
Submerged Eutectic-Assisted, Solvent-Free Mechanochemical Formation of a Propranolol Salt and Its Other Multicomponent Solids


 ,
, 

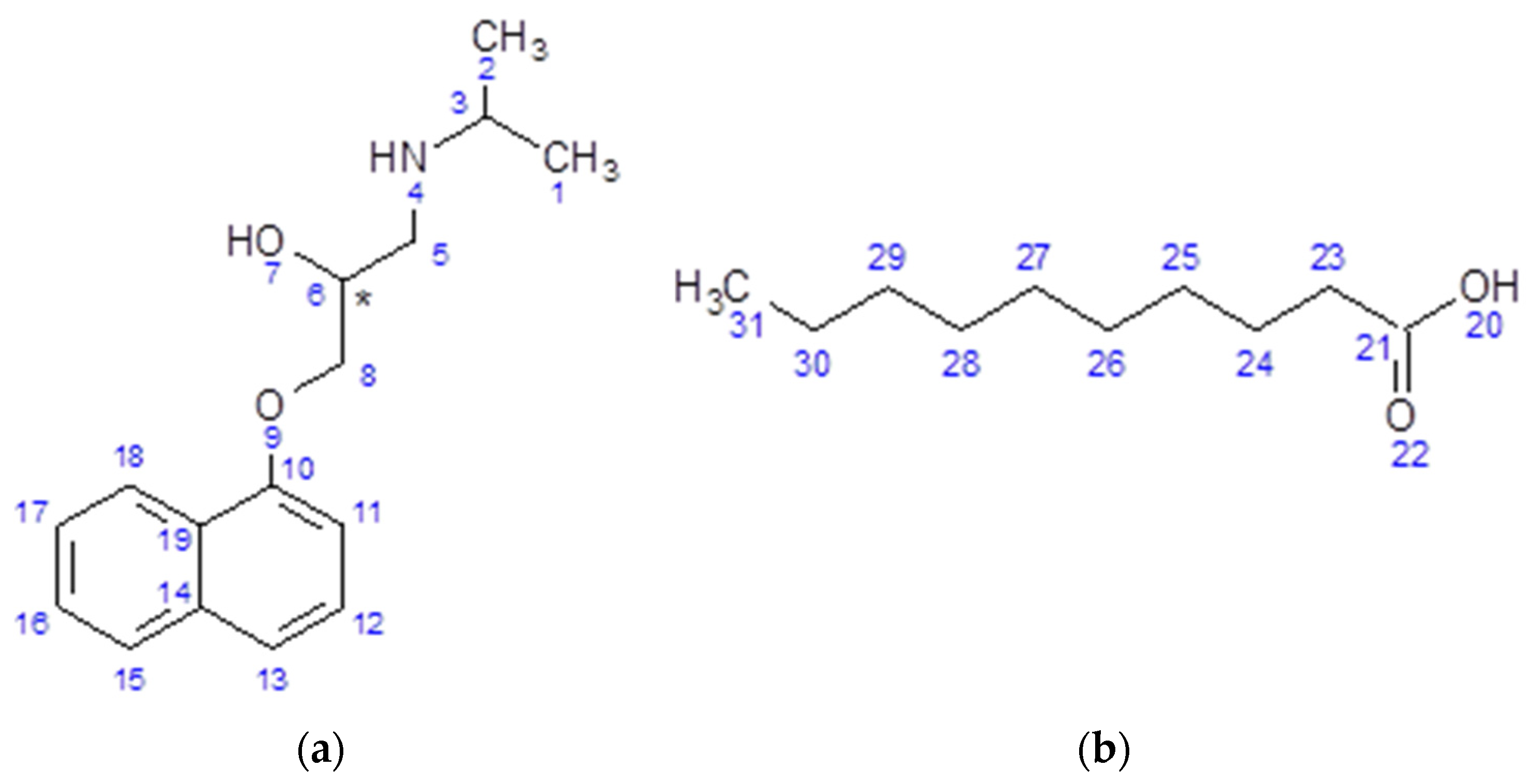{kind=link}
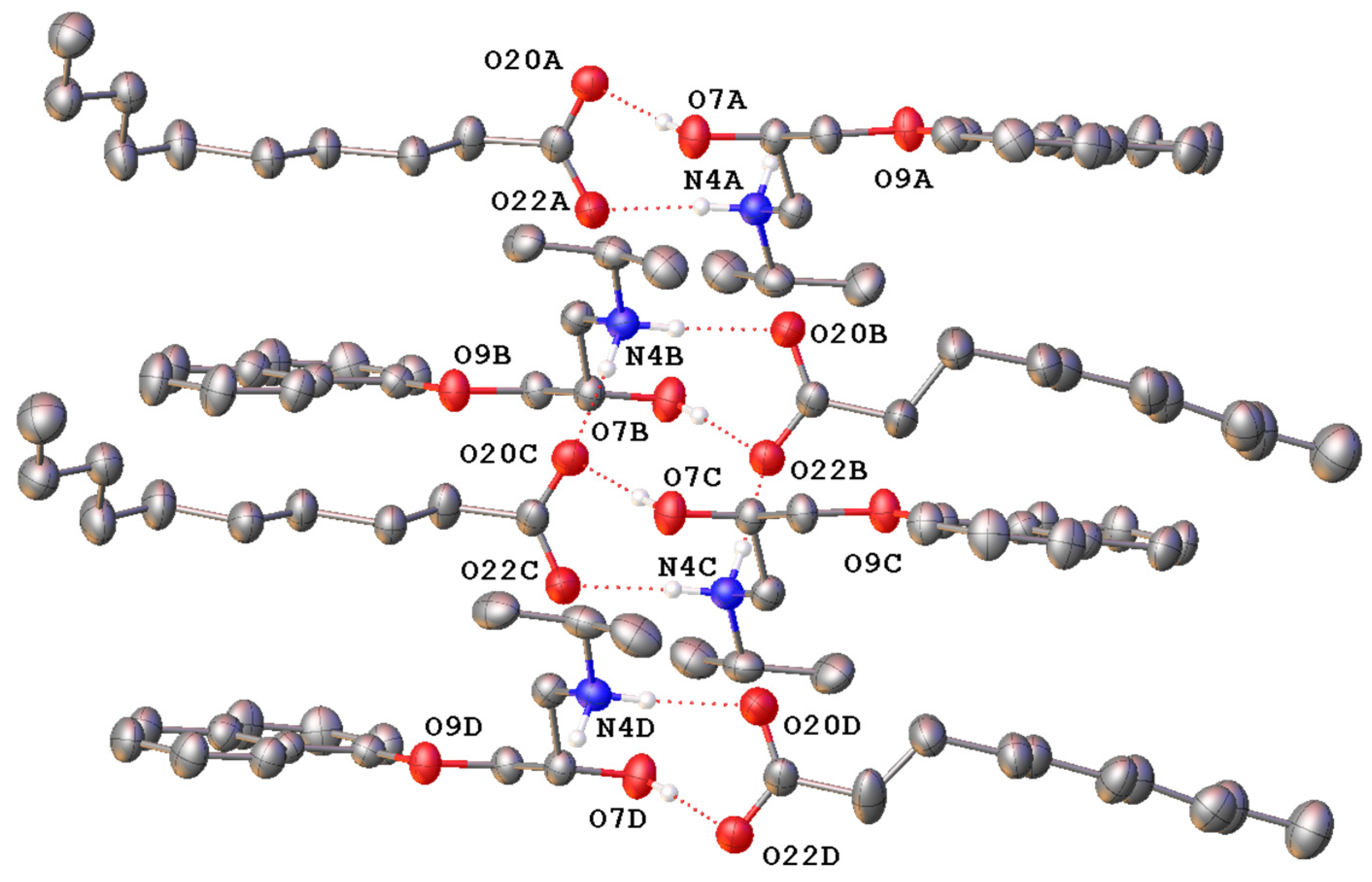{kind=link}
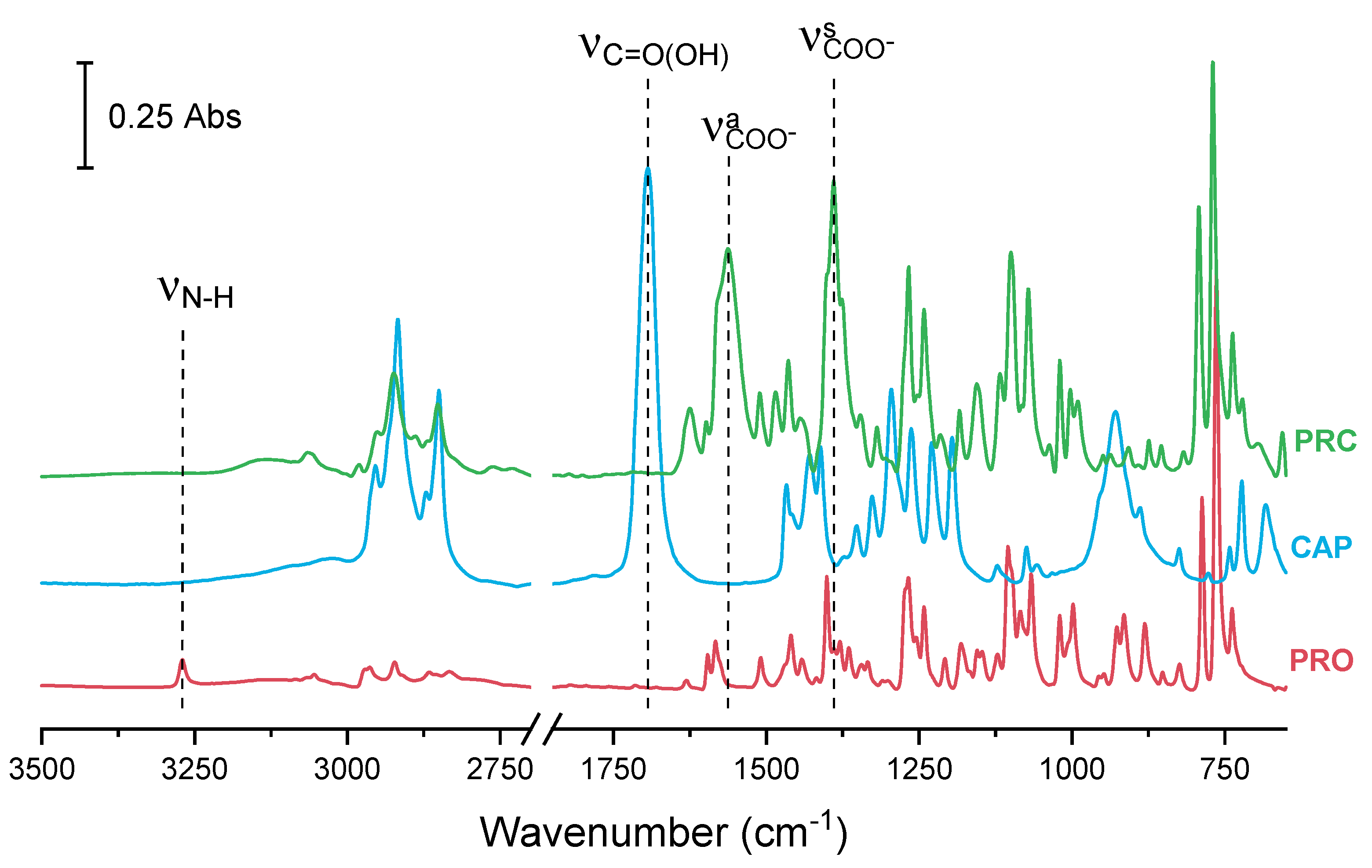{kind=link}
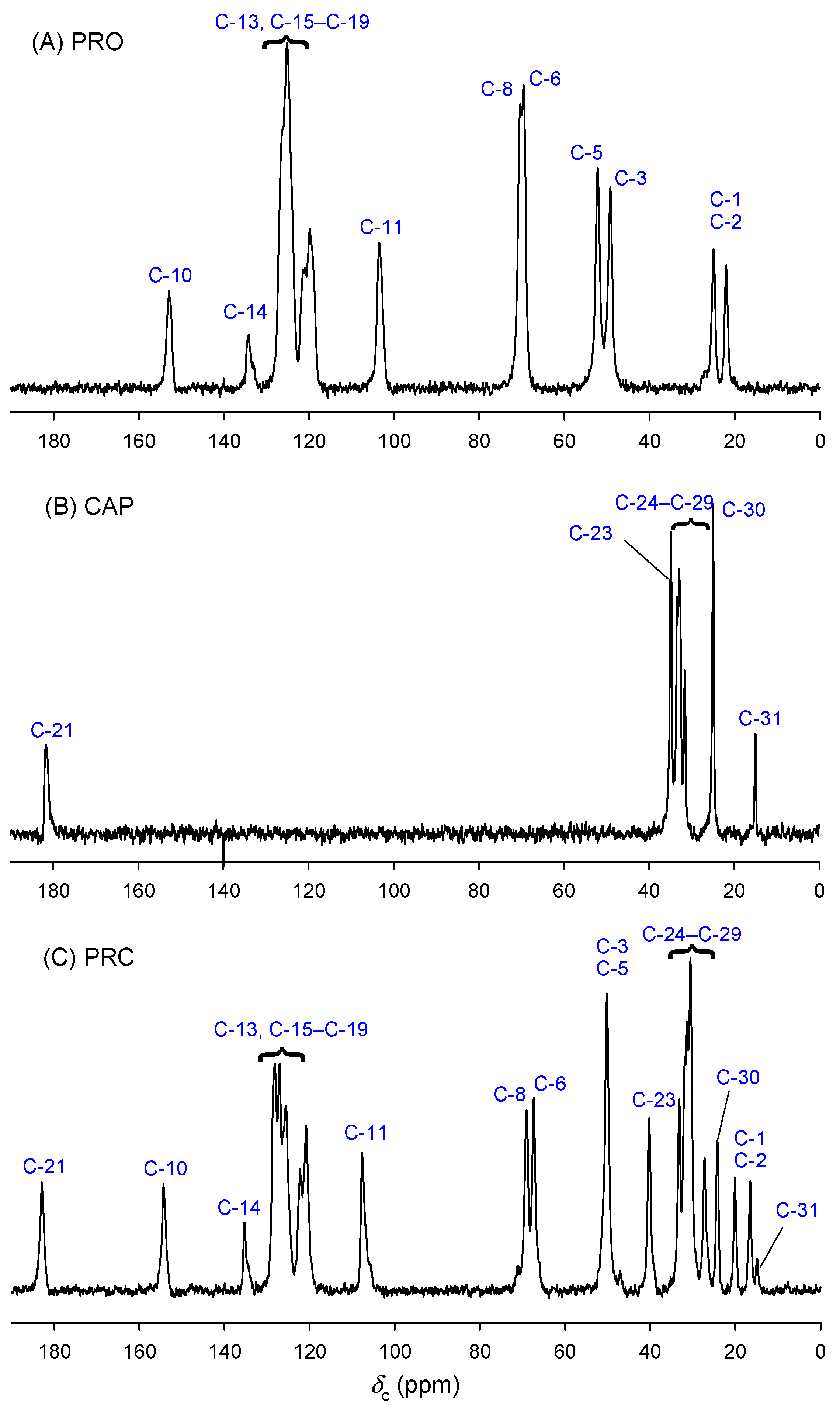{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Sample Preparation
PRO Free Base Synthesis
Synthesis of Propranolol Caprate (PRC) Salt
Preparation of PRO-CAP, PRC-CAP and PRC-PRO Physical Mixtures
Preparation of Samples for Contact and Grinding-Assisted Solid-State Reaction
2.2.2. Single Crystal X-ray Diffraction (SCXRD)
2.2.3. Fourier Transform Infrared Spectroscopy (FTIR)
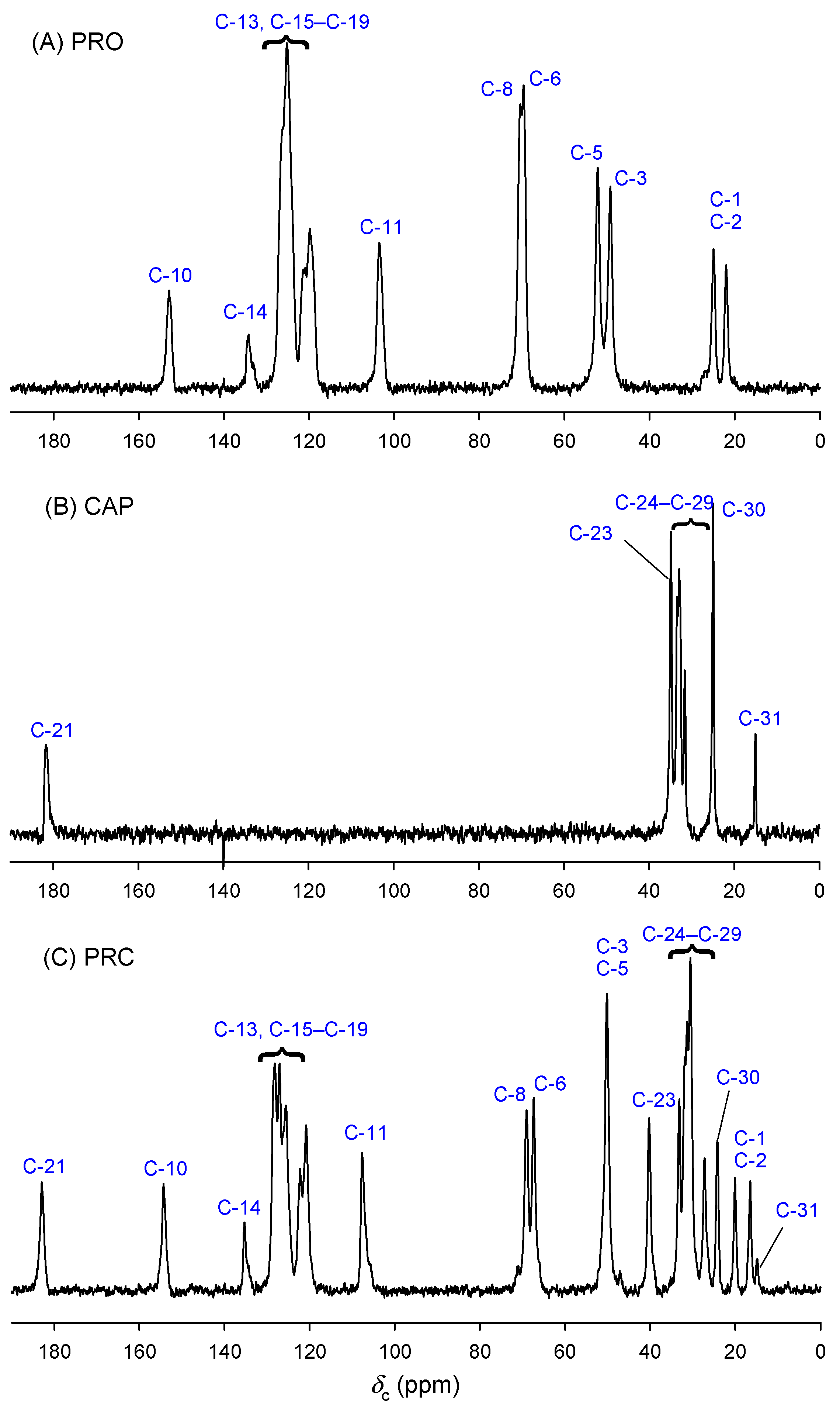
2.2.4. Solution and Solid-State Nuclear Magnetic Resonance (NMR)
2.2.5. Differential Scanning Calorimetry (DSC)
2.2.6. Thermogravimetry (TGA)
2.2.7. Polarised Light Microscopy
2.2.8. Powder X-ray Diffraction (PXRD)
2.2.9. Broadband Dielectric Spectroscopy (BDS)
3. Results
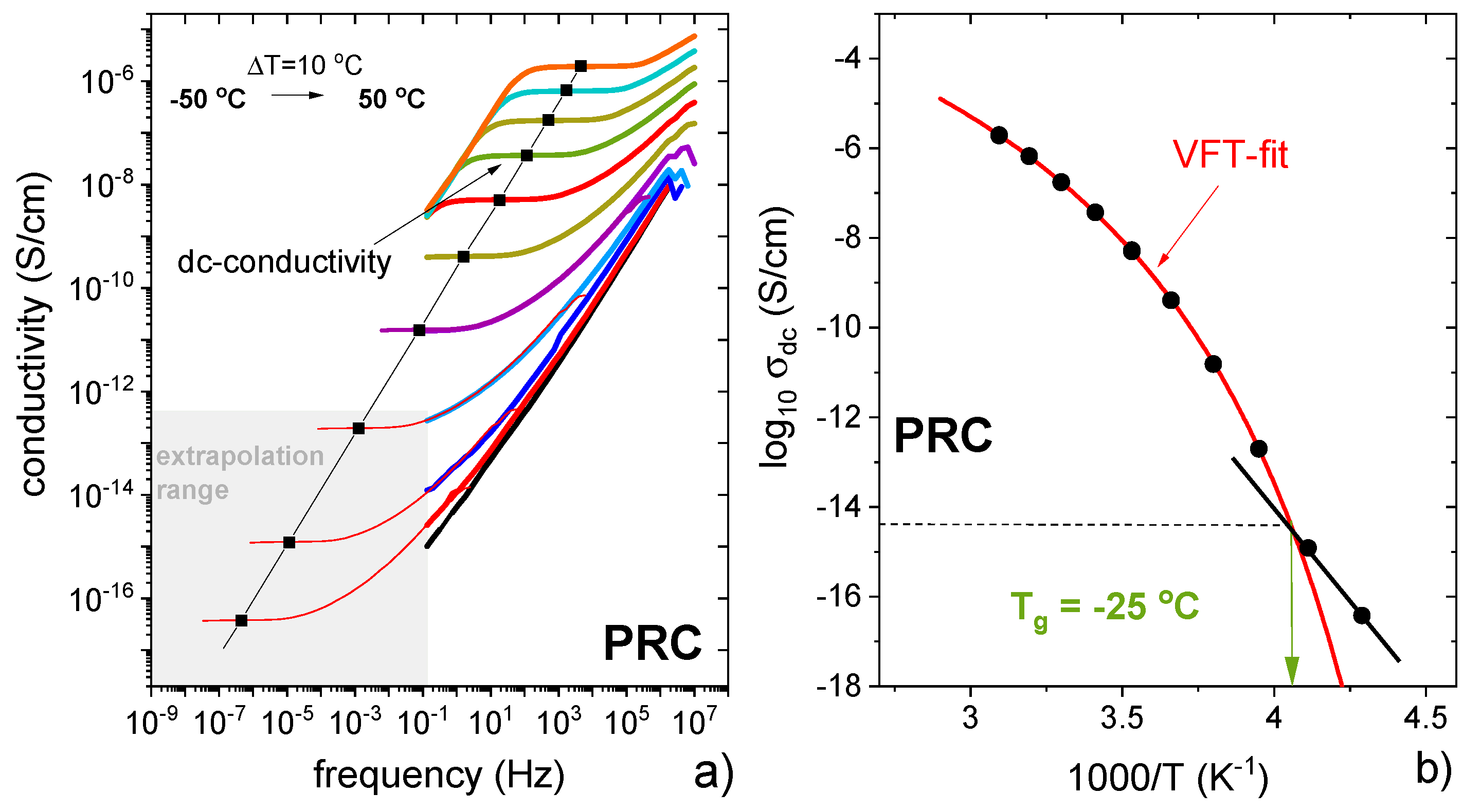
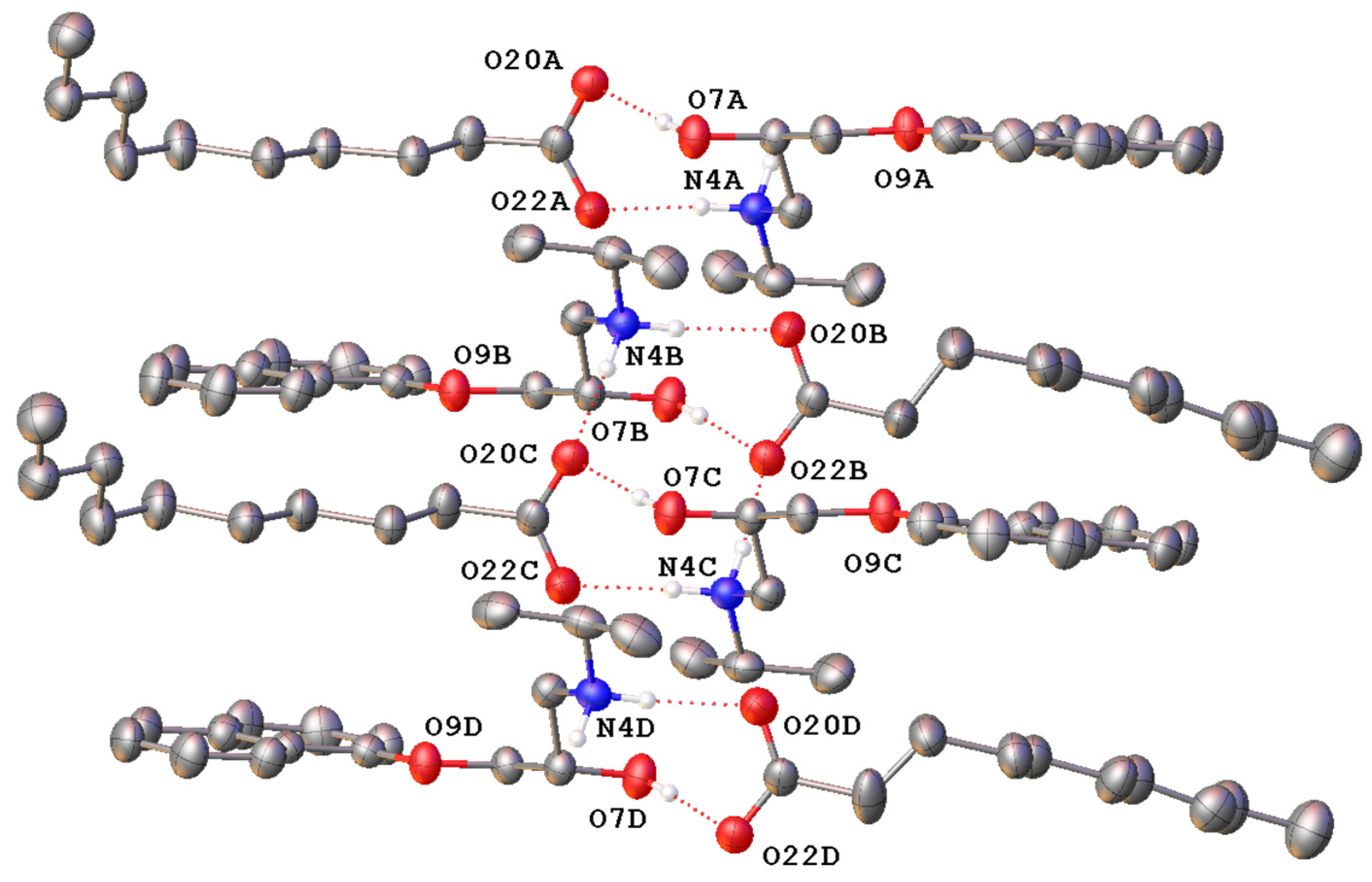
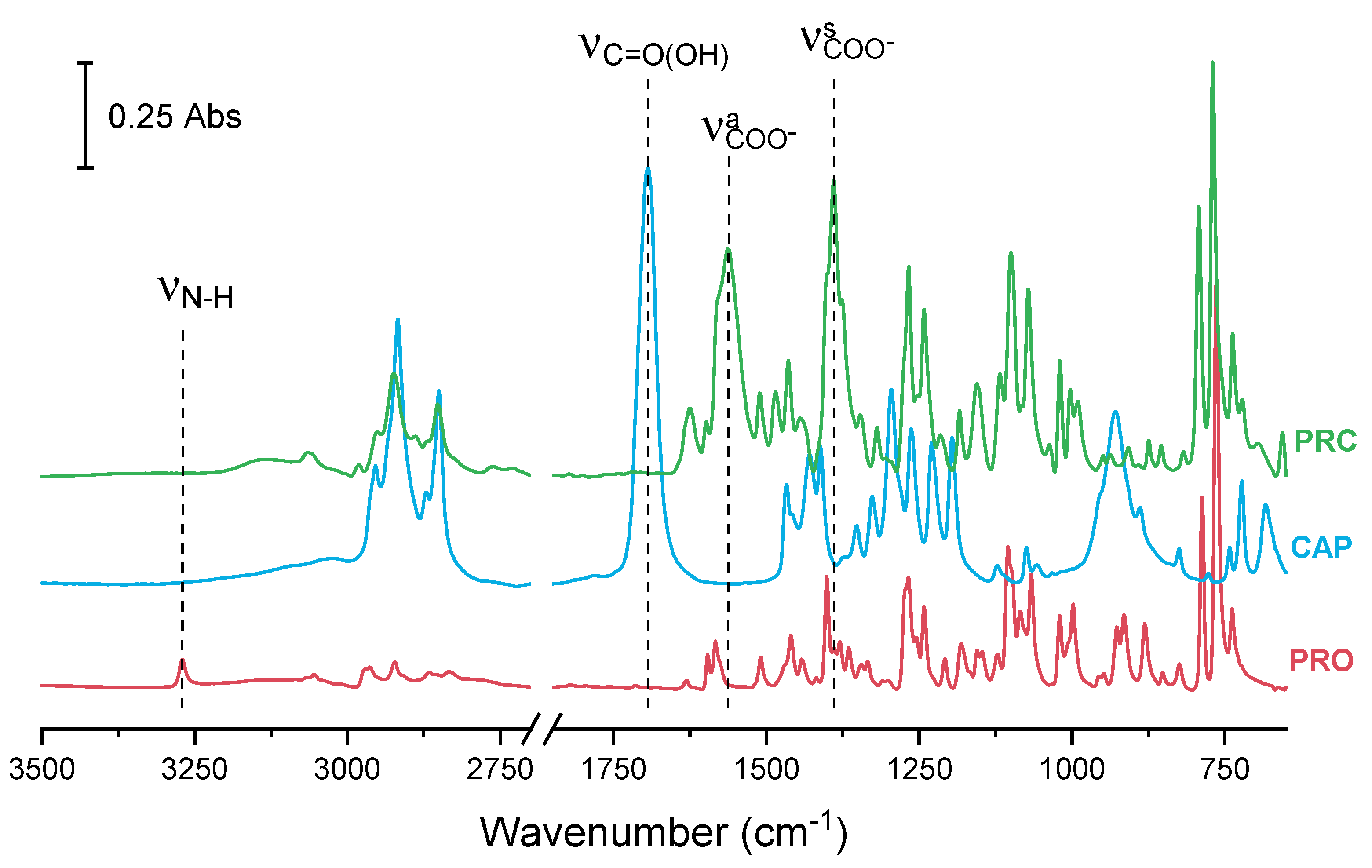
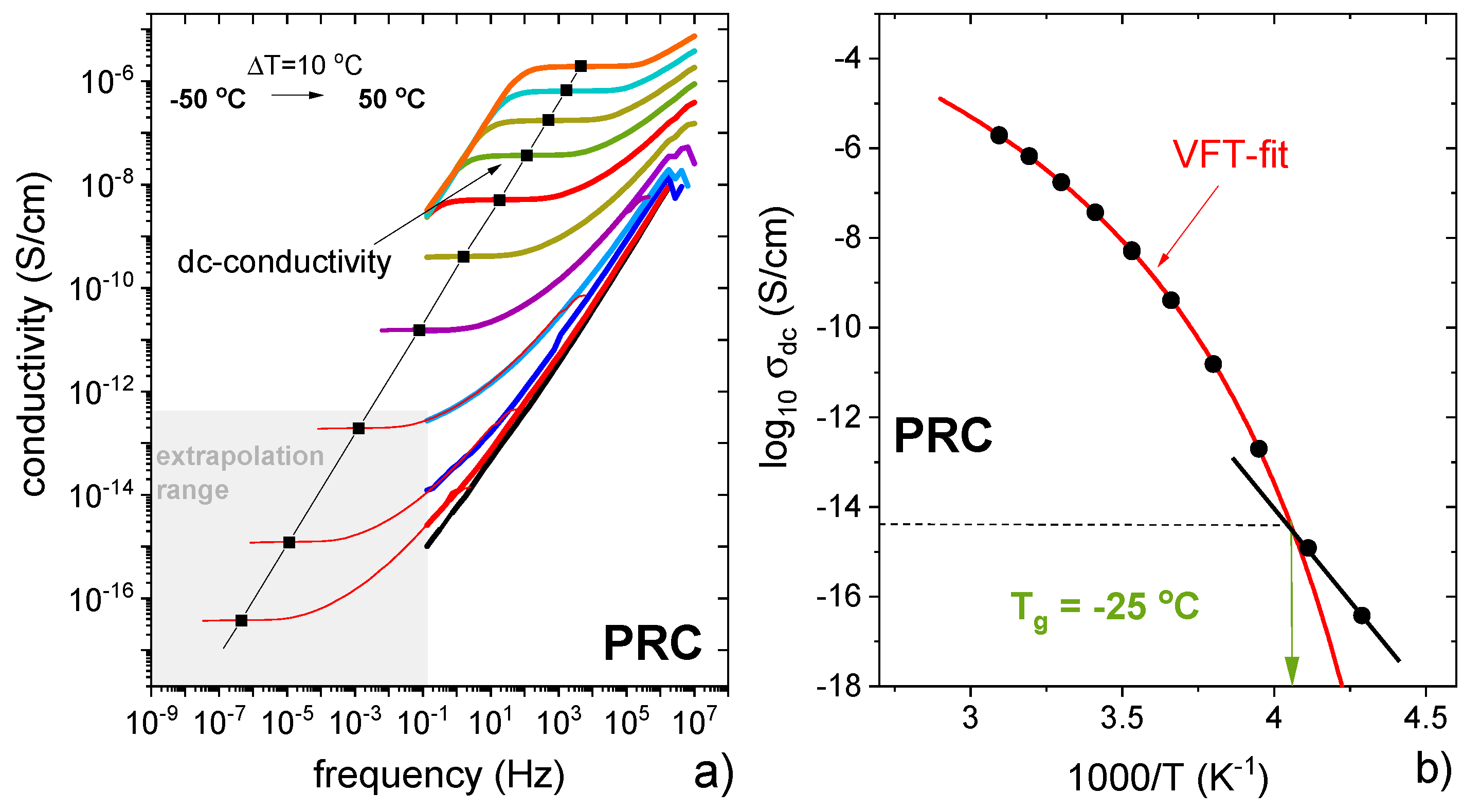
3.1. Characterisation of Propranolol Caprate Salt
3.2. Solvent-Free Formation of PRC and Other Solid-State Forms of PRO-CAP
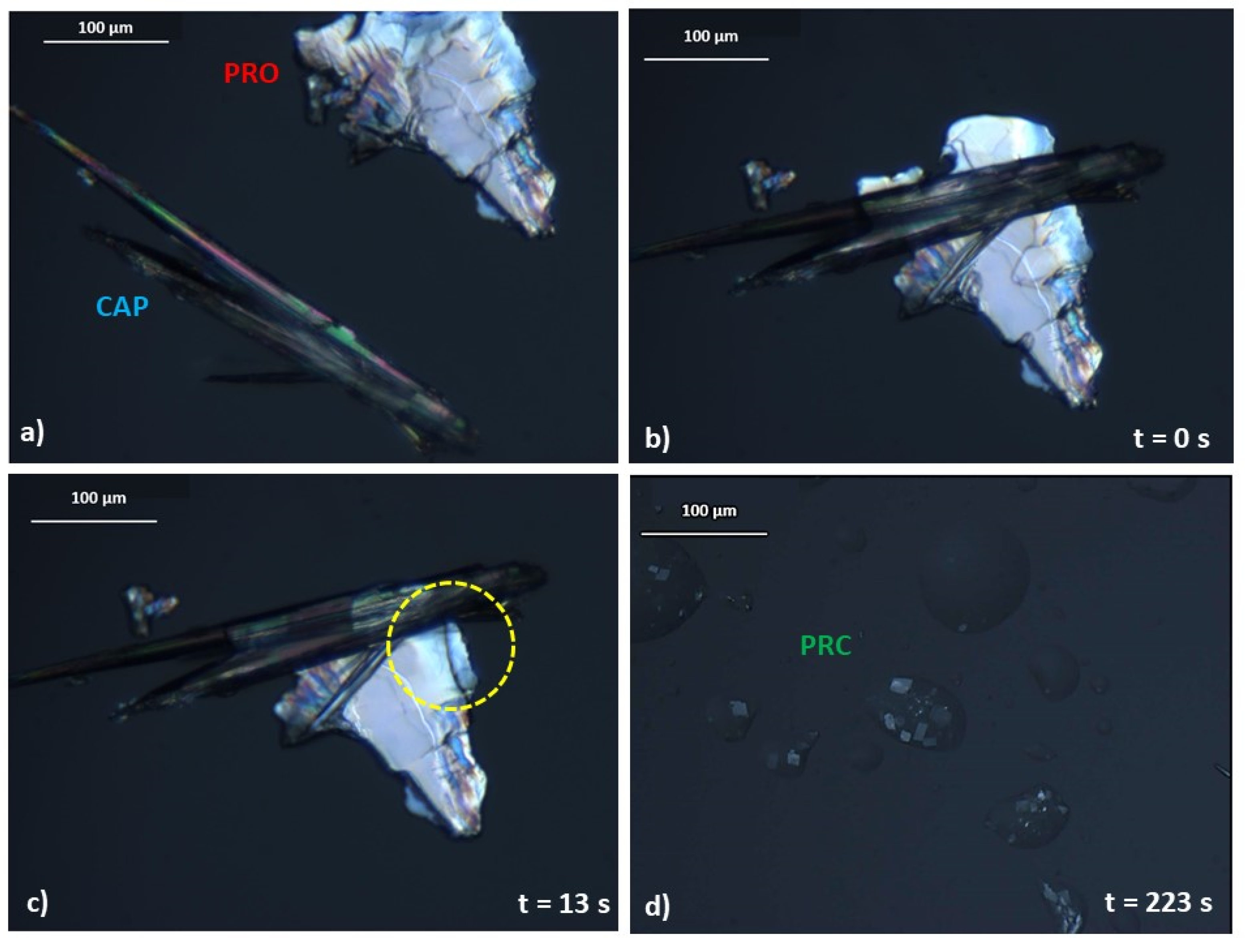
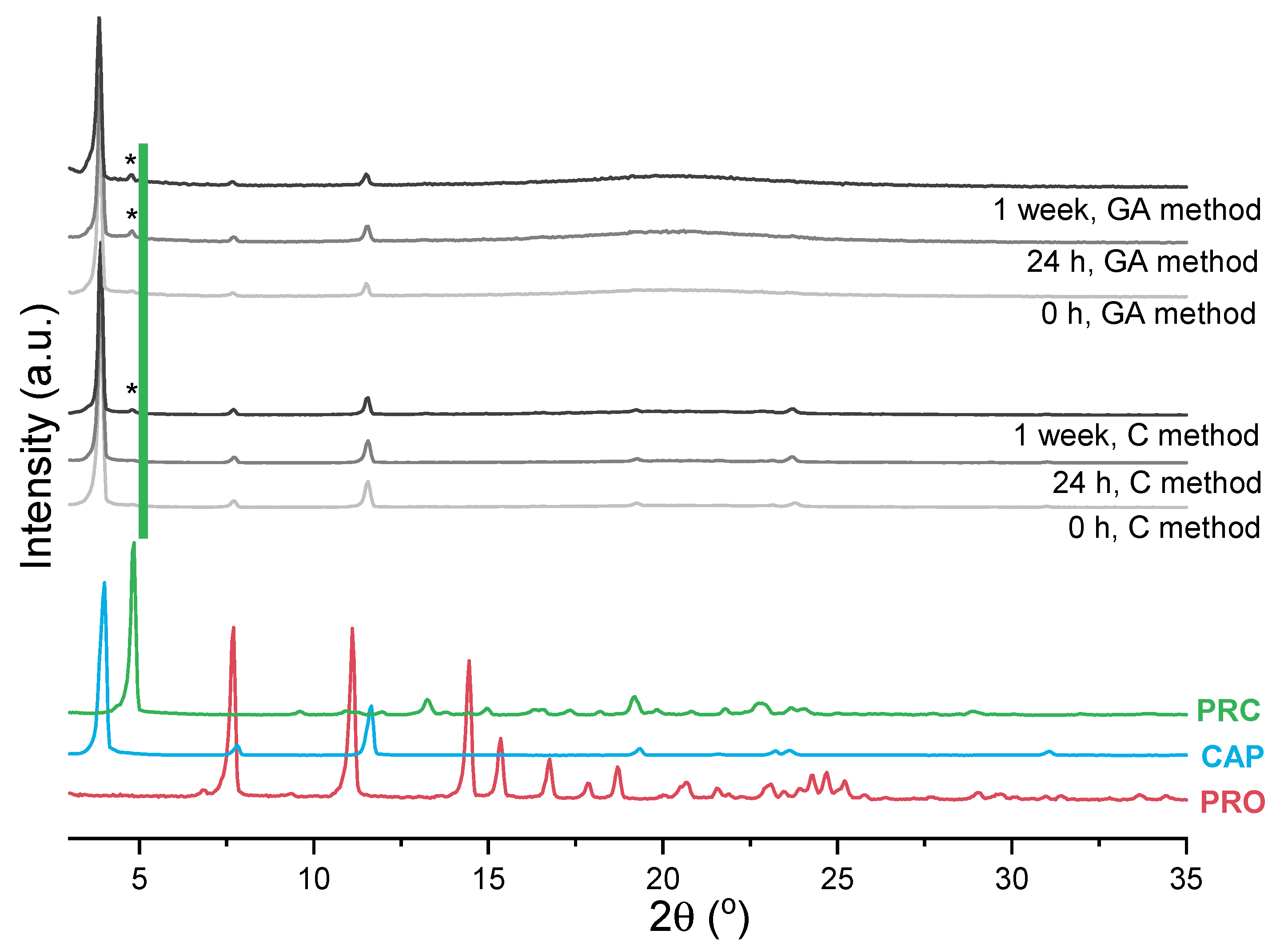
3.2.1. Impact of Temperature and Mechanism of PRC Formation
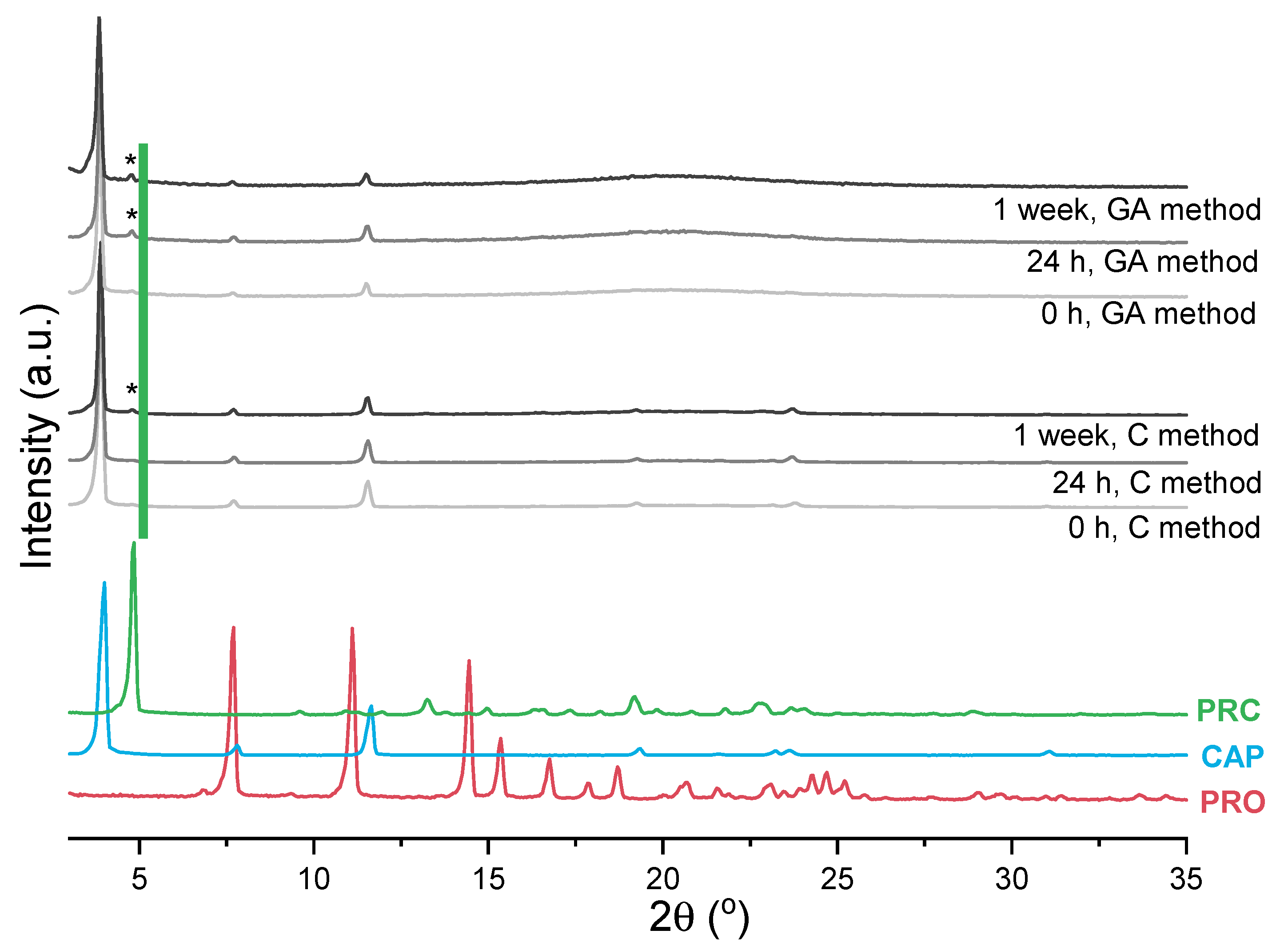
3.2.2. Mechanosynthesis of Propranolol Caprate Salt
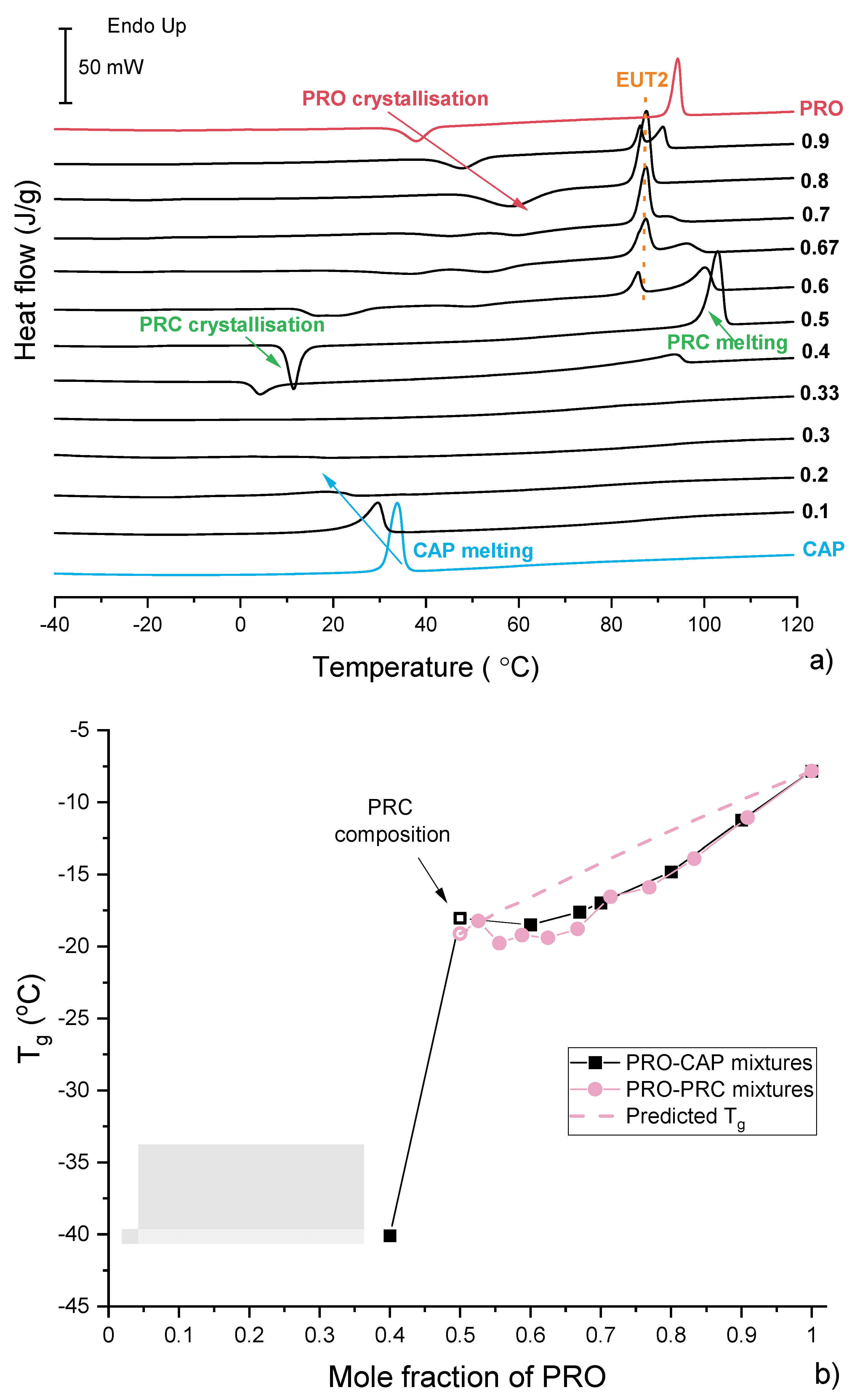
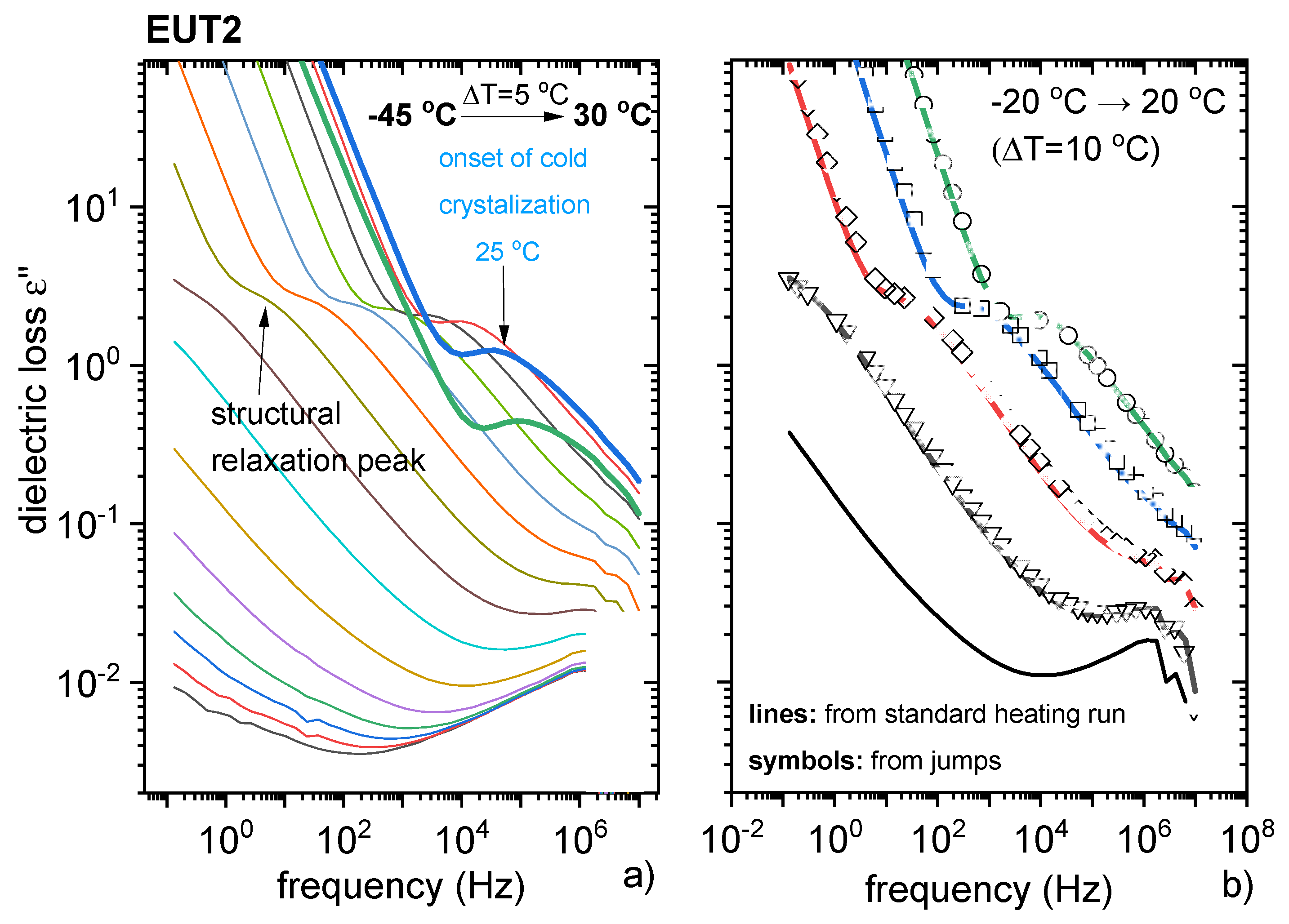
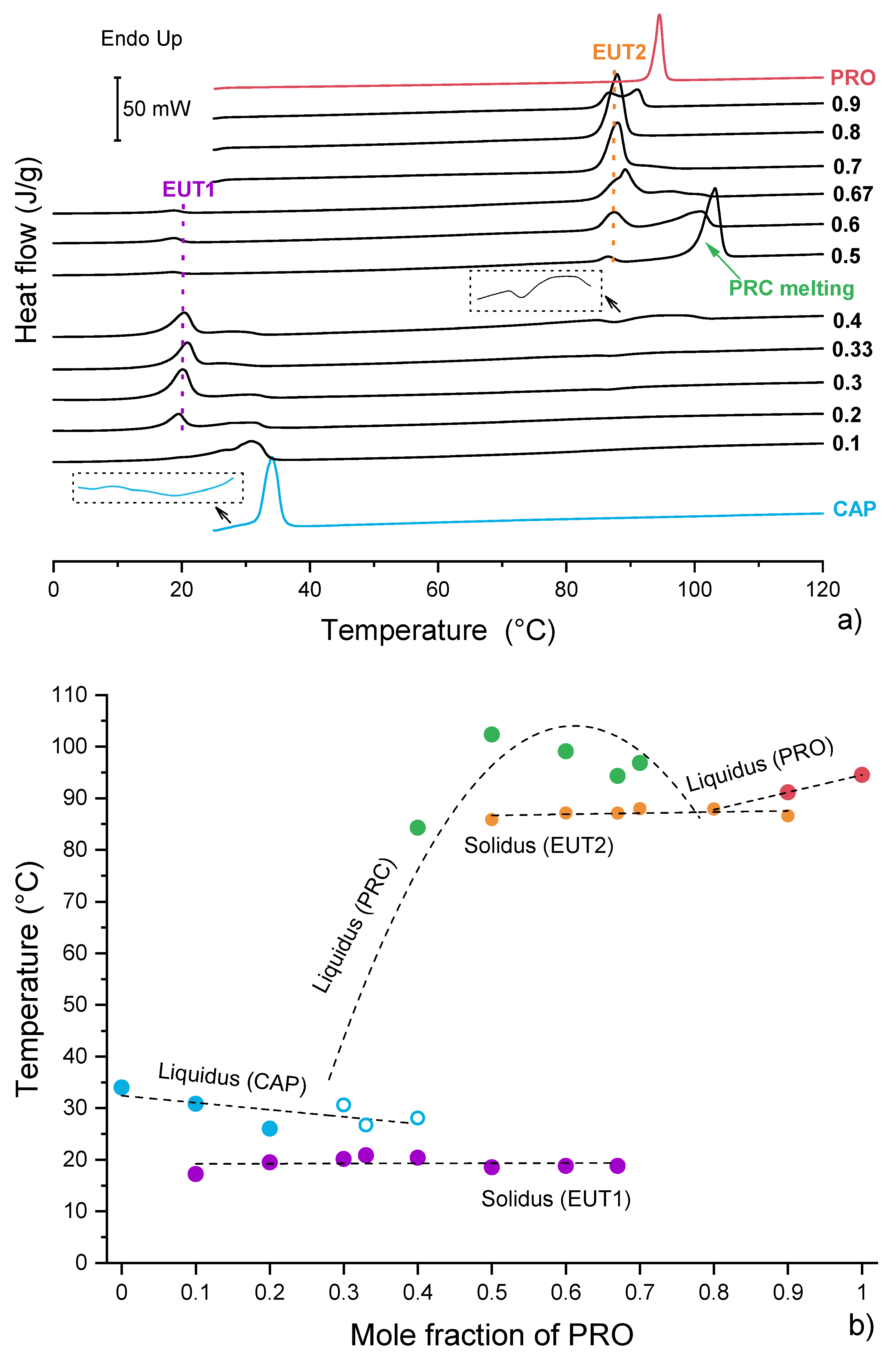
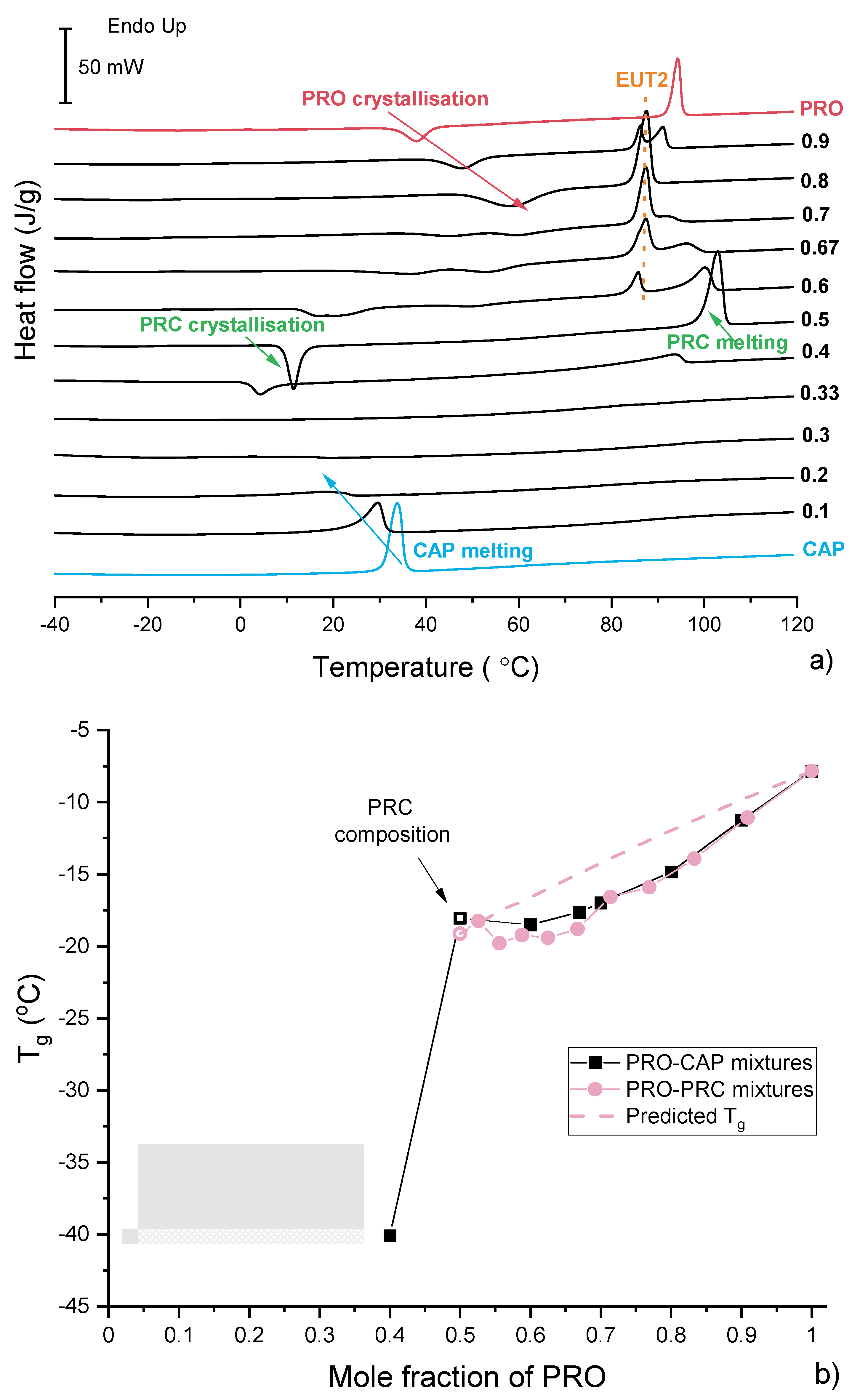
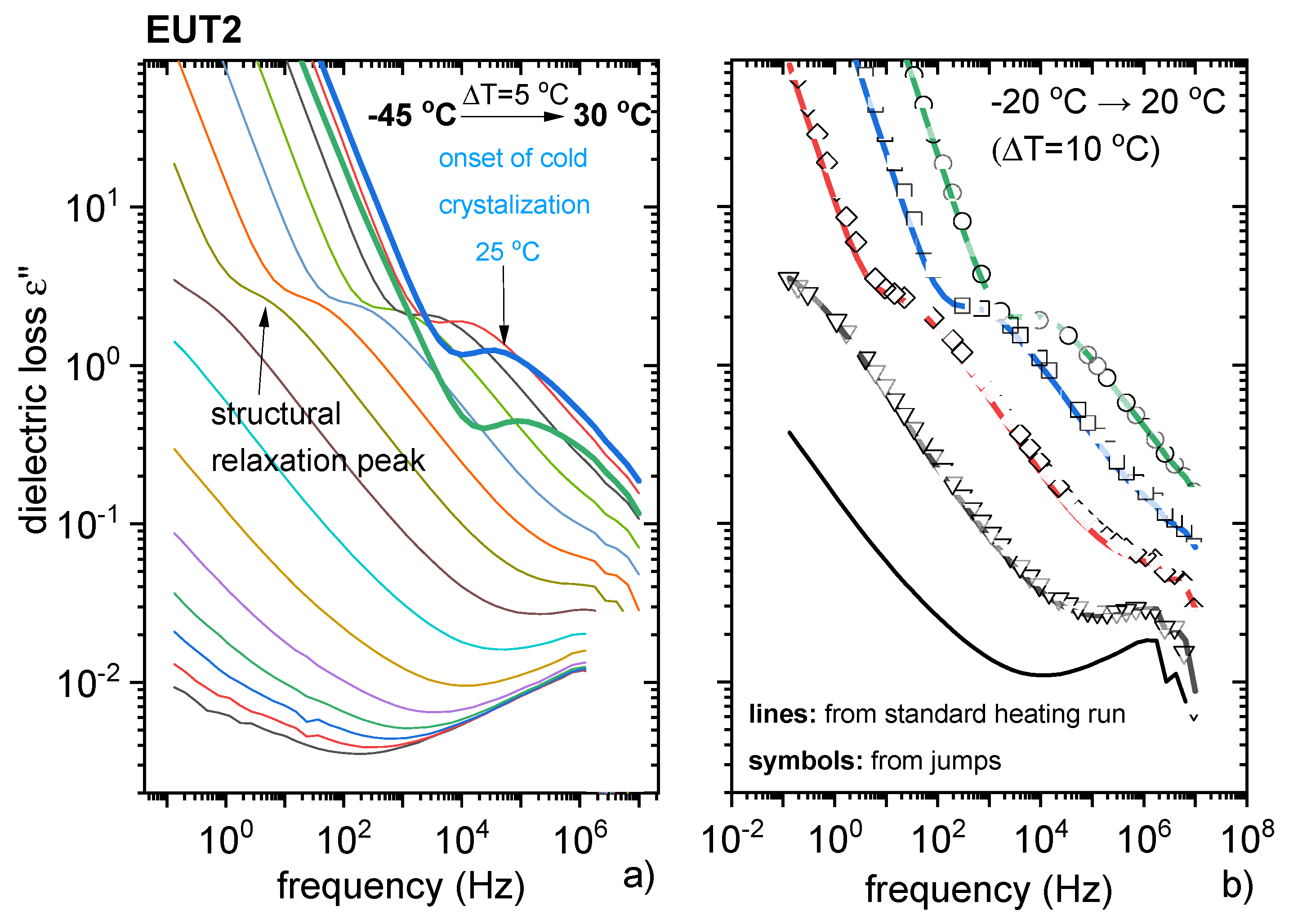
3.3. Characterisation of Supercooled PRO-CAP Systems
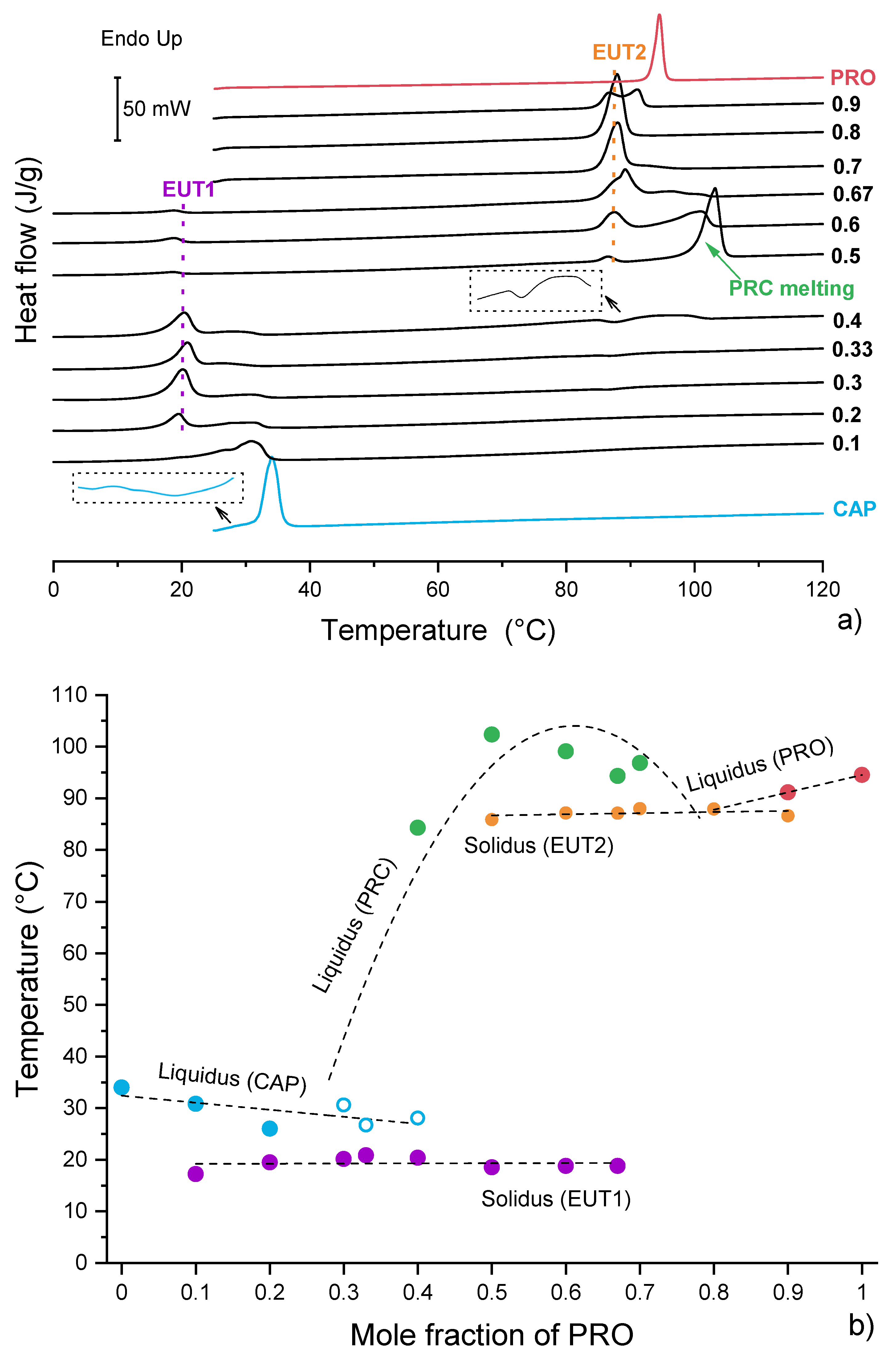
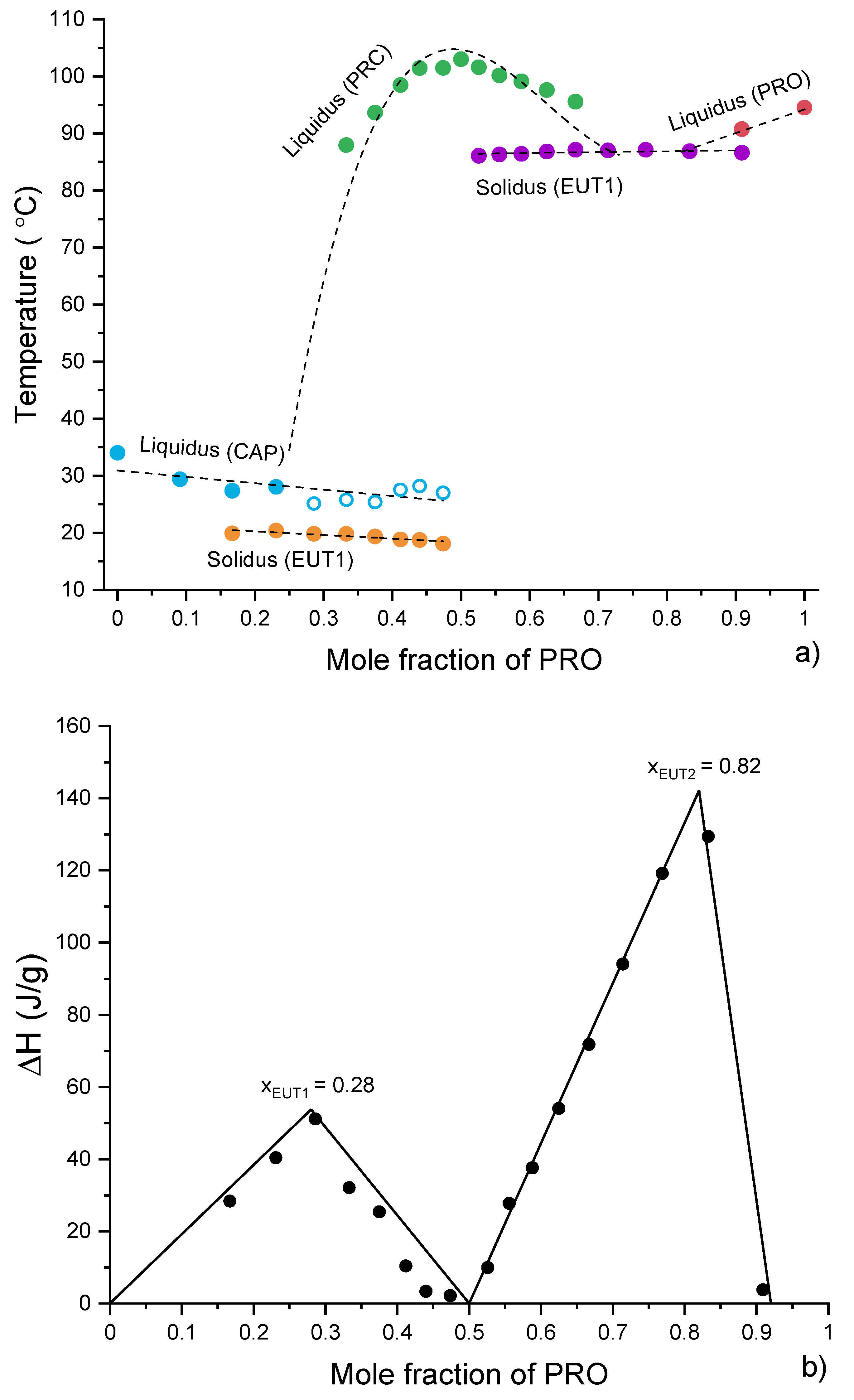
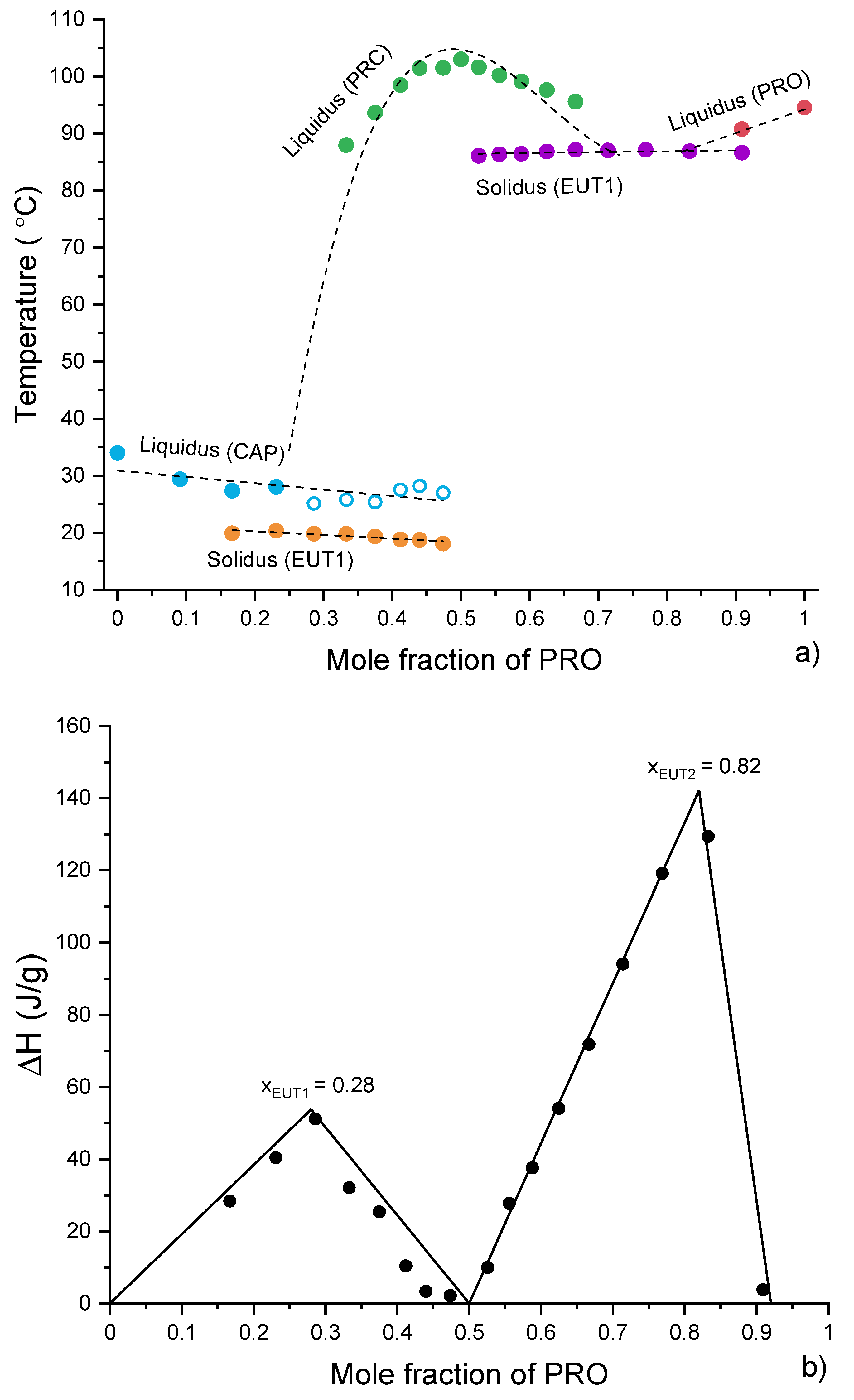
3.4. Analysis of Eutectic Compositions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Andreasen, C.; Andersson, C. Current use of beta-blockers in patients with coronary artery disease. Trends Cardiovasc. Med. 2018, 28, 382–389. [Google Scholar] [CrossRef]
- Bangalore, S.; Sawhney, S.; Messerli, F.H. Relation of Beta-Blocker-Induced Heart Rate Lowering and Cardioprotection in Hypertension. J. Am. Coll. Cardiol. 2008, 52, 1482–1489. [Google Scholar] [CrossRef] [Green Version]
- Wagner, M.J.; Cranmer, L.D.; Loggers, E.T.; Pollack, S.M. Propranolol for the treatment of vascular sarcomas. J. Exp. Pharmacol. 2018, 10, 51–58. [Google Scholar] [CrossRef] [Green Version]
- Kilcline, C.; Frieden, I.J. Infantile hemangiomas: How common are they? A systematic review of the medical literature. Pediatr. Dermatol. 2008, 25, 168–173. [Google Scholar] [CrossRef]
- Shayan, Y.R.; Prendiville, J.S.; Goldman, R.D. Use of propranolol in treating hemangiomas. Can. Fam. Physician 2011, 57, 302–303. [Google Scholar] [PubMed]
- Ferrari, B.; Mons, R.; Vollat, B.; Fraysse, B.; Paxéus, N.; Lo Giudice, R.; Pollio, A.; Garric, J. Environmental risk assessment of six human pharmaceuticals: Are the current environmental risk assessment procedures sufficient for the protection of the aquatic environment? Environ. Toxicol. Chem. 2004, 23, 1344–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, E.; Rubino, J. Solubility, melting point and salting-out relationships in a group of secondary amine hydrochloride salts. Int. J. Pharm. 1996, 130, 179–185. [Google Scholar] [CrossRef]
- Al Shaker, H.A.; Qinna, N.A.; Badr, M.; Al Omari, M.M.H.; Idkaidek, N.; Matalka, K.Z.; Badwan, A.A. Glucosamine modulates propranolol pharmacokinetics via intestinal permeability in rats. Eur. J. Pharm. Sci. 2017, 105, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, H.K.; Marriott, J.F. Formulations for children: Problems and solutions. Br. J. Clin. Pharmacol. 2015, 79, 405–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, F.L.; Ernest, T.B.; Tuleu, C.; Gul, M.O. Formulation approaches to pediatric oral drug delivery: Benefits and limitations of current platforms. Expert Opin. Drug Deliv. 2015, 12, 1727–1740. [Google Scholar] [CrossRef]
- Guan, Y.; Zuo, T.; Chang, M.; Zhang, F.; Wei, T.; Shao, W.; Lin, G. Propranolol hydrochloride-loaded liposomal gel for transdermal delivery: Characterization and in vivo evaluation. Int. J. Pharm. 2015, 487, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Kunzi-Rapp, K. Topical propranolol therapy for infantile hemangiomas. Pediatr. Dermatol. 2012, 29, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Stott, P.W.; Williams, A.C.; Barry, B.W. Mechanistic study into the enhanced transdermal permeation of a model β-blocker, propranolol, by fatty acids: A melting point depression effect. Int. J. Pharm. 2001, 219, 161–176. [Google Scholar] [CrossRef]
- Wang, Y.; Fan, Q.; Song, Y.; Michniak, B. Effects of Fatty Acids and Iontophoresis on the Delivery of Midodrine Hydrochloride and the Structure of Human Skin. Pharm. Res. 2003, 20, 1612–1618. [Google Scholar] [CrossRef]
- Mohammadi-Samani, S.; Yousefi, G.; Mohammadi, F.; Ahmadi, F. Meloxicam transdermal delivery: Effect of eutectic point on the rate and extent of skin permeation. Iran. J. Basic Med. Sci. 2014, 17, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Chadwick, K.; Davey, R.; Cross, W. How does grinding produce co-crystals? Insights from the case of benzophenone and diphenylamine. CrystEngComm 2007, 9, 732–734. [Google Scholar] [CrossRef]
- Mottillo, C.; Friščić, T. Advances in solid-state transformations of coordination bonds: From the ball mill to the aging chamber. Molecules 2017, 22, 144. [Google Scholar] [CrossRef] [Green Version]
- Cooke, C.L.; Davey, R.J.; Black, S.; Muryn, C.; Pritchard, R.G. Binary and ternary phase diagrams as routes to salt discovery: Ephedrine and pimelic acid. Cryst. Growth Des. 2010, 10, 5270–5278. [Google Scholar] [CrossRef]
- Paluch, K.J.; McCabe, T.; Müller-Bunz, H.; Corrigan, O.I.; Healy, A.M.; Tajber, L. Formation and physicochemical properties of crystalline and amorphous salts with different stoichiometries formed between ciprofloxacin and succinic acid. Mol. Pharm. 2013, 10, 3640–3654. [Google Scholar] [CrossRef] [PubMed]
- Trask, A.V.; Haynes, D.A.; Motherwell, W.D.S.; Jones, W. Screening for crystalline salts via mechanochemistry. Chem. Commun. 2006, 51–53. [Google Scholar] [CrossRef]
- Mesallati, H.; Conroy, D.; Hudson, S.; Tajber, L. Preparation and characterization of amorphous ciprofloxacin-amino acid salts. Eur. J. Pharm. Biopharm. 2017, 121, 73–89. [Google Scholar] [CrossRef] [Green Version]
- Michalchuk, A.A.L.; Tumanov, I.A.; Drebushchak, V.A.; Boldyreva, E.V. Advances in elucidating mechanochemical complexities via implementation of a simple organic system. Faraday Discuss. 2014, 170, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.L.; Vasoya, J.M.; De Lima Cirqueira, M.; Yeh, K.L.; Lee, T.; Serajuddin, A.T.M. Continuous preparation of 1:1 haloperidol-maleic acid salt by a novel solvent-free method using a twin screw melt extruder. Mol. Pharm. 2017, 14, 1278–1291. [Google Scholar] [CrossRef]
- Zotova, J.; Wojnarowska, Z.; Twamley, B.; Paluch, M.; Tajber, L. Green Synthesis of Lidocaine Ionic Liquids and Salts: Mechanisms of Formation and Interactions in the Crystalline and Supercooled States. ACS Sustain. Chem. Eng. 2020, 49, 18266–18276. [Google Scholar] [CrossRef]
- Bialek, K.; Wojnarowska, Z.; Twamley, B.; Tajber, L. Characterisation and fundamental insight into the formation of new solid state, multicomponent systems of propranolol. Int. J. Pharm. 2021, 602, 120605. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, C71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Van Rossum, B.J.; Förster, H.; De Groot, H.J.M. High-Field and High-Speed CP-MAS 13 C NMR Heteronuclear Dipolar-Correlation Spectroscopy of Solids with Frequency-Switched Lee-Goldburg Homonuclear Decoupling. J. Magn. Reson. 1997, 124, 516–519. [Google Scholar] [CrossRef]
- Zorin, V. Gsim—A Visualisation and Processing Program for Solid-State NMR. Available online: http://gsim.sourceforge.net/ (accessed on 1 December 2021).
- Umerska, A.; Bialek, K.; Zotova, J.; Skotnicki, M.; Tajber, L. Anticrystal engineering of ketoprofen and ester local anesthetics: Ionic liquids or deep eutectic mixtures? Pharmaceutics 2020, 12, 368. [Google Scholar] [CrossRef] [Green Version]
- Wojnarowska, Z.; Zotowa, J.; Knapik-Kowalczuk, J.; Tajber, L.; Paluch, M. Effect of electrostatic interactions on the relaxation dynamics of pharmaceutical eutectics. Eur. J. Pharm. Sci. 2019, 134, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Hodgkinson, P. NMR crystallography of molecular organics. Prog. Nucl. Magn. Reson. Spectrosc. 2020, 118–119, 10–53. [Google Scholar] [CrossRef]
- Strachan, C.; Saarinen, J.; Lipiäinen, T.; Vuorimaa-Laukkanen, E.; Rautaniemi, K.; Laaksonen, T.; Skotnicki, M.; Dračínský, M. Spectroscopic Methods in Solid-state Characterization. In Characterization of Pharmaceutical Nano and Microsystems; Wiley Online Books: Hoboken, NJ, USA, 2021; pp. 27–95. ISBN 9781119414018. [Google Scholar]
- Arita, T.; Yoo, J.; Adschiri, T. Relation between the solution-state behavior of self-assembled monolayers on nanoparticles and dispersion of nanoparticles in organic solvents. J. Phys. Chem. C 2011, 115, 3899–3909. [Google Scholar] [CrossRef]
- Sakaue, K. National Institute of Advanced Industrial Science and Technology. Available online: https://www.aist.go.jp/index_en.html (accessed on 1 December 2021).
- Crowley, K.J.; Forbes, R.T.; York, P.; Nyqvist, H.; Camber, O. Oleate salt formation and mesomorphic behavior in the propranolol/oleic acid binary system. J. Pharm. Sci. 1999, 88, 586–591. [Google Scholar] [CrossRef]
- Ogiso, T.; Shintani, M. Mechanism for the enhancement effect of fatty acids on the percutaneous absorption of propranolol. J. Pharm. Sci. 1990, 79, 1065–1071. [Google Scholar] [CrossRef] [PubMed]
- Zielińska-Pisklak, M.A.; Pisklak, D.M.I.; Wawer, I. 1H and 13C NMR characteristics of β-blockers. Magn. Reson. Chem. 2011, 49, 284–290. [Google Scholar] [CrossRef]
- Braga, D.; Maini, L.; De Sanctis, G.; Rubini, K.; Grepioni, F.; Chierotti, M.R.; Gobetto, R. Mechanochemical Preparation of Hydrogen-Bonded Adducts between the Diamine 1,4-Diazabicyclo [2.2.2]octane and Dicarboxylic Acids of Variable Chain Length: An X-ray Diffraction and Solid-State NMR Study. Chem.-A Eur. J. 2003, 9, 5538–5548. [Google Scholar] [CrossRef]
- Zotova, J.; Wojnarowska, Z.; Twamley, B.; Tajber, L. Formation of stoichiometric and non-stoichiometric ionic liquid and cocrystal multicomponent phases of lidocaine with azelaic acid by changing counterion ratios. J. Mol. Liq. 2021, 344, 117737. [Google Scholar] [CrossRef]
- Barr, M.R.; Dunell, B.A.; Grant, R.F. Premelting Phenomena in Long-Chain Fatty Acids. Can. J. Chem. 1963, 41, 1188–1196. [Google Scholar] [CrossRef]
- Zerbi, G.; Conti, G.; Minoni, G.; Pison, S.; Bigotto, A. Premelting phenomena in fatty acids: An infrared and Raman study. J. Phys. Chem. 1987, 91, 2386–2393. [Google Scholar] [CrossRef]
- Meltzer, V.; Pincu, E. Thermodynamic study of binary mixture of citric acid and tartaric acid. Cent. Eur. J. Chem. 2012, 10, 1584–1589. [Google Scholar] [CrossRef] [Green Version]
- Paul, I.C.; Curtin, D.Y. Thermally Induced Organic Reactions in the Solid State. Acc. Chem. Res. 1973, 6, 217–225. [Google Scholar] [CrossRef]
- Fox, T.G. Influence of diluent and of copolymer composition on the glass temperature of a polymer system. Bull. Am. Phys. Soc. 1956, 1, 123–135. [Google Scholar]
- Wojnarowska, Z.; Tajber, L.; Paluch, M. Density Scaling in Ionic Glass Formers Controlled by Grotthuss Conduction. J. Phys. Chem. B 2019, 123, 1156–1160. [Google Scholar] [CrossRef] [PubMed]
- Wojnarowska, Z.; Paluch, K.J.; Shoifet, E.; Schick, C.; Tajber, L.; Knapik, J.; Wlodarczyk, P.; Grzybowska, K.; Hensel-Bielowka, S.; Verevkin, S.P.; et al. Molecular origin of enhanced proton conductivity in anhydrous ionic systems. J. Am. Chem. Soc. 2015, 137, 1157–1164. [Google Scholar] [CrossRef] [Green Version]
- Wojnarowska, Z.; Thoms, E.; Blanchard, B.; Tripathy, S.N.; Goodrich, P.; Jacquemin, J.; Knapik-Kowalczuk, J.; Paluch, M. How is Charge Transport Different in Ionic Liquids?: Effect of High Pressure. Phys. Chem. Chem. Phys. 2017, 19, 14141–14147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, P.G.; Hadgraft, J. Facilitated transfer of cationic drugs across a lipoidal membrane by oleic acid and lauric acid. Int. J. Pharm. 1987, 37, 251–255. [Google Scholar] [CrossRef]
- Touitou, E.; Chow, D.D.; Lawter, J.R. Chiral β-blockers for transdermal delivery. Int. J. Pharm. 1994, 104, 19–28. [Google Scholar] [CrossRef]
- N’Da, D.D. Prodrug strategies for enhancing the percutaneous absorption of drugs. Molecules 2014, 19, 20780–20807. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bialek, K.; Wojnarowska, Z.; Skotnicki, M.; Twamley, B.; Paluch, M.; Tajber, L. Submerged Eutectic-Assisted, Solvent-Free Mechanochemical Formation of a Propranolol Salt and Its Other Multicomponent Solids. Pharmaceutics 2021, 13, 2125. https://doi.org/10.3390/pharmaceutics13122125
Bialek K, Wojnarowska Z, Skotnicki M, Twamley B, Paluch M, Tajber L. Submerged Eutectic-Assisted, Solvent-Free Mechanochemical Formation of a Propranolol Salt and Its Other Multicomponent Solids. Pharmaceutics. 2021; 13(12):2125. https://doi.org/10.3390/pharmaceutics13122125
Chicago/Turabian StyleBialek, Klaudia, Zaneta Wojnarowska, Marcin Skotnicki, Brendan Twamley, Marian Paluch, and Lidia Tajber. 2021. "Submerged Eutectic-Assisted, Solvent-Free Mechanochemical Formation of a Propranolol Salt and Its Other Multicomponent Solids" Pharmaceutics 13, no. 12: 2125. https://doi.org/10.3390/pharmaceutics13122125
APA StyleBialek, K., Wojnarowska, Z., Skotnicki, M., Twamley, B., Paluch, M., & Tajber, L. (2021). Submerged Eutectic-Assisted, Solvent-Free Mechanochemical Formation of a Propranolol Salt and Its Other Multicomponent Solids. Pharmaceutics, 13(12), 2125. https://doi.org/10.3390/pharmaceutics13122125