
Machine Learning Predicts Drug Metabolism and Bioaccumulation by Intestinal Microbiota


 ,
,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Dataset Curation and Labelling
2.2. Feature Generation
2.3. Data Preprocessing and Visualisation
2.4. Development of Machine Learning Models
2.5. Selection and Optimisation of Best Model
2.6. Data Analysis and Statistics
3. Results and Discussion
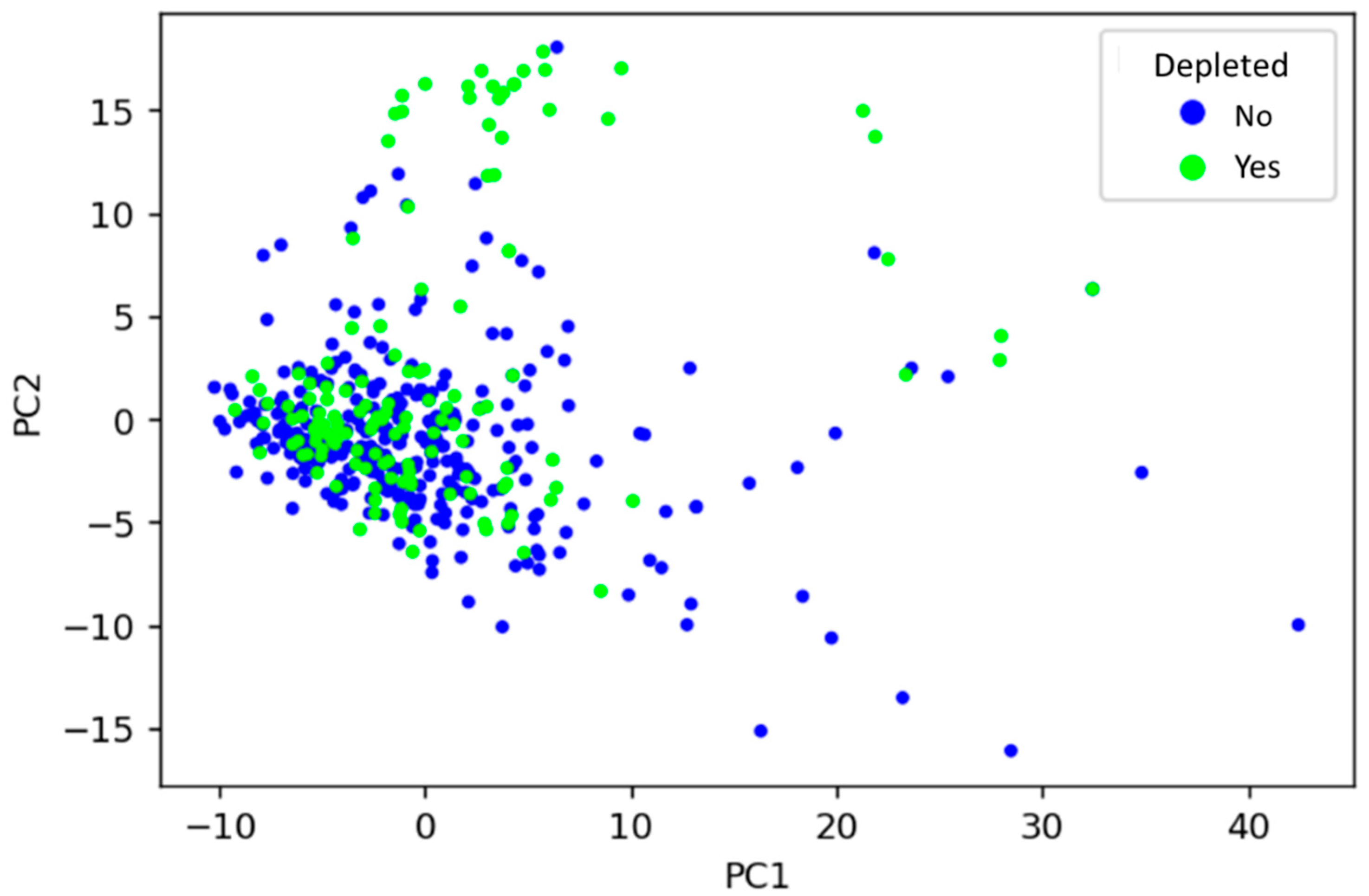
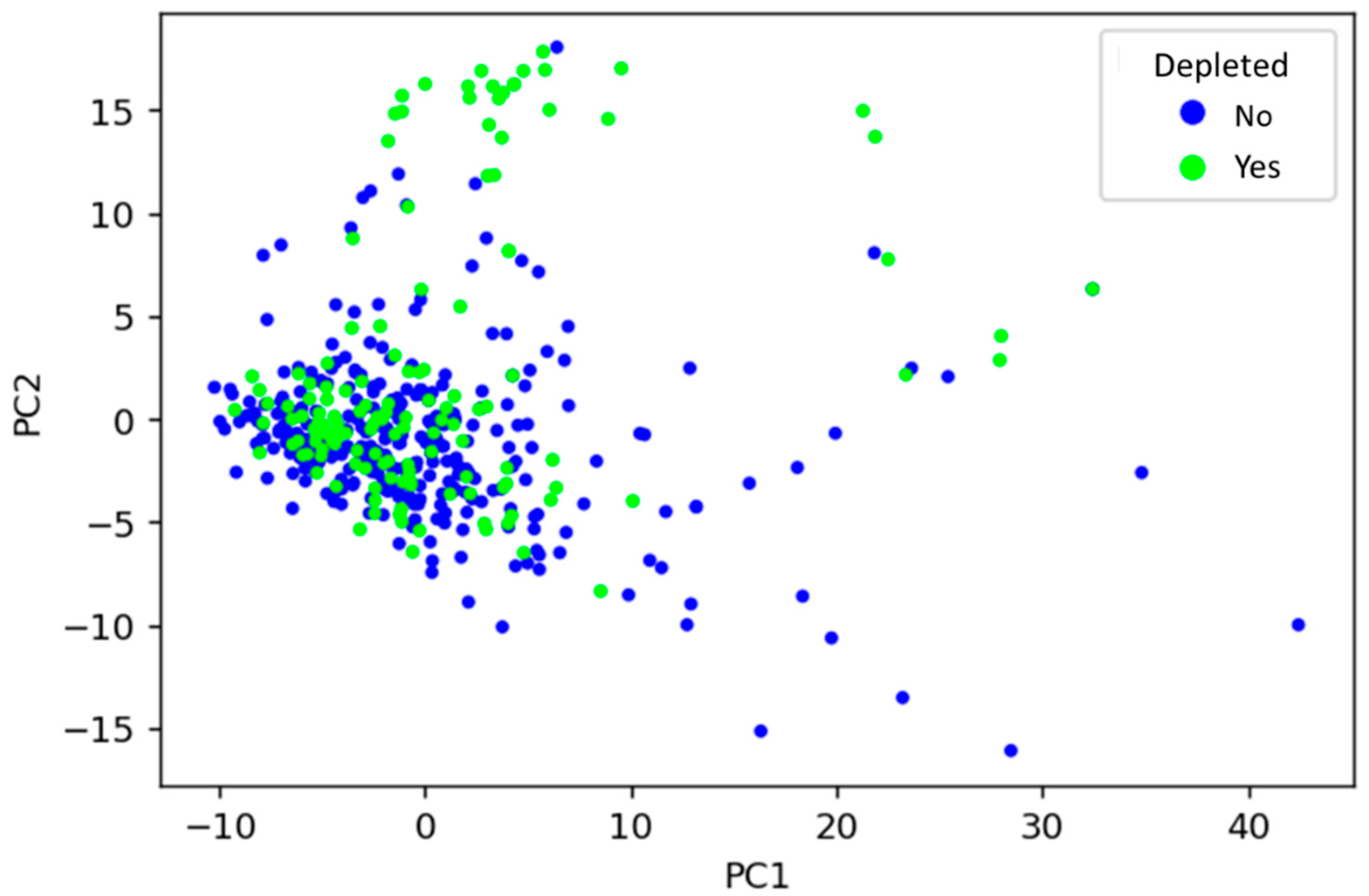
3.1. Unsupervised Learning
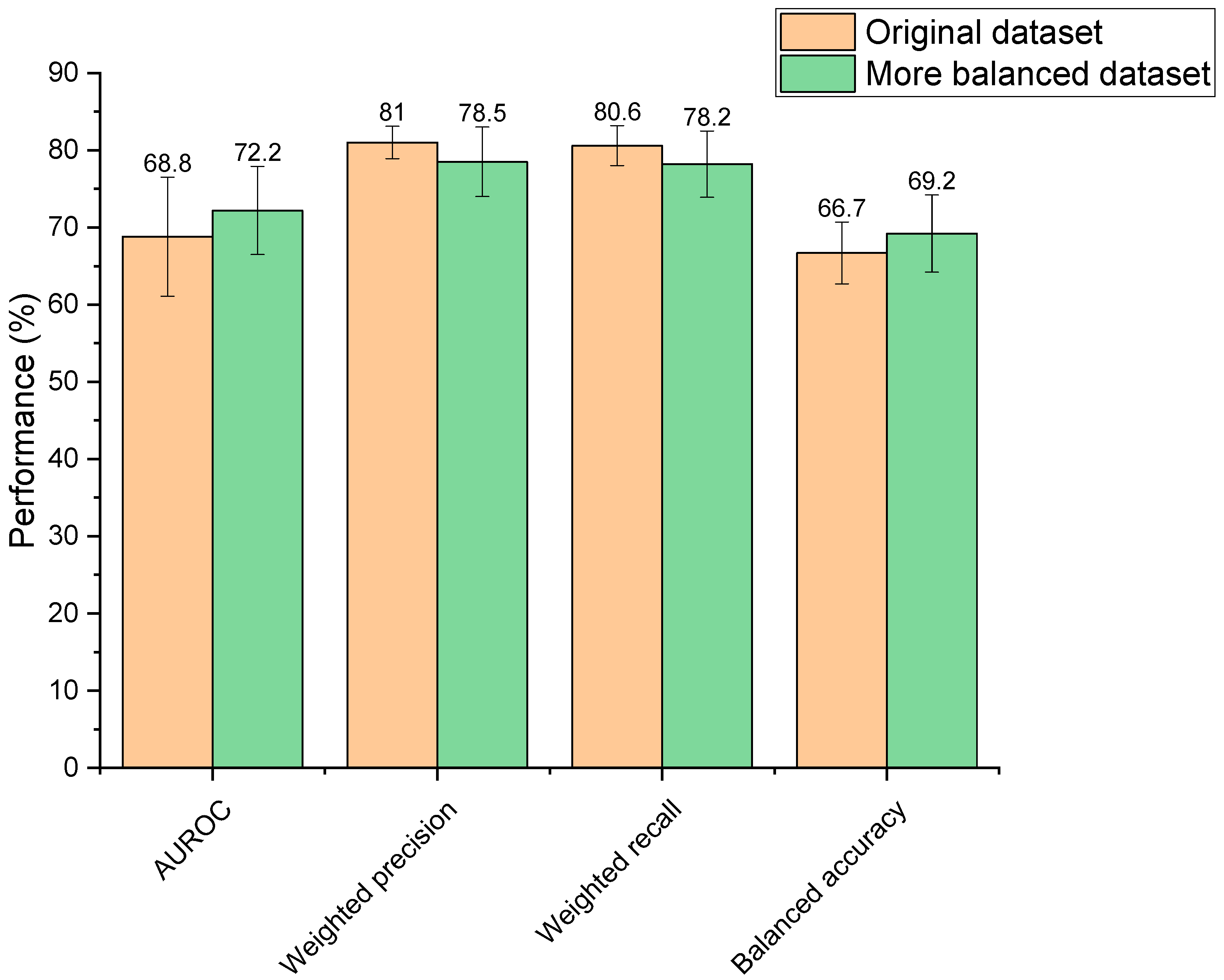
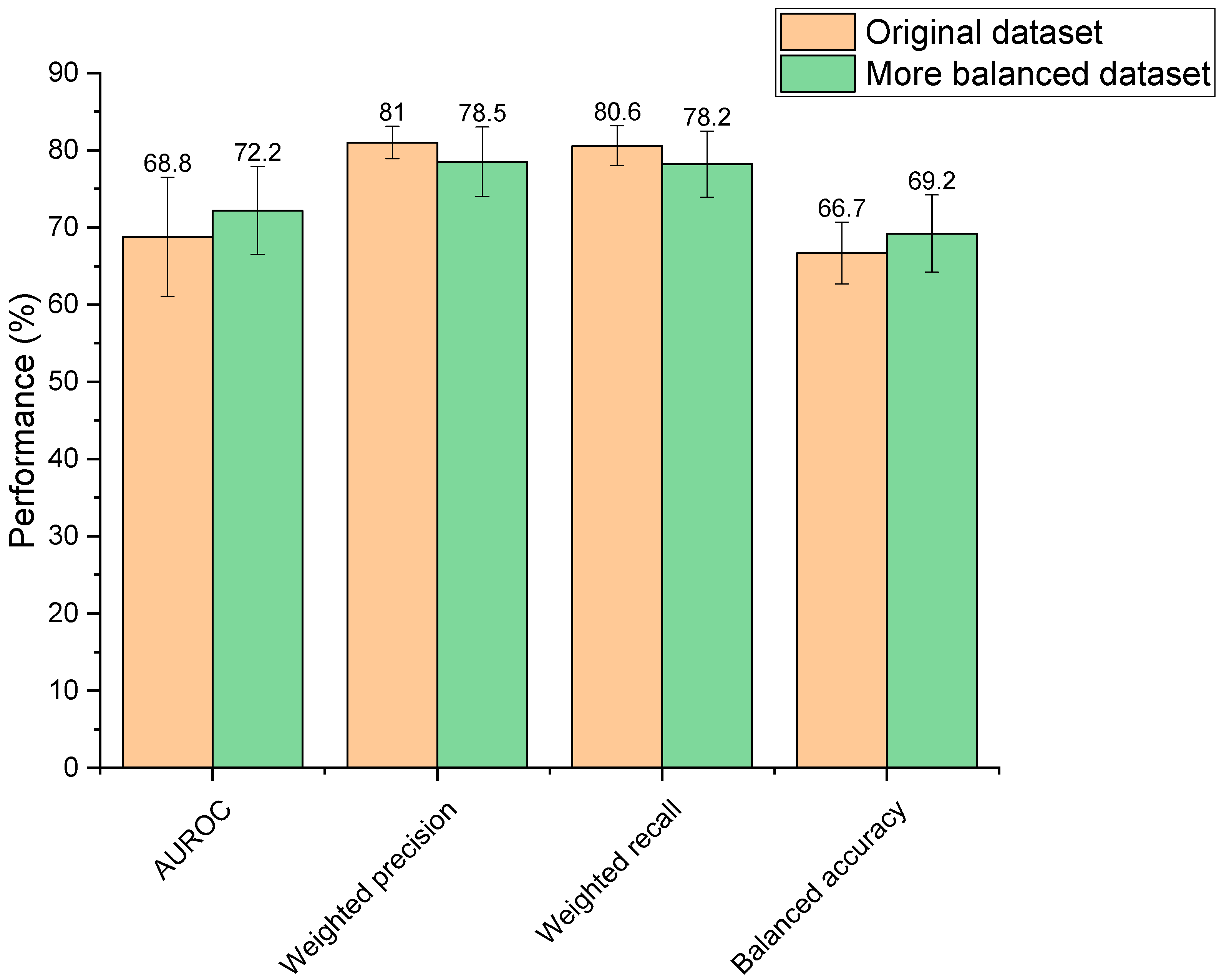
3.2. Dataset Balancing
3.3. Feature Selection
3.4. Supervised Machine Learning
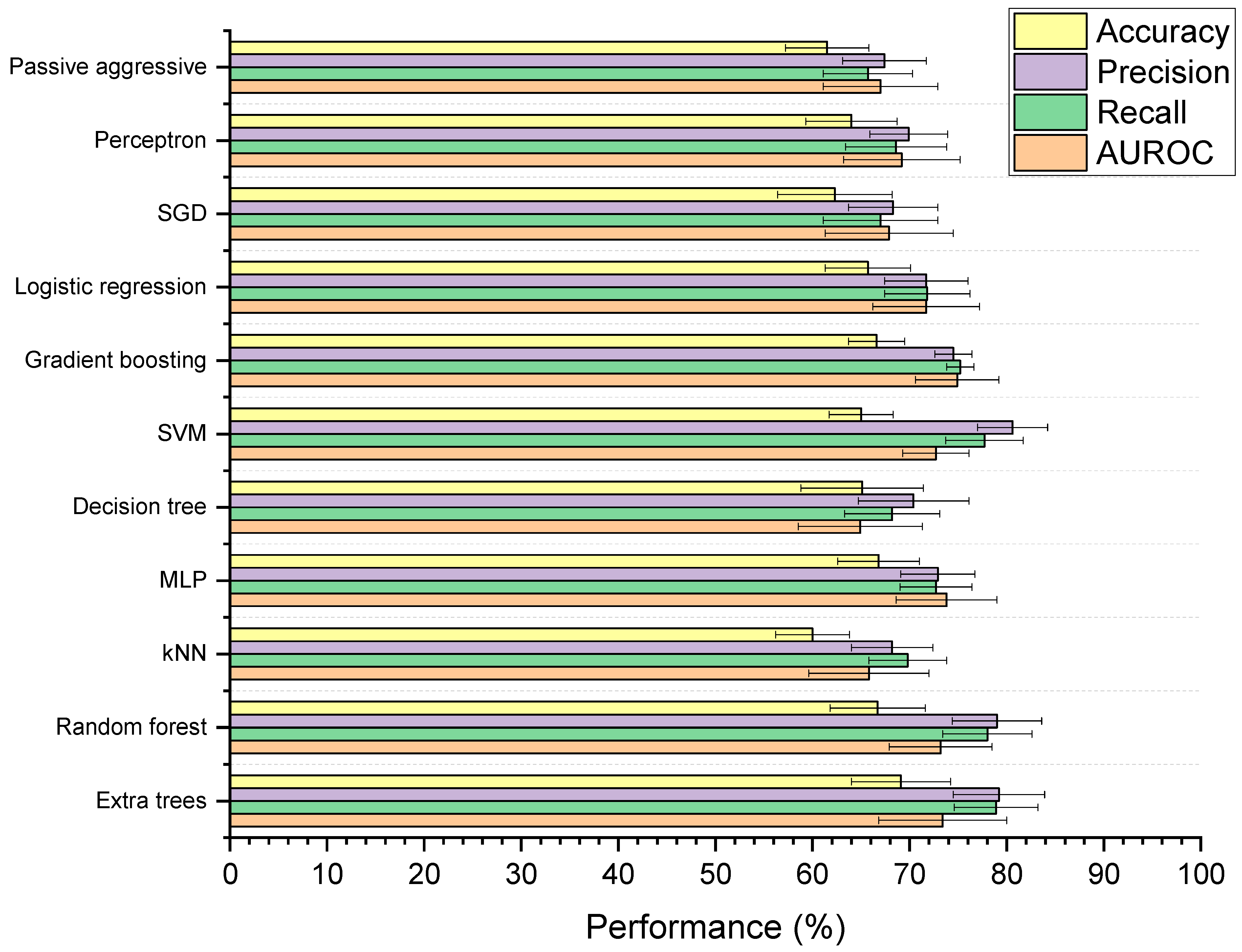
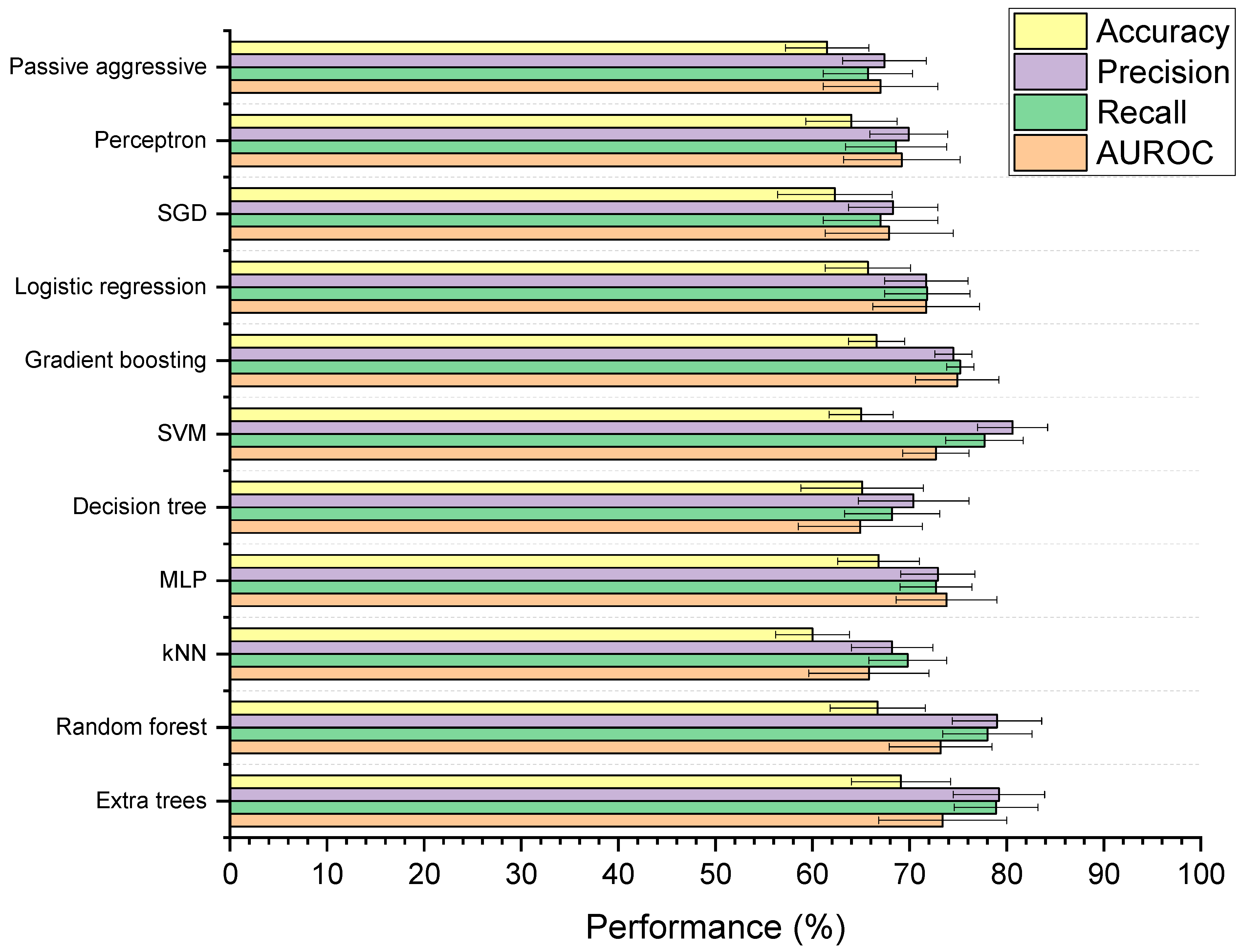
3.4.1. Baseline Models
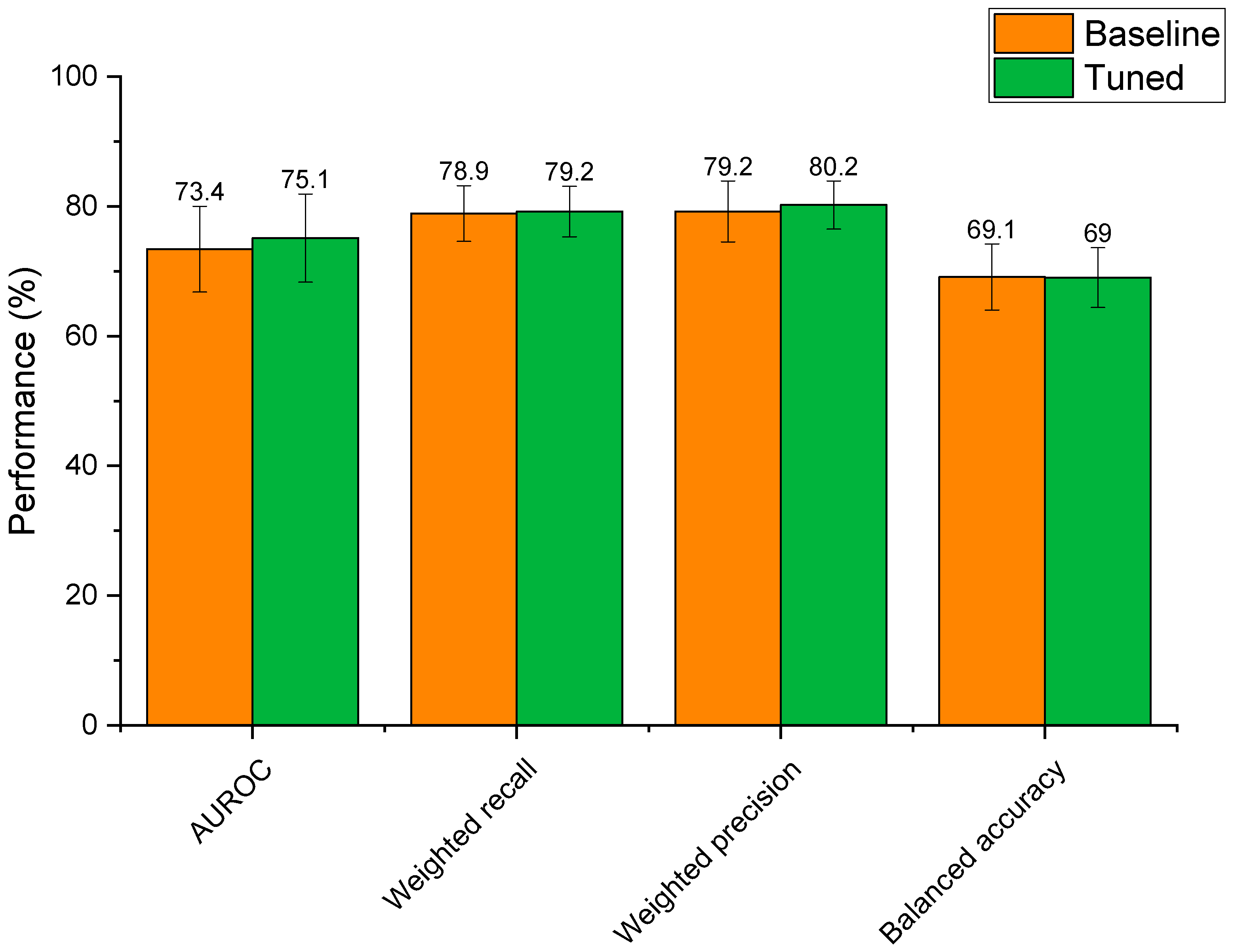
3.4.2. Hyperparameter Optimisation
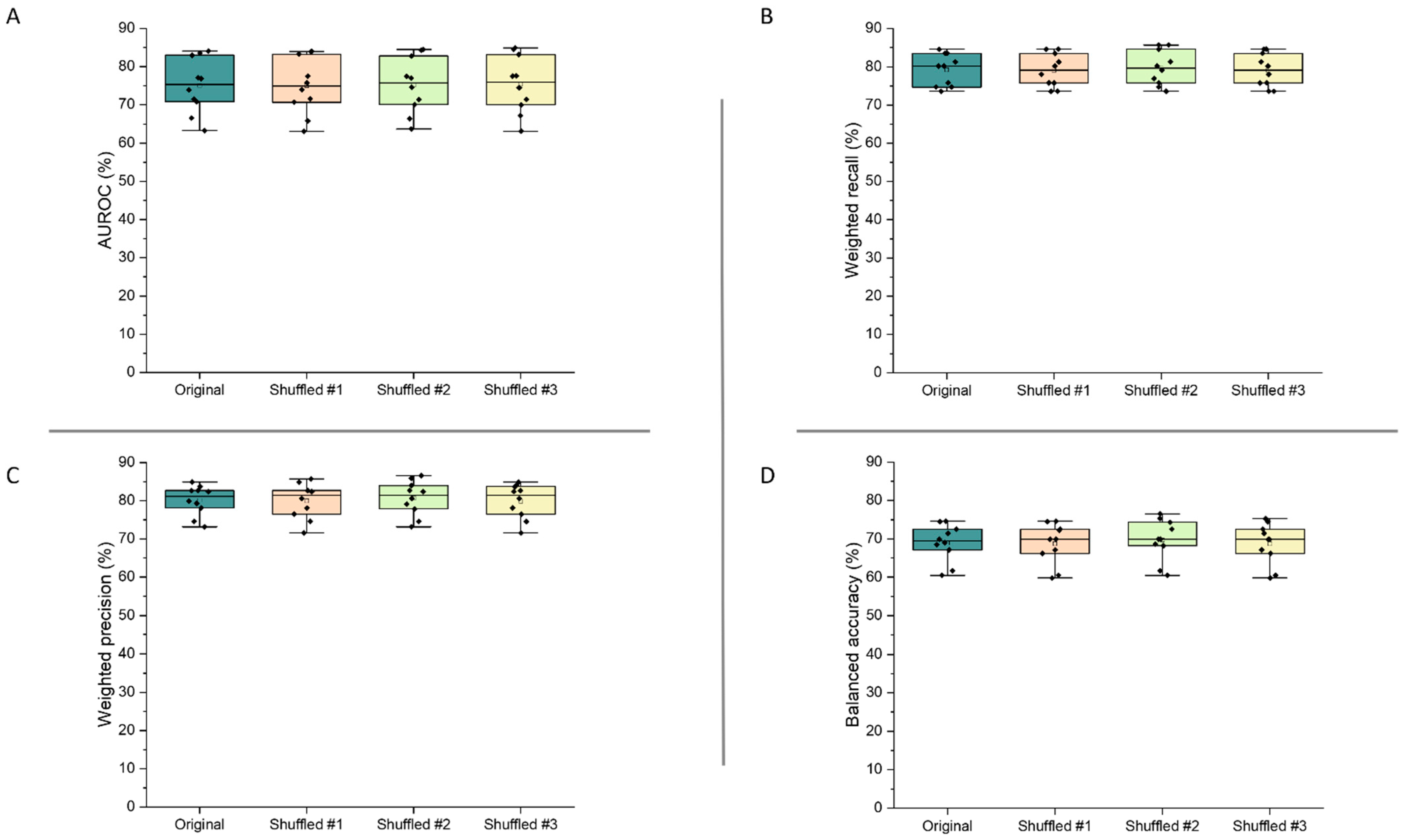
3.4.3. Feature Shuffling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zimmermann, M.; Zimmermann-Kogadeeva, M.; Wegmann, R.; Goodman, A.L. Mapping human microbiome drug metabolism by gut bacteria and their genes. Nature 2019, 570, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Javdan, B.; Lopez, J.G.; Chankhamjon, P.; Lee, Y.J.; Hull, R.; Wu, Q.; Wang, X.; Chatterjee, S.; Donia, M.S. Personalized Mapping of Drug Metabolism by the Human Gut Microbiome. Cell 2020, 181, 1661–1679. [Google Scholar] [CrossRef]
- Klünemann, M.; Andrejev, S.; Blasche, S.; Mateus, A.; Phapale, P.; Devendran, S.; Vappiani, J.; Simon, B.; Scott, T.A.; Kafkia, E.; et al. Bioaccumulation of therapeutic drugs by human gut bacteria. Nature 2021, 597, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Zimmermann-Kogadeeva, M.; Wegmann, R.; Goodman, A.L. Separating host and microbiome contributions to drug pharmacokinetics and toxicity. Science 2019, 363, eaat9931. [Google Scholar] [CrossRef]
- Zimmermann-Kogadeeva, M.; Zimmermann, M.; Goodman, A.L. Insights from pharmacokinetic models of host-microbiome drug metabolism. Gut Microbes 2019, 11, 587–596. [Google Scholar] [CrossRef] [Green Version]
- Coombes, Z.; Yadav, V.; McCoubrey, L.E.; Freire, C.; Basit, A.W.; Conlan, R.S.; Gonzalez, D. Progestogens Are Metabolized by the Gut Microbiota: Implications for Colonic Drug Delivery. Pharmaceutics 2020, 12, 760. [Google Scholar] [CrossRef]
- Yadav, V.; Mai, Y.; McCoubrey, L.E.; Wada, Y.; Tomioka, M.; Kawata, S.; Charde, S.; Basit, A.W. 5-Aminolevulinic Acid as a Novel Therapeutic for Inflammatory Bowel Disease. Biomedicines 2021, 9, 578. [Google Scholar] [CrossRef]
- Chankhamjon, P.; Javdan, B.; Lopez, J.; Hull, R.; Chatterjee, S.; Donia, M.S. Systematic mapping of drug metabolism by the human gut microbiome. bioRxiv 2019, 538215. [Google Scholar] [CrossRef]
- Yadav, V.; Varum, F.; Bravo, R.; Furrer, E.; Basit, A.W. Gastrointestinal stability of therapeutic anti-TNF alpha IgG1 monoclonal antibodies. Int. J. Pharm. 2016, 502, 181–187. [Google Scholar] [CrossRef]
- Yadav, V.; Gaisford, S.; Merchant, H.A.; Basit, A.W. Colonic bacterial metabolism of corticosteroids. Int. J. Pharm. 2013, 457, 268–274. [Google Scholar] [CrossRef]
- Wang, J.; Yadav, V.; Smart, A.L.; Tajiri, S.; Basit, A.W. Toward Oral Delivery of Biopharmaceuticals: An Assessment of the Gastrointestinal Stability of 17 Peptide Drugs. Mol. Pharm. 2015, 12, 966–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sousa, T.; Yadav, V.; Zann, V.; Borde, A.; Abrahamsson, B.; Basit, A.W. On the Colonic Bacterial Metabolism of Azo-Bonded Prodrugs of 5-Aminosalicylic Acid. J. Pharm. Sci. 2014, 103, 3171–3175. [Google Scholar] [CrossRef]
- Sousa, T.; Paterson, R.; Moore, V.; Carlsson, A.; Abrahamsson, B.; Basit, A.W. The gastrointestinal microbiota as a site for the biotransformation of drugs. Int. J. Pharm. 2008, 363, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Clarke, G.; Sandhu, K.V.; Griffin, B.T.; Dinan, T.G.; Cryan, J.F.; Hyland, N.P. Gut Reactions: Breaking Down Xenobiotic-Microbiome Interactions. Pharm. Rev. 2019, 71, 198–224. [Google Scholar] [CrossRef]
- Walsh, J.; Griffin, B.T.; Clarke, G.; Hyland, N.P. Drug-gut microbiota interactions: Implications for neuropharmacology. Br. J. Pharm. 2018, 175, 4415–4429. [Google Scholar] [CrossRef] [Green Version]
- Vinarov, Z.; Abdallah, M.; Agundez, J.A.G.; Allegaert, K.; Basit, A.W.; Braeckmans, M.; Ceulemans, J.; Corsetti, M.; Griffin, B.T.; Grimm, M.; et al. Impact of gastrointestinal tract variability on oral drug absorption and pharmacokinetics: An UNGAP review. Eur. J. Pharm. Sci. 2021, 162, 105812. [Google Scholar] [CrossRef]
- Rekdal, V.M.; Bess, E.N.; Bisanz, J.E.; Turnbaugh, P.J.; Balskus, E.P. Discovery and inhibition of an interspecies gut bacterial pathway for Levodopa metabolism. Science 2019, 364, eaau6323. [Google Scholar] [CrossRef] [Green Version]
- Wilson, I.D.; Nicholson, J.K. Gut microbiome interactions with drug metabolism, efficacy, and toxicity. Transl. Res. 2017, 179, 204–222. [Google Scholar] [CrossRef] [Green Version]
- Curro, D. The role of gut microbiota in the modulation of drug action: A focus on some clinically significant issues. Expert Rev. Clin. Pharm. 2018, 11, 171–183. [Google Scholar] [CrossRef]
- Flowers, S.A.; Bhat, S.; Lee, J.C. Potential Implications of Gut Microbiota in Drug Pharmacokinetics and Bioavailability. Pharmacotherapy 2020, 40, 704–712. [Google Scholar] [CrossRef]
- Taggart, A.J.; McDermott, B.J.; Roberts, S.D. The Effect of Age and Acetylator Phenotype on the Pharmacokinetics of Sulfasalazine in Patients with Rheumatoid Arthritis. Clin. Pharmacokinet. 1992, 23, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Wang, X.W.; Wu, A.K.; Fan, Y.; Friedman, J.; Dahlin, A.; Waldor, M.K.; Weinstock, G.M.; Weiss, S.T.; Liu, Y.Y. Deciphering functional redundancy in the human microbiome. Nat. Commun. 2020, 11, 6217. [Google Scholar] [CrossRef]
- Walsh, J.; Olavarria-Ramirez, L.; Lach, G.; Boehme, M.; Dinan, T.G.; Cryan, J.F.; Griffin, B.T.; Hyland, N.P.; Clarke, G. Impact of host and environmental factors on β-glucuronidase enzymatic activity: Implications for gastrointestinal serotonin. Am. J. Physiol. -Gastrointest. Liver Physiol. 2020, 318, G816–G826. [Google Scholar] [CrossRef] [PubMed]
- Haiser, H.J.; Seim, K.L.; Balskus, E.P.; Turnbaugh, P.J. Mechanistic insight into digoxin inactivation by Eggerthella lenta augments our understanding of its pharmacokinetics. Gut Microbes 2014, 5, 233–238. [Google Scholar] [CrossRef] [Green Version]
- Haiser, H.J.; Gootenberg, D.B.; Chatman, K.; Sirasani, G.; Balskus, E.P.; Turnbaugh, P.J. Predicting and manipulating cardiac drug inactivation by the human gut bacterium Eggerthella lenta. Science 2013, 341, 295–298. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Crnkovic, C.M.; Won, K.-J.; Yang, X.; Lee, J.R.; Orjala, J.; Lee, H.; Jeong, H. Commensal Gut Bacteria Convert the Immunosuppressant Tacrolimus to Less Potent Metabolites. Drug Metab. Dispos. 2019, 47, 194. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.R.; Muthukumar, T.; Dadhania, D.; Taur, Y.; Jenq, R.R.; Toussaint, N.C.; Ling, L.; Pamer, E.; Suthanthiran, M. Gut microbiota and tacrolimus dosing in kidney transplantation. PLoS ONE 2015, 10, e0122399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hitchings, R.; Kelly, L. Predicting and Understanding the Human Microbiome’s Impact on Pharmacology. Trends Pharm. Sci. 2019, 40, 495–505. [Google Scholar] [CrossRef]
- Ke, W.; Saba, J.A.; Yao, C.H.; Hilzendeger, M.A.; Drangowska-Way, A.; Joshi, C.; Mony, V.K.; Benjamin, S.B.; Zhang, S.; Locasale, J.; et al. Dietary serine-microbiota interaction enhances chemotherapeutic toxicity without altering drug conversion. Nat. Commun. 2020, 11, 2587. [Google Scholar] [CrossRef]
- Mager, L.F.; Burkhard, R.; Pett, N.; Cooke, N.C.A.; Brown, K.; Ramay, H.; Paik, S.; Stagg, J.; Groves, R.A.; Gallo, M.; et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 2020, 369, 1481. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. ICH Guideline M3(R2) on Non-Clinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorisation for Pharmaceuticals; European Medicines Agency: London, UK, 2009.
- McCoubrey, L.E.; Gaisford, S.; Orlu, M.; Basit, A.W. Predicting drug-microbiome interactions with machine learning. Biotechnol. Adv. 2021, 107797. [Google Scholar] [CrossRef] [PubMed]
- McCoubrey, L.E.; Elbadawi, M.; Orlu, M.; Gaisford, S.; Basit, A.W. Harnessing machine learning for development of microbiome therapeutics. Gut Microbes 2021, 13, 1–20. [Google Scholar] [CrossRef]
- Khan, S.; Hauptman, R.; Kelly, L. Engineering the Microbiome to Prevent Adverse Events: Challenges and Opportunities. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 159–179. [Google Scholar] [CrossRef] [PubMed]
- Koppel, N.; Maini Rekdal, V.; Balskus, E.P. Chemical transformation of xenobiotics by the human gut microbiota. Science 2017, 356, eaag2770. [Google Scholar] [CrossRef]
- Wang, W.; Ye, Z.; Gao, H.; Ouyang, D. Computational pharmaceutics—A new paradigm of drug delivery. J. Control. Release 2021, 338, 119–136. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.N.; Alexander, M.; Turnbaugh, P.J. Precision Medicine Goes Microscopic: Engineering the Microbiome to Improve Drug Outcomes. Cell Host Microbe 2019, 26, 22–34. [Google Scholar] [CrossRef]
- Sharma, A.K.; Jaiswal, S.K.; Chaudhary, N.; Sharma, V.K. A novel approach for the prediction of species-specific biotransformation of xenobiotic/drug molecules by the human gut microbiota. Sci. Rep. 2017, 7, 9751. [Google Scholar] [CrossRef] [Green Version]
- Elmassry, M.M.; Kim, S.; Busby, B. Predicting drug-metagenome interactions: Variation in the microbial beta-glucuronidase level in the human gut metagenomes. PLoS ONE 2021, 16, e0244876. [Google Scholar] [CrossRef]
- Hatton, G.B.; Yadav, V.; Basit, A.W.; Merchant, H.A. Animal Farm: Considerations in Animal Gastrointestinal Physiology and Relevance to Drug Delivery in Humans. J. Pharm. Sci. 2015, 104, 2747–2776. [Google Scholar] [CrossRef]
- Capecchi, A.; Probst, D.; Reymond, J.-L. One molecular fingerprint to rule them all: Drugs, biomolecules, and the metabolome. J. Cheminformatics 2020, 12, 43. [Google Scholar] [CrossRef]
- Morgan, H.L. The Generation of a Unique Machine Description for Chemical Structures-A Technique Developed at Chemical Abstracts Service. J. Chem. Doc. 1965, 5, 107–113. [Google Scholar] [CrossRef]
- Reker, D.; Shi, Y.; Kirtane, A.R.; Hess, K.; Zhong, G.J.; Crane, E.; Lin, C.H.; Langer, R.; Traverso, G. Machine Learning Uncovers Food- and Excipient-Drug Interactions. Cell Rep. 2020, 30, 3710–3716. [Google Scholar] [CrossRef]
- Badillo, S.; Banfai, B.; Birzele, F.; Davydov, I.I.; Hutchinson, L.; Kam-Thong, T.; Siebourg-Polster, J.; Steiert, B.; Zhang, J.D. An Introduction to Machine Learning. Clin. Pharmacol. Ther. 2020, 107, 871–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrynemackers, M.; Kuffner, R.; Geurts, P. On protocols and measures for the validation of supervised methods for the inference of biological networks. Front. Genet. 2013, 4, 262. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Liu, B.; Peng, S.; Sun, J.; Qiao, X. Computer-Aided Grading of Gliomas Combining Automatic Segmentation and Radiomics. Int. J. Biomed. Imaging 2018, 2018, 2512037. [Google Scholar] [CrossRef] [Green Version]
- Hsu, T.-Y. Machine learning applied to stock index performance enhancement. J. Bank. Financ. Technol. 2021, 5, 21–33. [Google Scholar] [CrossRef]
- McCoubrey, L.E.; Elbadawi, M.; Orlu, M.; Gaisford, S.; Basit, A.W. Machine Learning Uncovers Adverse Drug Effects on Intestinal Bacteria. Pharmaceutics 2021, 13, 1026. [Google Scholar] [CrossRef] [PubMed]
- Geurts, P.; Ernst, D.; Wehenkel, L. Extremely randomized trees. Mach. Learn. 2006, 63, 3–42. [Google Scholar] [CrossRef] [Green Version]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Liang, Y.; Zhang, S.; Qiao, H.; Yao, Y. iPromoter-ET: Identifying promoters and their strength by extremely randomized trees-based feature selection. Anal. Biochem. 2021, 630, 114335. [Google Scholar] [CrossRef]
- Andersson, S.; Bathula, D.R.; Iliadis, S.I.; Walter, M.; Skalkidou, A. Predicting women with depressive symptoms postpartum with machine learning methods. Sci. Rep. 2021, 11, 7877. [Google Scholar] [CrossRef]
- Tannergren, C.; Borde, A.; Borestrom, C.; Abrahamsson, B.; Lindahl, A. Evaluation of an in vitro faecal degradation method for early assessment of the impact of colonic degradation on colonic absorption in humans. Eur. J. Pharm. Sci. 2014, 57, 200–206. [Google Scholar] [CrossRef]
- Dong, J.; Gao, H.; Ouyang, D. PharmSD: A novel AI-based computational platform for solid dispersion formulation design. Int. J. Pharm. 2021, 604, 120705. [Google Scholar] [CrossRef] [PubMed]
- Reker, D. Practical considerations for active machine learning in drug discovery. Drug Discov. Today Technol. 2019, 32–33, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Awad, A.; Trenfield, S.J.; Pollard, T.D.; Jie Ong, J.; Elbadawi, M.; McCoubrey, L.E.; Goyanes, A.; Gaisford, S.; Basit, A.W. Connected Healthcare: Improving Patient Care using Digital Health Technologies. Adv. Drug Deliv. Rev. 2021, 178, 113958. [Google Scholar] [CrossRef]
- Muniz Castro, B.; Elbadawi, M.; Ong, J.J.; Pollard, T.; Song, Z.; Gaisford, S.; Perez, G.; Basit, A.W.; Cabalar, P.; Goyanes, A. Machine learning applied to over 900 3D printed drug delivery systems. J. Control. Release 2021, 590, 119837. [Google Scholar] [CrossRef]
- Elbadawi, M.; McCoubrey, L.E.; Gavins, F.K.H.; Ong, J.J.; Goyanes, A.; Gaisford, S.; Basit, A.W. Disrupting 3D printing of medicines with machine learning. Trends Pharmacol. Sci. 2021, 42, 745–757. [Google Scholar] [CrossRef]
- Schuhmacher, A.; Gatto, A.; Hinder, M.; Kuss, M.; Gassmann, O. The upside of being a digital pharma player. Drug Discov. Today 2020, 25, 1569–1574. [Google Scholar] [CrossRef]

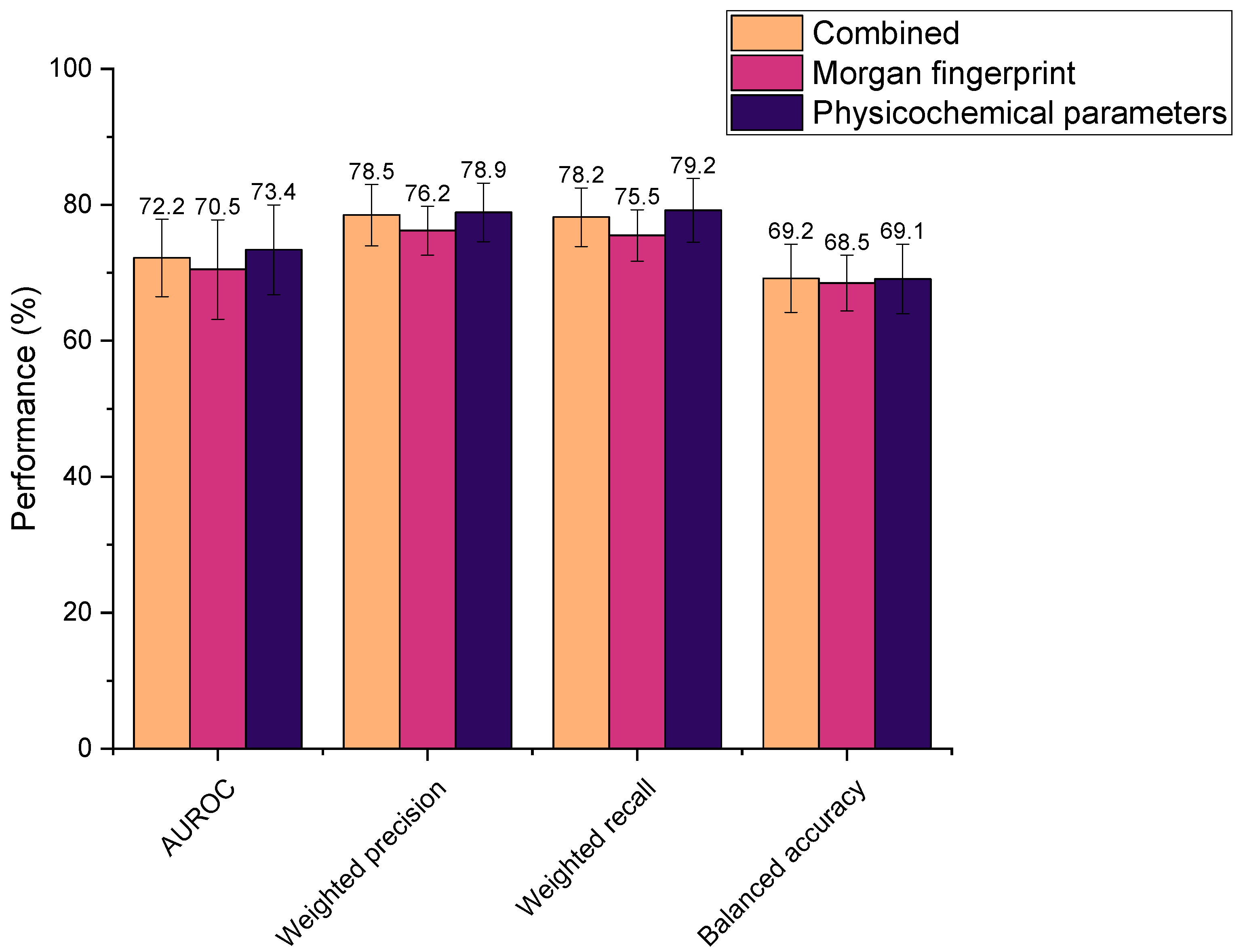
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCoubrey, L.E.; Thomaidou, S.; Elbadawi, M.; Gaisford, S.; Orlu, M.; Basit, A.W. Machine Learning Predicts Drug Metabolism and Bioaccumulation by Intestinal Microbiota. Pharmaceutics 2021, 13, 2001. https://doi.org/10.3390/pharmaceutics13122001
McCoubrey LE, Thomaidou S, Elbadawi M, Gaisford S, Orlu M, Basit AW. Machine Learning Predicts Drug Metabolism and Bioaccumulation by Intestinal Microbiota. Pharmaceutics. 2021; 13(12):2001. https://doi.org/10.3390/pharmaceutics13122001
Chicago/Turabian StyleMcCoubrey, Laura E., Stavriani Thomaidou, Moe Elbadawi, Simon Gaisford, Mine Orlu, and Abdul W. Basit. 2021. "Machine Learning Predicts Drug Metabolism and Bioaccumulation by Intestinal Microbiota" Pharmaceutics 13, no. 12: 2001. https://doi.org/10.3390/pharmaceutics13122001
APA StyleMcCoubrey, L. E., Thomaidou, S., Elbadawi, M., Gaisford, S., Orlu, M., & Basit, A. W. (2021). Machine Learning Predicts Drug Metabolism and Bioaccumulation by Intestinal Microbiota. Pharmaceutics, 13(12), 2001. https://doi.org/10.3390/pharmaceutics13122001