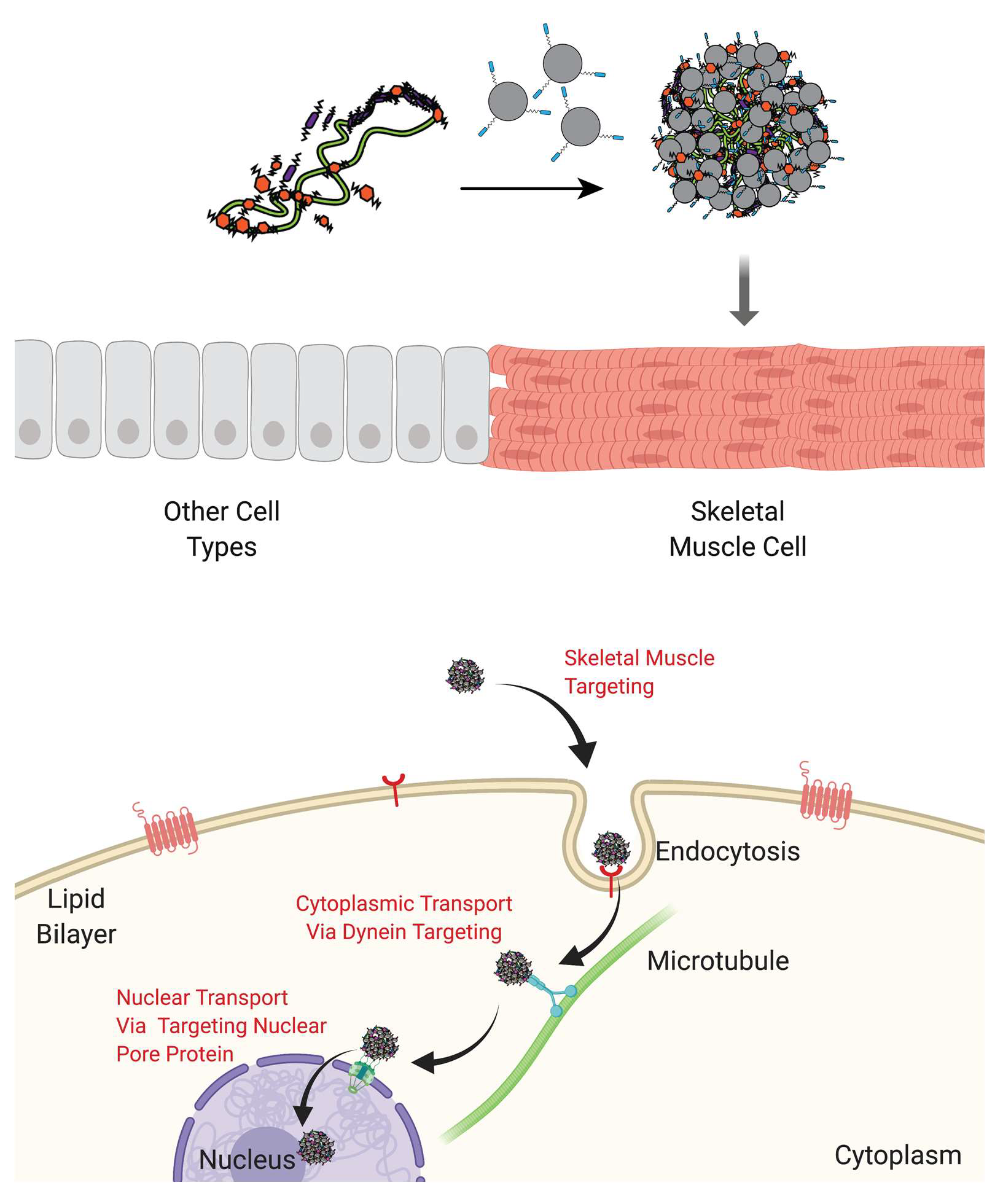{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
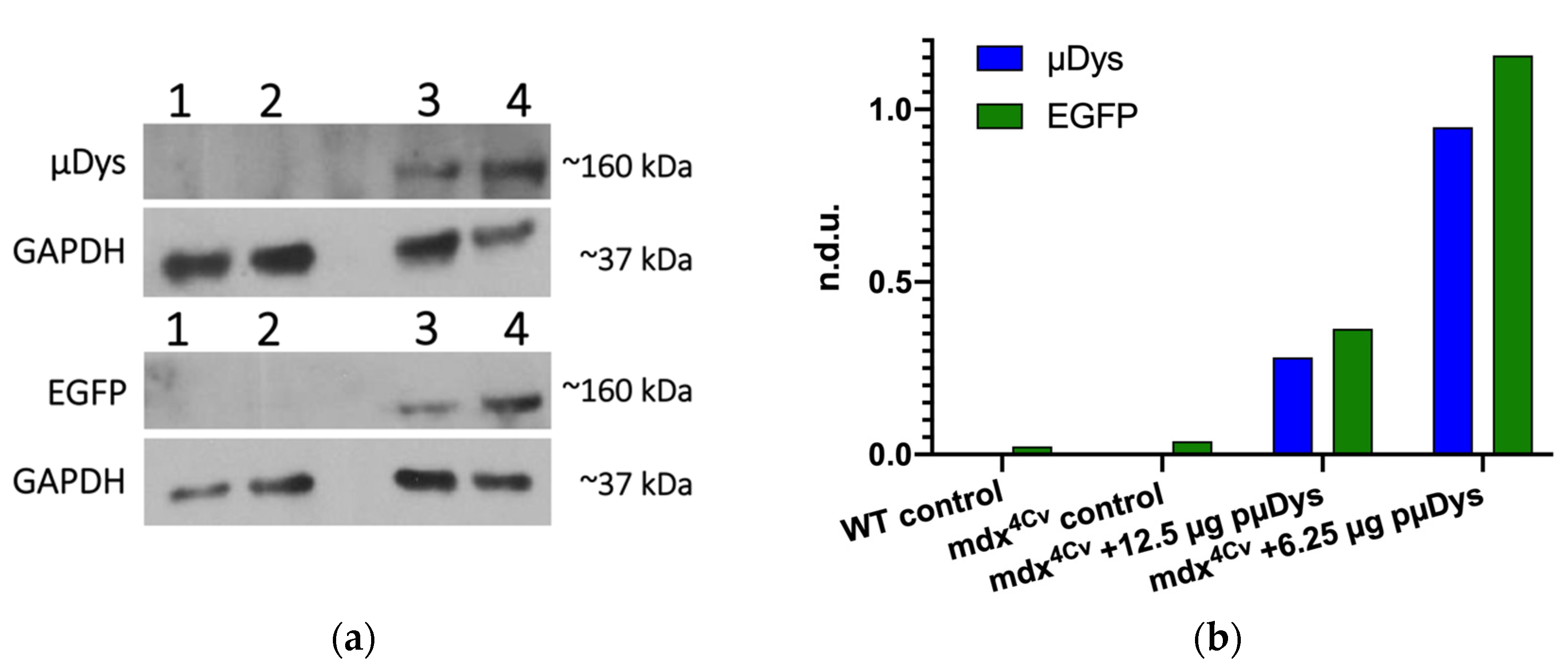{kind=link}
1. Introduction
2. Materials and Methods
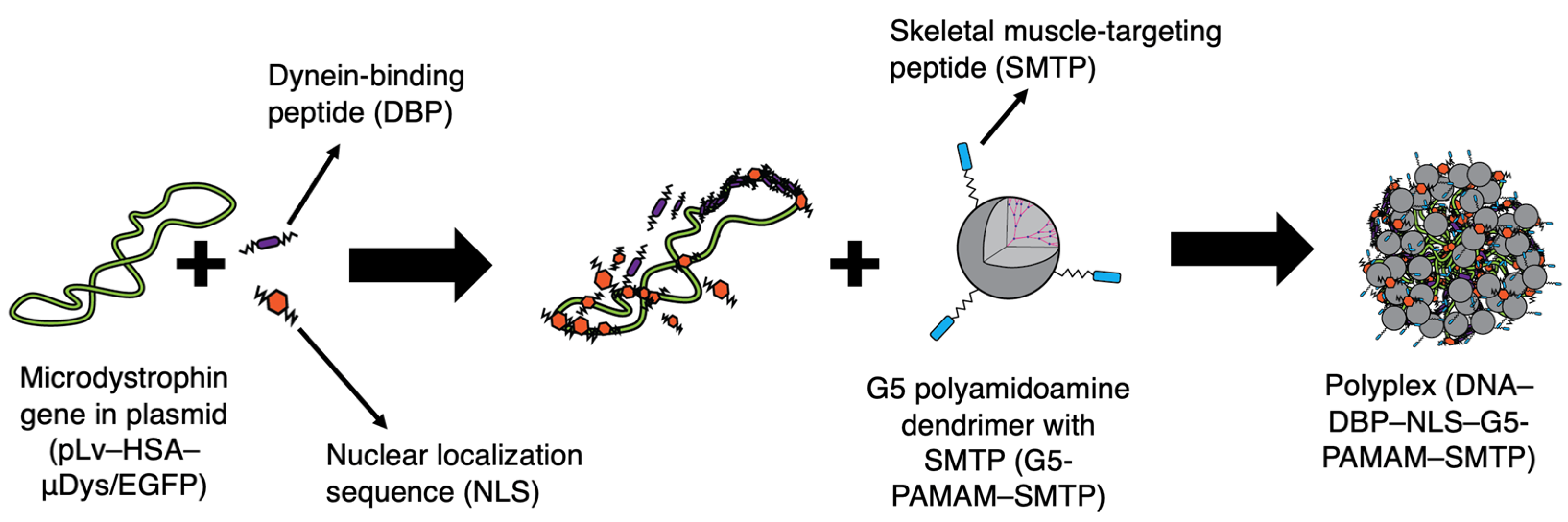
2.1. Dendrimer−DBP–NLS–Plasmid DNA Polyplex Formulation
2.1.1. Main Polyplex Formulation Scheme
2.1.2. Polyplex Formulation with a Fusion Peptide
2.1.3. Polyplex Storage
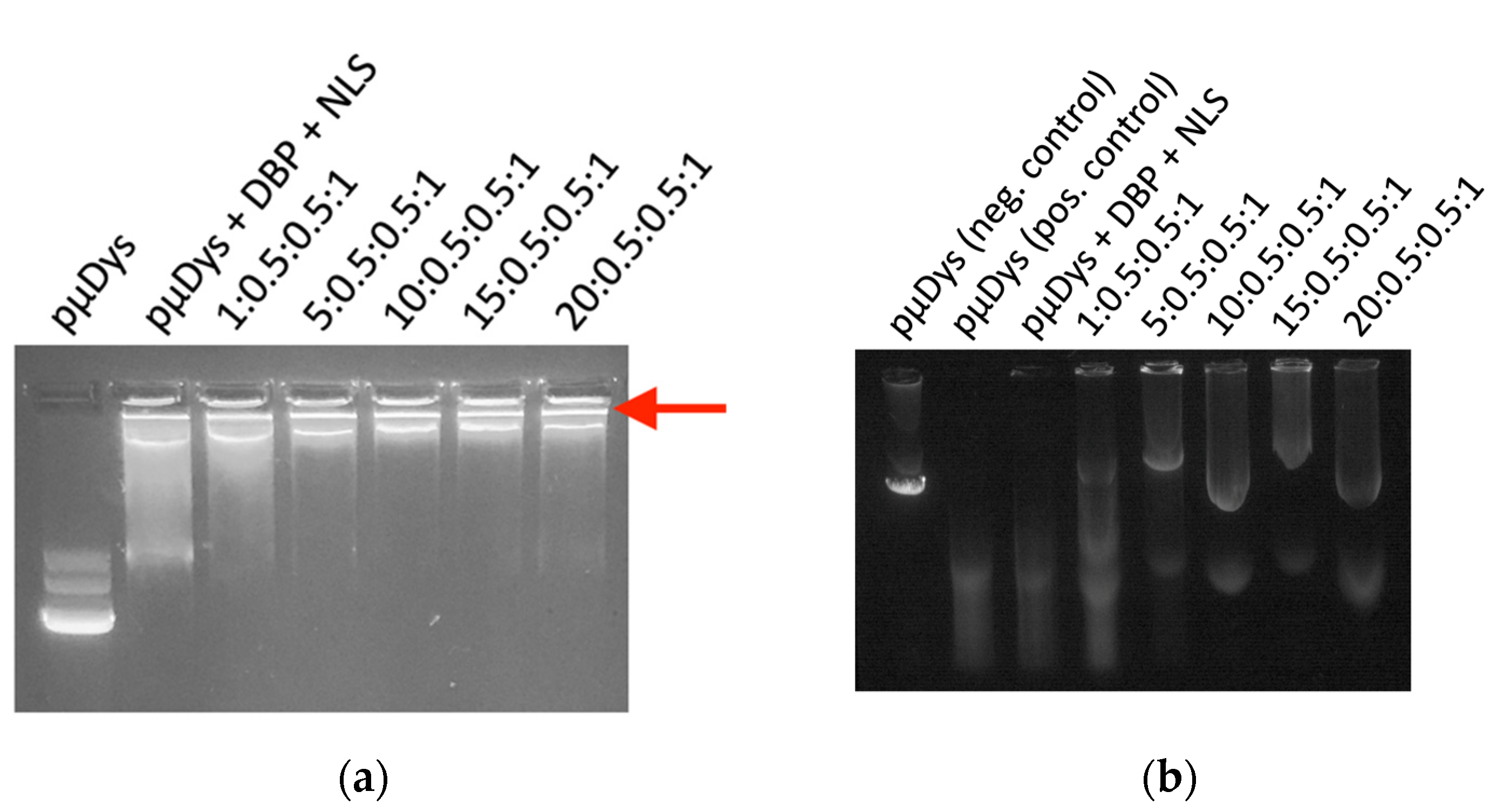
2.2. Gel Retention Assay
2.3. Protection from Serum Degradation Assay
2.4. Size and Zeta Potential Characterization
2.5. Cytotoxicity Assay
2.6. In Vitro Gene Delivery Studies
2.6.1. Cell Lines and Transfections
2.6.2. Live Cell Image Acquisition and Analysis
2.7. In Vivo Gene Delivery
2.8. Gene and Protein Expression
2.9. Statistical Analysis
3. Results and Discussion
3.1. Characterization of Nanocarrier Polyplexes
3.1.1. Protection of pµDys
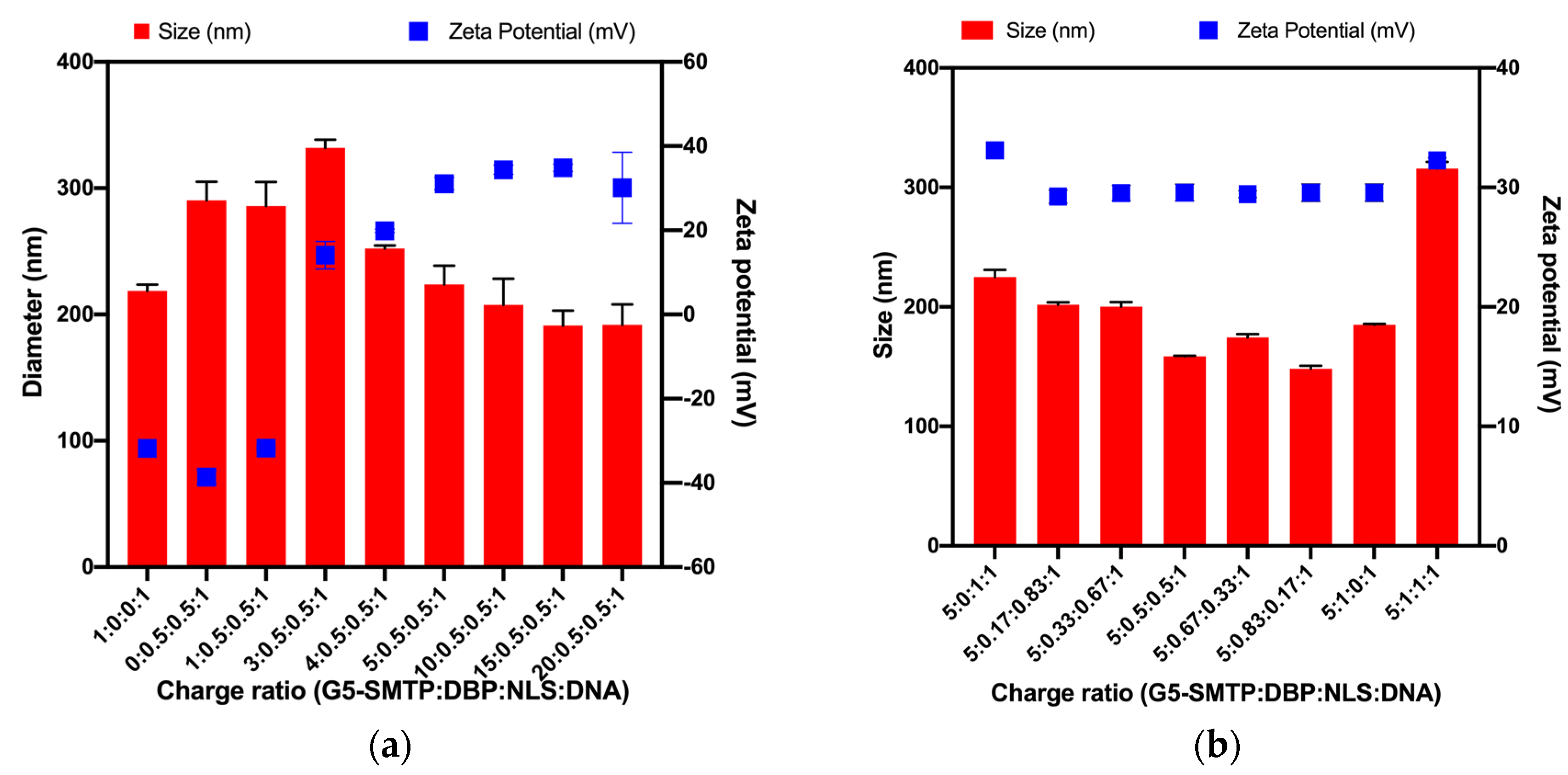
3.1.2. Size and Surface Charges of Polyplexes
3.2. In Vitro Performance of Nanocarriers
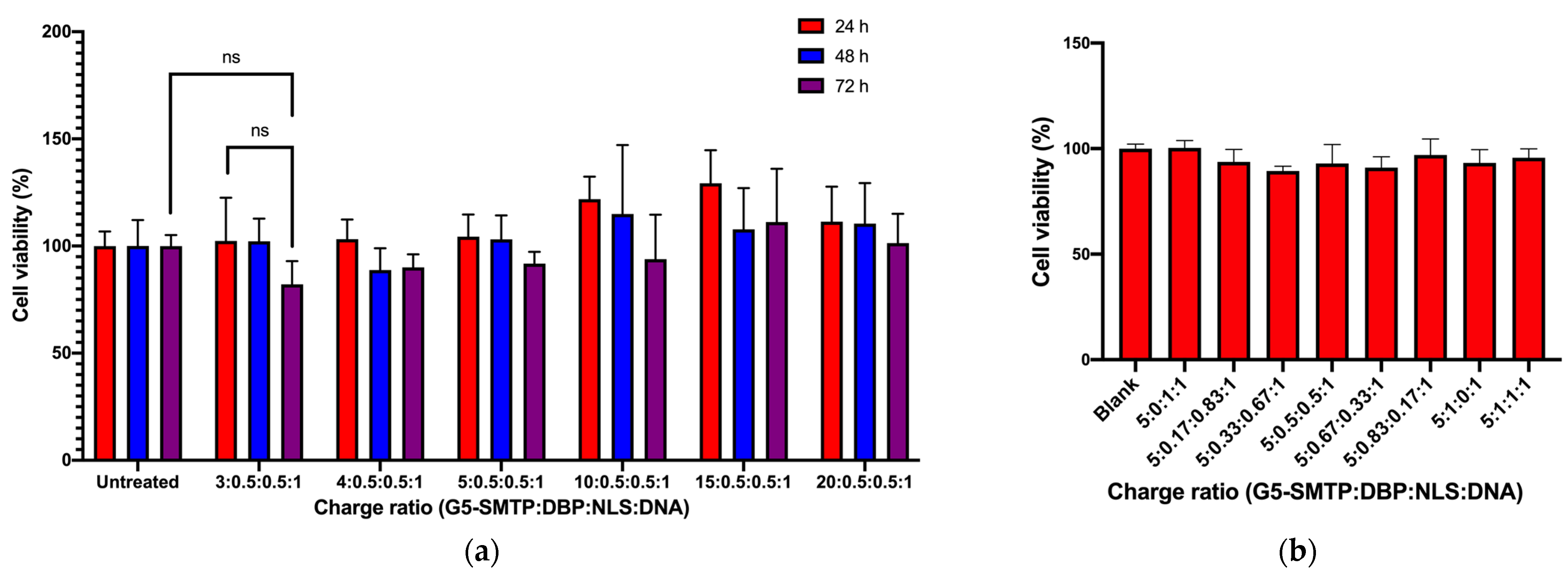
3.2.1. Cytotoxicity in C2C12 Mouse Skeletal Muscle Cells
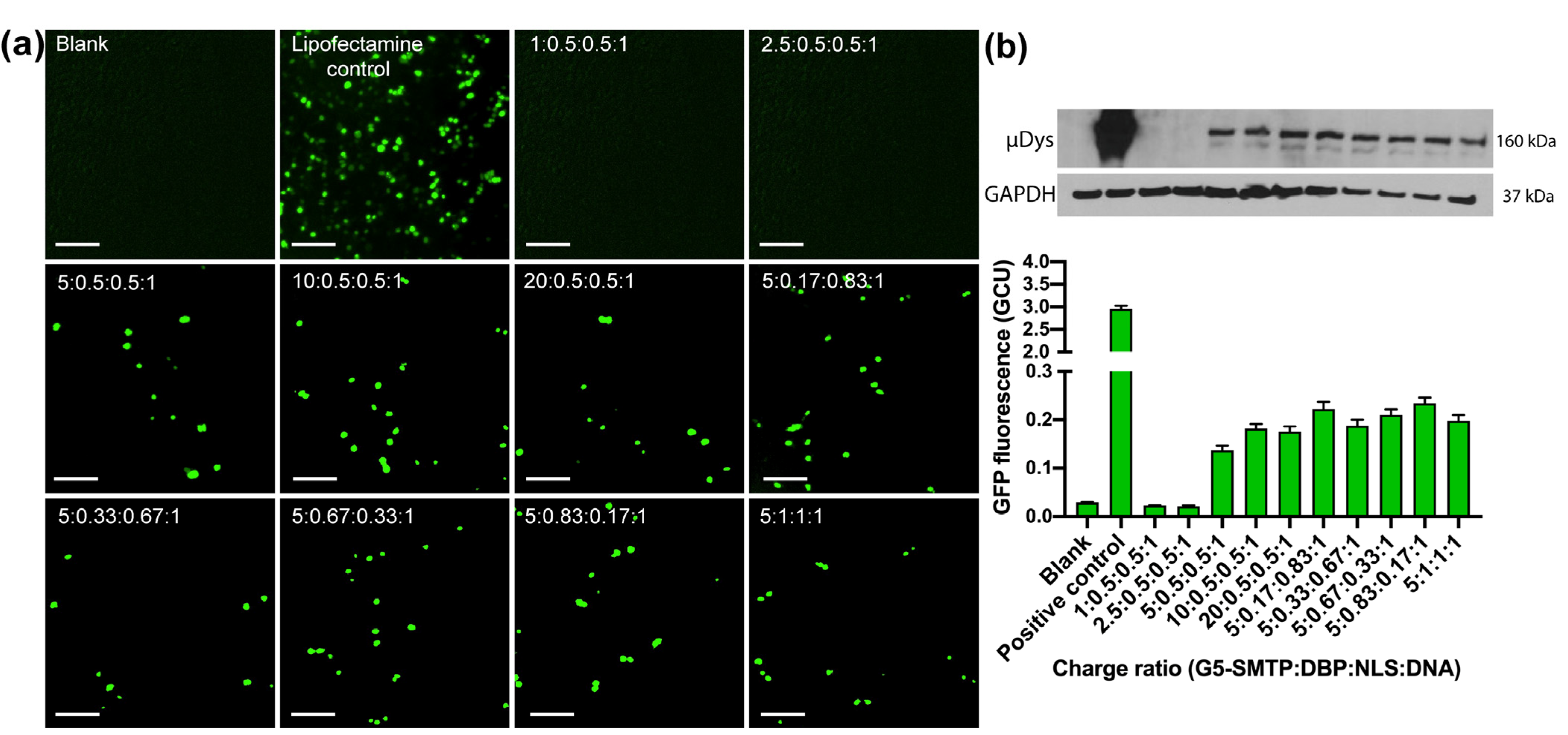
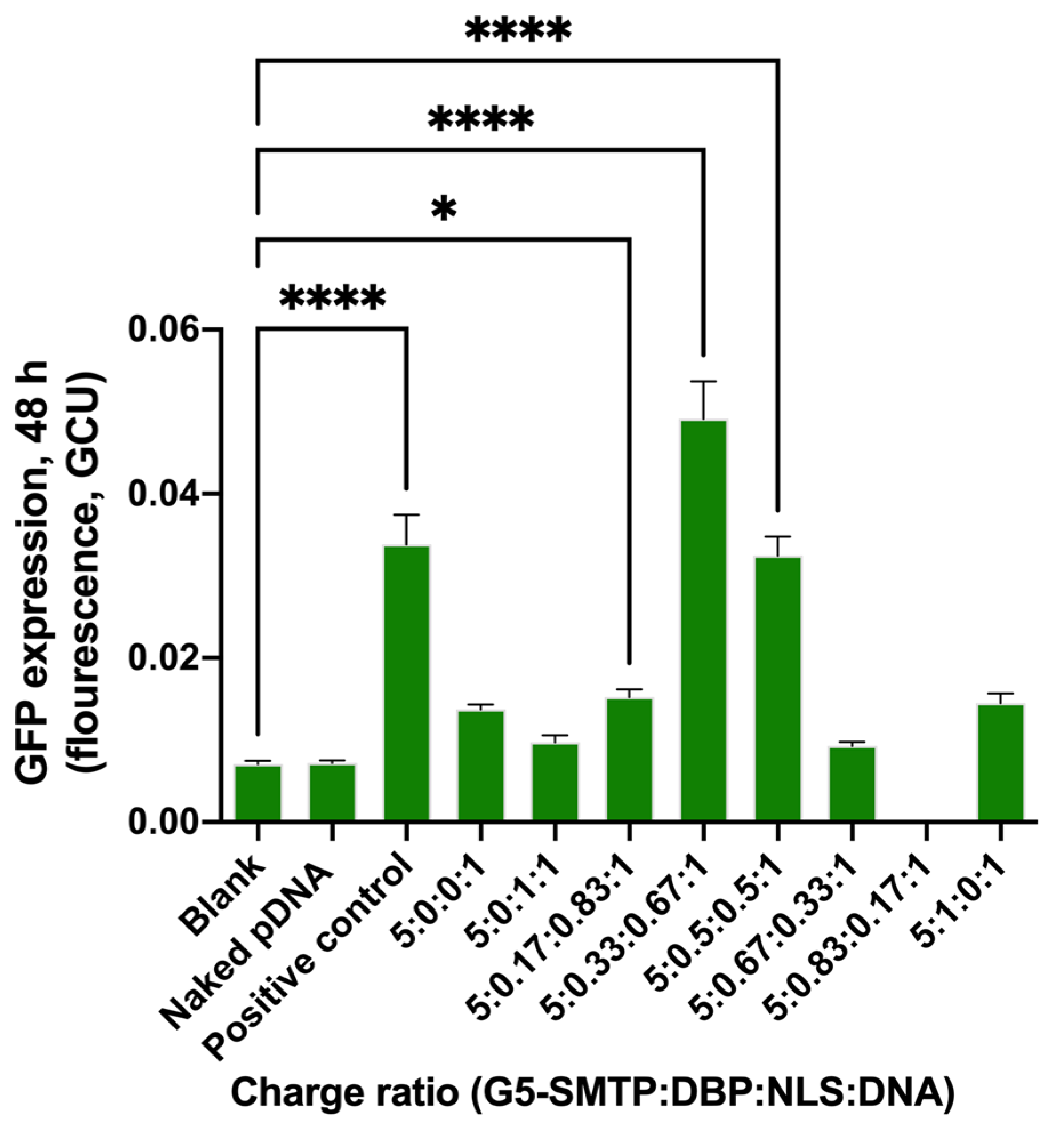
3.2.2. Transfection in HEK 293T and C2C12 Cells
3.3. In Vivo Performance of Nanocarriers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Genetic Alliance; District of Columbia Department of Health. Appendix G Single-Gene Disorders. In Understanding Genetics: A District of Columbia Guide for Patients and Health Professionals; Genetic Alliance: London, UK, 2010. [Google Scholar]
- Chial, H. Rare Genetic Disorders: Learning About Genetic Disease Through Gene Mapping, SNPs, and Microarray Data. Nat. Educ. 2008, 1, 192. [Google Scholar]
- Boland, B.J.; Silbert, P.L.; Groover, R.V.; Wollan, P.C.; Silverstein, M.D. Skeletal, cardiac, and smooth muscle failure in Duchenne muscular dystrophy. Pediatr. Neurol. 1996, 14, 7–12. [Google Scholar] [CrossRef]
- Landfeldt, E.; Thompson, R.; Sejersen, T.; McMillan, H.J.; Kirschner, J.; Lochmüller, H. Life expectancy at birth in Duchenne muscular dystrophy: A systematic review and meta-analysis. Eur. J. Epidemiol. 2020, 35, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Campbell, K.; Rodino-Klapac, L.; Sahenk, Z.; Shilling, C.; Lewis, S.; Bowles, D.; Gray, S.; Li, C.; Galloway, G.; et al. Dystrophin Immunity in Duchenne’s Muscular Dystrophy. N. Engl. J. Med. 2010, 363, 1429–1437. [Google Scholar] [CrossRef]
- Angelini, C.; Peterle, E. Old and new therapeutic developments in steroid treatment in Duchenne muscular dystrophy. Acta Myol. 2012, 31, 9–15. [Google Scholar] [PubMed]
- Petrich, J.; Marchese, D.; Jenkins, C.; Storey, M.; Blind, J. Gene Replacement Therapy: A Primer for the Health-system Pharmacist. J. Pharm. Pract. 2020, 33, 846–855. [Google Scholar] [CrossRef]
- Takefman, D.; Bryan, W. The state of gene therapies: The FDA perspective. Mol. Ther. 2012, 20, 877–878. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gonçalves, G.A.R.; de Paiva, R.M.A. Gene therapy: Advances, challenges and perspectives. Einstein 2017, 15, 369–375. [Google Scholar] [CrossRef]
- Lim, K.R.Q.; Maruyama, R.; Yokota, T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des. Devel. Ther. 2017, 11, 533–545. [Google Scholar] [CrossRef]
- Gao, Q.Q.; McNally, E.M. The Dystrophin Complex: Structure, Function, and Implications for Therapy. In Comprehensive Physiology; Wiley: Hoboken, NJ, USA, 2015; Volume 5, pp. 1223–1239. [Google Scholar]
- Duan, D. Systemic AAV Micro-dystrophin Gene Therapy for Duchenne Muscular Dystrophy. Mol. Ther. 2018, 26, 2337–2356. [Google Scholar] [CrossRef]
- Benabdallah, B.F.; Duval, A.; Rousseau, J.; Chapdelaine, P.; Holmes, M.C.; Haddad, E.; Tremblay, J.P.; Beauséjour, C.M. Targeted gene addition of microdystrophin in mice skeletal muscle via human myoblast transplantation. Mol. Ther.-Nucleic Acids 2013, 2, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Pan, X.; Hakim, C.H.; Yang, H.T.; Yue, Y.; Zhang, K.; Terjung, R.L.; Duan, D. Microdystrophin ameliorates muscular dystrophy in the canine model of duchenne muscular dystrophy. Mol. Ther. 2013, 21, 750–757. [Google Scholar] [CrossRef]
- Vulin, A.; Barthélémy, I.; Goyenvalle, A.; Thibaud, J.L.; Beley, C.; Griffith, G.; Benchaouir, R.; Le Hir, M.; Unterfinger, Y.; Lorain, S.; et al. Muscle function recovery in golden retriever muscular dystrophy after AAV1-U7 exon skipping. Mol. Ther. 2012, 20, 2120–2133. [Google Scholar] [CrossRef] [PubMed]
- Bish, L.T.; Sleeper, M.M.; Forbes, S.C.; Wang, B.; Reynolds, C.; Singletary, G.E.; Trafny, D.; Morine, K.J.; Sanmiguel, J.; Cecchini, S.; et al. Long-term restoration of cardiac dystrophin expression in golden retriever muscular dystrophy following raav6-mediated exon skipping. Mol. Ther. 2012, 20, 580–589. [Google Scholar] [CrossRef]
- Wang, Z.; Storb, R.; Halbert, C.L.; Banks, G.B.; Butts, T.M.; Finn, E.E.; Allen, J.M.; Miller, A.D.; Chamberlain, J.S.; Tapscott, S.J. Successful regional delivery and long-term expression of a dystrophin gene in canine muscular dystrophy: A preclinical model for human therapies. Mol. Ther. 2012, 20, 1501–1507. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Sahenk, Z.; Lehman, K.; Nease, C.; Lowes, L.P.; Miller, N.F.; Iammarino, M.A.; Alfano, L.N.; Nicholl, A.; Al-Zaidy, S.; et al. Assessment of Systemic Delivery of rAAVrh74.MHCK7.micro-dystrophin in Children with Duchenne Muscular Dystrophy: A Nonrandomized Controlled Trial. JAMA Neurol. 2020, 77, 1122–1131. [Google Scholar] [CrossRef] [PubMed]
- Linden, R. Gene therapy: What it is, what it is not and what it will be. Estud. Avançados 2010, 24, 31–69. [Google Scholar] [CrossRef]
- Daftarian, P.M.; Stone, G.W.; Kovalski, L.; Kumar, M.; Vosoughi, A.; Urbieta, M.; Blackwelder, P.; Dikici, E.; Serafini, P.; Duffort, S.; et al. A targeted and adjuvanted nanocarrier lowers the effective dose of liposomal amphotericin B and enhances adaptive immunity in murine cutaneous leishmaniasis. J. Infect. Dis. 2013, 208, 1914–1922. [Google Scholar] [CrossRef]
- Jativa, S.D.; Thapar, N.; Broyles, D.; Dikici, E.; Daftarian, P.; Jiménez, J.J.; Daunert, S.; Deo, S.K. Enhanced Delivery of Plasmid DNA to Skeletal Muscle Cells using a DLC8-Binding Peptide and ASSLNIA-Modified PAMAM Dendrimer. Mol. Pharm. 2019, 16, 2376–2384. [Google Scholar] [CrossRef]
- Zhong, T.; Ai, P.; Zhou, J. Structures and properties of PAMAM dendrimer: A multi-scale simulation study. Fluid Phase Equilib. 2011, 302, 43–47. [Google Scholar] [CrossRef]
- Labieniec-Watala, M.; Watala, C. PAMAM dendrimers: Destined for success or doomed to fail? Plain and modified PAMAM dendrimers in the context of biomedical applications. J. Pharm. Sci. 2015, 104, 2–14. [Google Scholar] [CrossRef]
- Liu, Z.-J.J.; Daftarian, P.; Kovalski, L.; Wang, B.; Tian, R.; Castilla, D.M.; Dikici, E.; Perez, V.L.; Deo, S.; Daunert, S.; et al. Directing and Potentiating Stem Cell-Mediated Angiogenesis and Tissue Repair by Cell Surface E-Selectin Coating. PLoS ONE 2016, 11, e0154053. [Google Scholar] [CrossRef]
- Mandal, A.K. Dendrimers in targeted drug delivery applications: A review of diseases and cancer. Int. J. Polym. Mater. Polym. Biomater. 2021, 70, 287–297. [Google Scholar] [CrossRef]
- Hersh, J.; Broyles, D.; Capcha, J.M.C.; Dikici, E.; Shehadeh, L.A.; Daunert, S.; Deo, S. Peptide-Modified Biopolymers for Biomedical Applications. ACS Appl. Bio Mater. 2021, 4, 229–251. [Google Scholar] [CrossRef]
- Daftarian, P.; Kaifer, A.E.; Li, W.; Blomberg, B.B.; Frasca, D.; Roth, F.; Chowdhury, R.; Berg, E.A.; Fishman, J.B.; Al Sayegh, H.A.; et al. Peptide-conjugated PAMAM dendrimer as a universal DNA vaccine platform to target antigen-presenting cells. Cancer Res. 2011, 71, 7452–7462. [Google Scholar] [CrossRef]
- Raza, F.; Zafar, H.; Zhu, Y.; Ren, Y.; Ullah, A.; Khan, A.U.; He, X.; Han, H.; Aquib, M.; Boakye-Yiadom, K.O.; et al. A review on recent advances in stabilizing peptides/proteins upon fabrication in hydrogels from biodegradable polymers. Pharmaceutics 2018, 10, 16. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.P.; Petrenko, V.A. Phage display. Chem. Rev. 1997, 97, 391–410. [Google Scholar] [CrossRef]
- Koivunen, E.; Arap, W.; Rajotte, D.; Lahdenranta, J.; Pasqualini, R. Identification of receptor ligands with phage display peptide libraries. J. Nucl. Med. 1999, 40, 883–888. [Google Scholar]
- Pasqualini, R.; Ruoslahti, E. Organ targeting in vivo using phage display peptide libraries. Nature 1996, 380, 364–366. [Google Scholar] [CrossRef]
- Rajendran, L.; Knölker, H.J.; Simons, K. Subcellular targeting strategies for drug design and delivery. Nat. Rev. Drug Discov. 2010, 9, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Ebner, D.C.; Bialek, P.; El-Kattan, A.F.; Ambler, C.M.; Tu, M.H. Strategies for Skeletal Muscle Targeting in Drug Discovery. Curr. Pharm Des. 2015, 21, 27–1336. [Google Scholar] [CrossRef]
- Samoylova, T.I.; Smith, B.F. Elucidation of muscle-binding peptides by phage display screening. Muscle Nerve 1999, 22, 460–466. [Google Scholar] [CrossRef]
- Favaro, M.T.P.; de Toledo, M.A.S.; Alves, R.F.; Santos, C.A.; Beloti, L.L.; Janissen, R.; de la Torre, L.G.; Souza, A.P.; Azzoni, A.R. Development of a non-viral gene delivery vector based on the dynein light chain Rp3 and the TAT peptide. J. Biotechnol. 2014, 173, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Toledo, M.A.S.; Favaro, M.T.P.; Alves, R.F.; Santos, C.A.; Beloti, L.L.; Crucello, A.; Santiago, A.S.; Mendes, J.S.; Horta, M.A.C.; Aparicio, R.; et al. Characterization of the human dynein light chain Rp3 and its use as a non-viral gene delivery vector. Appl. Microbiol. Biotechnol. 2014, 98, 3591–3602. [Google Scholar] [CrossRef] [PubMed]
- Ray, M.; Tang, R.; Jiang, Z.; Rotello, V.M. Quantitative Tracking of Protein Trafficking to the Nucleus Using Cytosolic Protein Delivery by Nanoparticle-Stabilized Nanocapsules. Bioconjug. Chem. 2015, 26, 1004–1007. [Google Scholar] [CrossRef] [PubMed]
- Zanta, M.A.; Belguise-Valladier, P.; Behr, J.P. Gene delivery: A single nuclear localization signal peptide is sufficient to carry DNA to the cell nucleus. Proc. Natl. Acad. Sci. USA 1999, 96, 91–96. [Google Scholar] [CrossRef]
- Pipe, S.; Leebeek, F.W.G.; Ferreira, V.; Sawyer, E.K.; Pasi, J. Clinical Considerations for Capsid Choice in the Development of Liver-Targeted AAV-Based Gene Transfer. Mol. Ther.-Methods Clin. Dev. 2019, 15, 170–178. [Google Scholar] [CrossRef]
- Chand, D.; Mohr, F.; McMillan, H.; Tukov, F.F.; Montgomery, K.; Kleyn, A.; Sun, R.; Tauscher-Wisniewski, S.; Kaufmann, P.; Kullak-Ublick, G. Hepatotoxicity following administration of onasemnogene abeparvovec (AVXS-101) for the treatment of spinal muscular atrophy. J. Hepatol. 2021, 74, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Kimura, E.; Han, J.J.; Li, S.; Fall, B.; Ra, J.; Haraguchi, M.; Tapscott, S.J.; Chamberlain, J.S. Cell-lineage regulated myogenesis for dystrophin replacement: A novel therapeutic approach for treatment of muscular dystrophy. Hum. Mol. Genet. 2008, 17, 2507–2517. [Google Scholar] [CrossRef]
- Riss, T.L.; Moravec, R.A.; Niles, A.L.; Duellman, S.; Benink, H.A.; Worzella, T.J.; Minor, L. Cell Viability Assays. Assay Guid. Man. 2004, 1–25, updated 2016 July 1. [Google Scholar]
- Ritter, P.; Yousefi, K.; Ramirez, J.; Dykxhoorn, D.M.; Mendez, A.J.; Shehadeh, L.A. LDL Cholesterol Uptake Assay Using Live Cell Imaging Analysis with Cell Health Monitoring. Physiol. Behav. 2020, 176, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Condor Capcha, J.M.; Lambert, G.; Dykxhoorn, D.M.; Salerno, A.G.; Hare, J.M.; Whitt, M.A.; Pahwa, S.; Jayaweera, D.T.; Shehadeh, L.A. Generation of SARS-CoV-2 Spike Pseudotyped Virus for Viral Entry and Neutralization Assays: A 1-Week Protocol. Front. Cardiovasc. Med. 2021, 7, 1–12. [Google Scholar] [CrossRef]
- Chapman, V.M.; Miller, D.R.; Armstrong, D.; Caskey, C.T. Recovery of induced mutations for X chromosome-linked muscular dystrophy in mice. Proc. Natl. Acad. Sci. USA 1989, 86, 1292–1296. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.-H.; Hakim, C.H.; Zhang, K.; Duan, D. Genotyping mdx, mdx3cv, and mdx4cv mice by primer competition polymerase chain reaction. Muscle Nerve 2011, 43, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Fox, L.J.; Richardson, R.M.; Briscoe, W.H. PAMAM dendrimer—Cell membrane interactions. Adv. Colloid Interface Sci. 2018, 257, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Taghavi Pourianazar, N.; Mutlu, P.; Gunduz, U. Bioapplications of poly(amidoamine) (PAMAM) dendrimers in nanomedicine. J. Nanopart. Res. 2014, 16, 2342. [Google Scholar] [CrossRef]
- Rizvi, S.A.A.; Saleh, A.M. Applications of nanoparticle systems in drug delivery technology. Saudi Pharm. J. 2018, 26, 64–70. [Google Scholar] [CrossRef]
- Hotze, E.M.; Phenrat, T.; Lowry, G.V. Nanoparticle Aggregation: Challenges to Understanding Transport and Reactivity in the Environment. J. Environ. Qual. 2010, 39, 1909–1924. [Google Scholar] [CrossRef]
- Kallay, N.; Žalac, S. Stability of nanodispersions: A model for kinetics of aggregation of nanoparticles. J. Colloid Interface Sci. 2002, 253, 70–76. [Google Scholar] [CrossRef]
- Zhang, W. Nanoparticle aggregation: Principles and modeling. Adv. Exp. Med. Biol. 2014, 811, 20–43. [Google Scholar] [CrossRef]
- Dobrovolskaia, M.A.; Patri, A.K.; Simak, J.; Hall, J.B.; Semberova, J.; De Paoli Lacerda, S.H.; McNeil, S.E. Nanoparticle Size and Surface Charge Determine Effects of PAMAM Dendrimers on Human Platelets in Vitro. Mol. Pharm. 2012, 9, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Pandita, D.; Poonia, N.; Kumar, S.; Lather, V.; Madaan, K. Dendrimers in drug delivery and targeting: Drug-dendrimer interactions and toxicity issues. J. Pharm. Bioallied Sci. 2014, 6, 139. [Google Scholar] [CrossRef] [PubMed]
- Biswas, A.K.; Islam, M.R.; Choudhury, Z.S.; Mostafa, A.; Kadir, M.F. Nanotechnology based approaches in cancer therapeutics. Adv. Nat. Sci. Nanosci. Nanotechnol. 2014, 5, 043001. [Google Scholar] [CrossRef]
- Cardarelli, F.; Digiacomo, L.; Marchini, C.; Amici, A.; Salomone, F.; Fiume, G.; Rossetta, A.; Gratton, E.; Pozzi, D.; Caracciolo, G. The intracellular trafficking mechanism of Lipofectamine-based transfection reagents and its implication for gene delivery. Sci. Rep. 2016, 6, 25879. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Larcher, L.M.; Ma, L.; Veedu, R.N. Systematic screening of commonly used commercial transfection reagents towards efficient transfection of single-stranded oligonucleotides. Molecules 2018, 23, 2564. [Google Scholar] [CrossRef]
- Ge, Y.; Zhang, Y.; Xia, J.; Ma, M.; He, S.; Nie, F.; Gu, N. Effect of surface charge and agglomerate degree of magnetic iron oxide nanoparticles on KB cellular uptake in vitro. Colloids Surf. B Biointerfaces 2009, 73, 294–301. [Google Scholar] [CrossRef]
- Yue, Z.G.; Wei, W.; Lv, P.P.; Yue, H.; Wang, L.Y.; Su, Z.G.; Ma, G.H. Surface charge affects cellular uptake and intracellular trafficking of chitosan-based nanoparticles. Biomacromolecules 2011, 12, 2440–2446. [Google Scholar] [CrossRef]
- Na, H.K.; Kim, H.; Son, J.G.; Lee, J.H.; Kim, J.K.; Park, J.; Lee, T.G. Facile synthesis and direct characterization of surface-charge-controlled magnetic iron oxide nanoparticles and their role in gene transfection in human leukemic T cell. Appl. Surf. Sci. 2019, 483, 1069–1080. [Google Scholar] [CrossRef]
- Erbacher, P.; Bettinger, T.; Belguise-Valladier, P.; Zou, S.; Coll, J.L.; Behr, J.P.; Remy, J.S. Transfection and Physical Properties of Various Saccharide, Poly(ethylene glycol), and Antibody-Derivatized Polyethylenimines (PEI). J. Gene Med. 1999, 1, 210–222. [Google Scholar] [CrossRef]
- Lee, H.; Jeong, J.H.; Park, T.G. PEG grafted polylysine with fusogenic peptide for gene delivery: High transfection efficiency with low cytotoxicity. J. Control. Release 2002, 79, 283–291. [Google Scholar] [CrossRef]
- Balci, B.; Dinçer, P. Efficient transfection of mouse-derived C2C12 myoblasts using a matrigel basement membrane matrix. Biotechnol. J. 2009, 4, 1042–1045. [Google Scholar] [CrossRef]
- Neuhuber, B.; Huang, D.I.; Daniels, M.P.; Torgan, C.E. High efficiency transfection of primary skeletal muscle cells with lipid-based reagents. Muscle Nerve 2002, 26, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.F.; Hoversten, K.E.; Powers, J.M.; Trobridge, G.D.; Rodgers, B.D. Genetic manipulation of myoblasts and a novel primary myosatellite cell culture system: Comparing and optimizing approaches. FEBS J. 2013, 280, 827–839. [Google Scholar] [CrossRef]
- Liang, K.W.; Hoffman, E.P.; Huang, L. Targeted Delivery of Plasmid DNA to Myogenic Cells via Transferrin-Conjugated Peptide Nucleic Acid. Mol. Ther. 2000, 1, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Danko, I.; Chapman, V.; Wolff, J.A. The frequency of revertants in mdx mouse genetic models for duchenne muscular dystrophy. Pediatr. Res. 1992, 32, 128–131. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuis, B.; Haenzi, B.; Hilton, S.; Carnicer-Lombarte, A.; Hobo, B.; Verhaagen, J.; Fawcett, J.W. Optimization of adeno-associated viral vector-mediated transduction of the corticospinal tract: Comparison of four promoters. Gene Ther. 2021, 28, 56–74. [Google Scholar] [CrossRef]
- Haery, L.; Deverman, B.E.; Matho, K.S.; Cetin, A.; Woodard, K.; Cepko, C.; Guerin, K.I.; Rego, M.A.; Ersing, I.; Bachle, S.M.; et al. Adeno-Associated Virus Technologies and Methods for Targeted Neuronal Manipulation. Front. Neuroanat. 2019, 13, 93. [Google Scholar] [CrossRef]
- Vorburger, S.A.; Hunt, K.K. Adenoviral Gene Therapy. Oncologist 2002, 7, 46–59. [Google Scholar] [CrossRef]
- Nance, M.E.; Hakim, C.H.; Yang, N.N.; Duan, D. Nanotherapy for Duchenne muscular dystrophy. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2018, 10, e1472. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.S.; Prabhakaran, V.; Verma, R.J. Design of non-viral vector with improved regulatory features towards therapeutic application. Bioinformation 2020, 16, 307–313. [Google Scholar] [CrossRef]
- Muses, S.; Morgan, J.E.; Wells, D.J. Restoration of dystrophin expression using the Sleeping Beauty transposon. PLoS Curr. 2011, 3, RRN1296. [Google Scholar] [CrossRef] [PubMed]
- Weisbart, R.H.; Hansen, J.E.; Nishimura, R.N.; Chan, G.; Wakelin, R.; Chang, S.S.; Baresi, L.; Chamberlain, J.S. An intracellular delivery vehicle for protein transduction of micro-dystrophin. J. Drug Target. 2005, 13, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Xiong, F.; Xu, Y.; Zheng, H.; Lu, X.; Feng, S.; Shang, Y.; Li, Y.; Zhang, Y.; Jin, S.; Zhang, C. Microdystrophin delivery in dystrophin-deficient (mdx) mice by genetically-corrected syngeneic MSCs transplantation. Transplant. Proc. 2010, 42, 2731–2739. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hersh, J.; Condor Capcha, J.M.; Iansen Irion, C.; Lambert, G.; Noguera, M.; Singh, M.; Kaur, A.; Dikici, E.; Jiménez, J.J.; Shehadeh, L.A.; et al. Peptide-Functionalized Dendrimer Nanocarriers for Targeted Microdystrophin Gene Delivery. Pharmaceutics 2021, 13, 2159. https://doi.org/10.3390/pharmaceutics13122159
Hersh J, Condor Capcha JM, Iansen Irion C, Lambert G, Noguera M, Singh M, Kaur A, Dikici E, Jiménez JJ, Shehadeh LA, et al. Peptide-Functionalized Dendrimer Nanocarriers for Targeted Microdystrophin Gene Delivery. Pharmaceutics. 2021; 13(12):2159. https://doi.org/10.3390/pharmaceutics13122159
Chicago/Turabian StyleHersh, Jessica, José Manuel Condor Capcha, Camila Iansen Irion, Guerline Lambert, Mauricio Noguera, Mohit Singh, Avinash Kaur, Emre Dikici, Joaquín J. Jiménez, Lina A. Shehadeh, and et al. 2021. "Peptide-Functionalized Dendrimer Nanocarriers for Targeted Microdystrophin Gene Delivery" Pharmaceutics 13, no. 12: 2159. https://doi.org/10.3390/pharmaceutics13122159
APA StyleHersh, J., Condor Capcha, J. M., Iansen Irion, C., Lambert, G., Noguera, M., Singh, M., Kaur, A., Dikici, E., Jiménez, J. J., Shehadeh, L. A., Daunert, S., & Deo, S. K. (2021). Peptide-Functionalized Dendrimer Nanocarriers for Targeted Microdystrophin Gene Delivery. Pharmaceutics, 13(12), 2159. https://doi.org/10.3390/pharmaceutics13122159