
Functionalized Nanoparticles Targeting Tumor-Associated Macrophages as Cancer Therapy

 ,
,
Abstract
:1. Introduction
2. Macrophages in Tumorigenesis
2.1. Tumor-Associated Macrophages (TAMs)
2.2. Macrophage Polarization and Tumor Development
3. Macrophages in Tumor Therapy
3.1. Tumor Therapy Targeting Tumor-Associated Macrophages (TAMs)
3.1.1. CSF-1/IL-34-CSF-1R
3.1.2. CCL2-CCR2
3.1.3. CCL5-CCR5
3.1.4. CXCL12-CXCR4
3.1.5. CD47-SIRPα
3.1.6. PI-3 Kinase γ (PI3Kγ)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Action | NCT Number | Tumor Type | Status | Target | Agent Name | Phase | Effect |
|---|---|---|---|---|---|---|---|
| Targeting signal transduction pathways | NCT02404844 | Breast cancer | Completed | PI3K pathway | BKM120 Tamoxifen | Phase 2 | Well-tolerated, preliminary activity in advanced cancers [128,129] |
| NCT02384239 | Breast cancer | Active, not recruiting | PI3K pathway | Palbociclib | Phase 2 | NA | |
| NCT02058381 | Breast cancer | Completed | PI3K pathway | Alpelisib (BYL719) Buparlisib (BKM120) | Phase 1b | Well-tolerated, preliminary antitumor activity [130] | |
| NCT01298713 | Breast neoplasms | Completed | PI3K-Akt- mTOR pathway | Tamoxifen Everolimus | Phase 2 | Well-tolerated, positive correlation [131] | |
| NCT01971515 | Solid tumor | Completed | p70S6K/Akt | MSC2363318A | Phase 1 | NA | |
| NCT02322853 | Metastatic breast cancer | Terminated | MAK pathway | Tamoxifen, ralimetinib (LY2228820) | Phase 2 | NA | |
| NCT03458221 | Ovarian carcinoma | Not yet recruiting | ST pathway | Itraconazole | Phase 3 | NA | |
| NCT00189358 | Ovarian cancer, cancer of the fallopian tube and peritoneal cancer | Completed | the EGFR pathway | ZD1839 Tamoxifen | Phase 2 | No side effects Ineffective for refractory/drug resistant cancers [132] | |
| NCT02040857 | Breast cancer | Active, not recruiting | CDK 4/6 | Palbociclib | Phase 2 | Well-tolerated [133] | |
| NCT01920061 | Neoplasm | Completed | PI3K/mTOR pathway | PF-05212384 (gedatolisib) | Phase 1 | NA | |
| NCT02228681 | Endometrial cancer | Active, not recruiting | mTOR pathway | Tamoxifen, everolimus, letrozole and medroxyprogesterone acetate | Phase 2 | Enhanced anticancer efficacy | |
| NCT04318223 | Metastatic breast cancer and locally advanced breast cancer | Recruiting | CDK4/6 | Palbociclib | Phase 2 | NA | |
| NCT02868268 | Neuroblastoma | Recruiting | PD-1/PD-L1 pathway | Gene panel sequencing of tumor specimens | Phase NA | NA | |
| NCT03382340 | Pancreatic cancer, breast cancer and ovarian cancer | Recruiting | STAT3/NF-kB | Imx-110 | Phase 1, 2 | NA | |
| NCT04504552 | Oral premalignant lesions | Not yet recruiting | PD-1/PD-L1 pathway | Avelumab | Phase 2 | NA | |
| TAM recruitment | NCT02471716 | Pigmented villonodular synovitis and tenosynovial giant-cell tumor | Completed | CSF-1R | FPA008 | Phase 1, 2 | NA |
| NCT02526017 | Advanced solid tumors, not limited to lung cancer, head and neck cancer, pancreatic cancer and ovarian cancer | Completed | CSF-1R | FPA008 | Phase 1, 2 | NA | |
| NCT02323191 | Solid tumor | Completed | CSF-1 | RO5509554 | Phase 1 | NA | |
| NCT02760797 | Neoplasms | Completed | CSF-1 | RO5509554 | Phase 1 | NA | |
| NCT03153410 | Pancreatic cancer | Recruiting | CSF-1 | IMC-CS4 | Early phase 1 | NA | |
| NCT04066244 | Amyotrophic lateral sclerosis | Recruiting | CSF-1 | BLZ945 | Phase 2 | NA | |
| NCT02829723 | Advanced solid tumors | Recruiting | CSF-1 | BLZ945 | Phase 1, 2 | NA | |
| NCT02880371 | Advanced solid tumors | Completed | CSF-1 | ARRY-382 | Phase 1, 2 | NA | |
| NCT01316822 | Metastatic cancer | Completed | CSF-1 | ARRY-382 | Phase 1 | NA | |
| NCT02777710 | Colorectal cancer, pancreatic cancer, metastatic cancer and advanced cancer | Completed | CSF-1 | PLX3397 | Phase 1 | NA | |
| NCT02584647 | Sarcoma and malignant peripheral nerve sheath tumors | Recruiting | CSF-1 | PLX3397 | Phase 1 | NA | |
| NCT02371369 | Pigmented villonodular synovitis, giant-cell tumors of the tendon sheath and tenosynovial giant-cell tumor | Active, not recruiting | CSF-1 | PLX3397 | Phase 3 | Mild side effects, improved patient symptoms and functional outcome [134,135,136,137] | |
| NCT01804530 | Solid tumor | Terminated | CSF-1 | PLX7486 TsOH | Phase 1 | NA | |
| NCT03320330 | Recurrent malignant solid neoplasm and osteosarcoma Refractory malignant solid neoplasm and osteosarcoma | Active, not recruiting | SEMA4D or CD100 | Pepinemab (VX15/2503) | Phase 1 Phase 2 | NA | |
| NCT03557970 | Recurrent acute myeloid leukemia and refractory acute myeloid leukemia | Active, not recruiting | CSF-1 | JNJ-40346527 | Phase 2 | NA | |
| NCT03690986 | Squamous cell carcinoma of the head and neck | Recruiting | Recruiting | VX15/2503 (ipilimumab) Nivolumab | Phase 1 | NA | |
| NCT03769155 | Pathologic stage IIIB/C/D cutaneous melanoma AJCC v8 | Recruiting | Recruiting | VX15/2503 (pepinemab) | Phase 1 | NA | |
| NCT03177460 | Prostate adenocarcinoma | Active, not recruiting | CSF-1 | JNJ-40346527 | Phase1 | NA | |
| NCT01572519 | Relapsed or refractory Hodgkin lymphoma | Completed | CSF-1 | JNJ-40346527 | Phase 1 | NA | |
| NCT01597739 | Arthritis, rheumatoid | Completed | CSF-1 | JNJ-40346527 | Phase 2 | NA | |
| NCT02732938 | Metastatic pancreatic ductal adenocarcinoma | Terminated | CCR-2 | PF-04136309 | Phase 2 | Safe and tolerable [86] | |
| NCT01413022 | Pancreatic neoplasms | Completed | CCR-2 | PF-04136309 | Phase 1 | NA | |
| NCT01785810 | Hematologic malignancy | Completed | CCR5 | Maraviroc | Phase 2 | NA | |
| NCT02826486 | Metastatic pancreatic adenocarcinoma | Active, not recruiting | CXCR4 | BL-8040 | Phase 2 | Safety, efficacy and immunobiological effects [101] | |
| NCT00992186 | Prostate cancer | Completed | CCL2 | Carlumab | Phase 2 | NA | |
| NCT01494688 | Solid tumor | Completed | CSF1-R | RG7155 | Phase 1 | NA | |
| Targeting TAM activation | NCT04331067 | Triple-negative breast cancer | Not yet recruiting | TIL and TAM | Paclitaxel, carboplatin Nivolumab, cabiralizumab | Phase 1, 2 | NA |
| NCT03285607 | Breast cancer | Withdrawn | CSF-1 | MCS110, cyclophosphamide and paclitaxel | Phase 1 | NA | |
| NCT02435680 | Triple-negative breast cancer | Completed | CSF-1 | MCS110, carboplatin and gemcitabine | Phase 2 | NA | |
| NCT01643850 | Giant-cell tumor of the tendon sheath | Completed | CSF-1 | MCS110 Placebo | Phase 2 | NA | |
| NCT00757757 | Prostate cancer and bone metastases | Terminated | CSF-1 | MCS110 | Phase 1, 2 | NA | |
| NCT01346358 | Neoplasms | Completed | CSF-1R | IMC-CS4 | Phase 1 | NA | |
| NCT03153410 | Pancreatic cancer | Recruiting | CSF1-R | IMC-CS4 | Phase 1 | NA | |
| NCT01309230 | Ovarian cancer | Active, not recruiting | Bi-shRNA furin and GMCSF | Vigil™ | Phase 2 | NA | |
| NCT02390752 | Neurofibroma, plexiform, precursor cell lymphoblastic, leukemia–lymphoma, leukemia and acute sarcoma | Recruiting | PLX3397 | Phase 1, 2 | NA | ||
| NCT04079712 | Metastatic large-cell neuroendocrine carcinoma, metastatic neuroendocrine carcinoma, metastatic neuroendocrine neoplasm and metastatic small-cell carcinoma | Recruiting | PD-L1 | Cabozantinib Cabozantinib S-malate Ipilimumab Nivolumab | Phase 2 | NA | |
| NCT02265536 | Neoplasms and neoplasm metastasis | Completed | LY3022855 | Phase 1 | NA | ||
| NCT04550624 | Advanced cholangiocarcinoma | Not yet recruiting | Pembrolizumab injection Lenvatinib mesylate | Phase 2 | NA | ||
| NCT01444404 | Advanced malignancy, advanced solid tumors, cancer, oncology and tumors | Completed | CSF-1R | AMG 820 | Phase 1 | NA | |
| NCT02223312 | Cancer Hematologic malignancies | Withdrawn | DC | TAPA-pulsed DC vaccine | Phase 1, 2 | NA | |
| NCT01217229 | Hodgkin lymphoma | Completed | PLX3397 | Phase 2 | NA | ||
| NCT01804530 | Solid tumor, tenosynovial giant-cell tumor | Terminated | CSF-1R | PLX7486 TsOH | Phase 1 | NA | |
| NCT01596751 | Metastatic breast cancer | Completed | DC | PLX3397 Eribulin | Phase 1, 2 | NA | |
| NCT00637390 | Ovarian cancer, fallopian tube cancer and peritoneal cancer | Terminated | CD52 | Alemtuzumab | Phase 1 | NA | |
| NCT01525602 | Solid tumors | Completed | PLX3397 Paclitaxel | Phase 1b | NA | ||
| Reprogramming TAMs | NCT03285607 | Breast cancer | Withdrawn | CSF-1 | MCS110, doxorubicin, cyclophosphamide and paclitaxel | Phase 1 | NA |
| NCT00492167 | Neuroblastoma | Active, not recruiting | Beta-glucan monoclonal antibody 3F8 | Phase 1 | NA | ||
| NCT03954691 | Cancer of head and neck | Not yet recruiting | Microglia, macrophages and NK cells. | Brain-tumor-immune cell communication | NA | NA | |
| NCT02953782 | Colorectal neoplasms Solid tumors | Completed | CD47 | Hu5F9-G4 | Phase 1, 2 | NA | |
| NCT03558139 | Solid tumor and ovarian cancer | Active, not recruiting | CD47 | Hu5F9-G4 | Phase 1 | NA | |
| NCT02216409 | Solid tumor | Completed | CD47 | Hu5F9-G4 | Safe and tolerable [108,138] | ||
| NCT01561911 | Cancer, neoplasms and lymphoma | Completed | CD40 | Chi Lob 7/4 (a chimeric monoclonal antibody) | Phase 1 | Activated B and NK cells [139] | |
| NCT03922477 | Acute myeloid leukemia | Recruiting | CD47 | Hu5F9-G4 | Phase 1 | NA | |
| NCT00912327 | Colorectal cancer | Completed | Imprime PGG | NA | NA | ||
| NCT03248479 | Acute myeloid leukemia and myelodysplastic syndromes | Recruiting | CD47 | Magrolimab Azacitidine | Phase 1 | NA | |
| NCT01433172 | Lung cancer and adenocarcinoma | Completed | CD40 and CCL21 | GM.CD40LCCL21 Vaccinations | Phase 1, 2 | Well-tolerated | |
| NCT01904123 | Metastatic malignant neoplasm in the brain, metastatic melanoma, recurrent brain neoplasm, recurrent glioblastoma and recurrent malignant glioma | Recruiting | STAT3 | STAT3 inhibitor WP1066 | Phase 1 | NA | |
| NCT02953509 | Lymphoma, non-Hodgkin’s; lymphoma, large B cell, diffuse; and indolent lymphoma | Recruiting | CD47 | Hu5F9-G4 | Phase 1, 2 | NA | |
| NCT00911560 | Neuroblastoma | Recruiting | Adjuvant OPT-821 in a vaccine containing two antigens (GD2L and GD3L) covalently linked to KLH | Phase 1, 2 | NA | ||
| NCT01103635 | Recurrent melanoma and stage IV melanoma | Completed | CD40 | CD40 agonist monoclonal antibody CP-870893, tremelimumab | Phase 1 | NA | |
| NCT03527147 | NHL, DLBCL, non-Hodgkin’s lymphoma and diffuse large B cell lymphoma | Recruiting | CD47 | Hu5F9-G4 | Phase 1 | NA | |
| NCT02157831 | Solid tumors | Completed | CD40 | CP-870893 | Phase 1 | NA | |
| NCT02225002 | Advanced solid tumors | Completed | CD40 | CP-870893 | Phase 1 | NA | |
| NCT00711191 | Pancreatic neoplasm | Completed | CD40 | CP-870893 | Phase 1 | Tumoricidal, the depletion of tumor stroma [140] | |
| NCT00607048 | Neoplasms | Completed | CD40 | CP-870893 | Phase 1 | Safe and no long-term [141] | |
| NCT01456585 | Adenocarcinoma pancreas | Completed | CD40 | CP-870893 | Phase 1 | NA | |
| NCT01103635 | Recurrent melanoma and stage IV melanoma | Completed | CD40 | CP-870893 | Phase 1 | NA | |
| NCT01008527 | Melanoma | Completed | CD40 | CP-870893 | Phase 1 | NA | |
| NCT02482168 | Solid tumors | Completed | CD40 | APX005M | Phase 1 | NA | |
| NCT01839604 | Advanced adult hepatocellular carcinoma and hepatocellular carcinoma metastatic | Completed | AZD9150 | Phase 1 | Safe and tolerable | ||
| NCT02367196 | Hematologic neoplasms | Active, not recruiting | CD47 | CC-90002 | Phase 1 | NA | |
| NCT02641002 | Leukemia, myeloid, acute and myelodysplastic syndromes | Terminated | CD47 | CC-90002 | Phase 1 | NA | |
| NCT02663518 | Hematologic malignancies and solid tumor | Recruiting | SIRPα | TTI-621 | Phase 1 | NA | |
| NCT02890368 | Solid tumors, mycosis fungoides, melanoma, merkel cell carcinoma, squamous cell carcinoma, breast carcinoma, and papillomavirus-related malignant neoplasm | Terminated | SIRPα | TTI-621 | Phase 1 | NA | |
| NCT02665416 | Advanced/metastatic solid tumors | Completed | CD40 | RO7009789 | Phase 1 | NA | |
| NCT02760797 | Neoplasms | Completed | CD40 | RO7009789 | Phase 1 | NA | |
| NCT02304393 | Solid tumors | Completed | CD40 | RO7009789 | Phase 1 | NA | |
| NCT02588443 | Pancreatic cancer | Completed | CD40 | RO7009789 | Phase 1 | NA | |
| NCT03329950 | Advanced malignancies | Recruiting | CD40 | CDX-301/CDX-1140 | Phase 1 | NA | |
| NCT00899574 | Breast cancer Breast neoplasms | Completed | TLR7 | Imiquimod | Phase 2 | NA | |
| NCT01421017 | Breast cancer, metastatic breast cancer and recurrent breast cancer | Completed | TLR7 | Imiquimod | Phase 1, 2 | Supplemented the response evaluation criteria in solid tumors [142,143] | |
| NCT04116320 | Melanoma, breast cancer, squamous cell cancer, non-small-cell lung cancer, cervical cancer, urothelial carcinoma, ovarian cancer, small-cell lung cancer and esophageal cancer | Recruiting | TLR7 | Imiquimod | Phase 1 | NA | |
| NCT03196180 | Cervical intraepithelial neoplasia | Active, not recruiting | TLR7 | Imiquimod | Phase 1 | NA | |
| NCT00319748 | Breast cancer, ovarian cancer, endometrial cancer and cervical cancer | Completed | TLR7 | 852A | Phase 2 | NA | |
| NCT00719199 | Colorectal cancer | Terminated | TLR9 | IMO-2055 | Phase 1 | NA | |
| NCT01040832 | Squamous cell carcinoma of the head and neck | Completed | TLR9 | IMO-2055 | Phase 2 | NA | |
| NCT01360827 | Squamous cell carcinoma of the head and neck | Terminated | TLR9 | IMO-2055 | Phase 1 | NA | |
| NCT02829723 | Advanced solid tumors | Recruiting | CSF1R | BLZ945 | Phase 1, 2 | NA |
4. Interaction of Macrophages with NPs
4.1. NP-Based Cancer Therapies Targeting TAMs
4.2. NP Uptake by Macrophages
4.3. Effect of NP Physical and Chemical Properties on Cellular Uptake
4.3.1. Size
4.3.2. Shape
4.3.3. Surface Charge
4.3.4. Surface Hydrophobicity
4.4. Effect of Macrophage Polarization Status on NP Uptake
4.5. Modulation of TAMs by NPs via Passive Targeting In Vivo
4.6. Targeting NPs to TAMs via Specific Surface Receptors
4.6.1. CD163
4.6.2. Mannose Receptor
4.6.3. C-Type Lectin
4.6.4. Scavenger Receptor B Type 1
4.6.5. Sialic Acid Binding Receptors
4.6.6. Legumain
| Category | NP Materials | Payload | Targeted Receptor | TAM-Targeting Strategy | Ref. |
|---|---|---|---|---|---|
| Polymeric NPs | Hyaluronic-acid-coated Pei-PLGA NPs | Methotrexate (MTX) | CD44 | Reprogramming TAMs | [117] |
| Chitosan NPs | Gal-1 siRNA | Passive targeting | Modulation of TME | [219] | |
| PEG-sheddable, mannose-modified PLGA NPs | Mannose receptor | Targeting TAMs | [231] | ||
| Cationic polymeric NPs | siCCR2 | Passive targeting | Inhibition of monocyte recruitment | [209] | |
| Carboxyl- and amino-functionalized polystyrene NPs | Passive targeting | Reprogramming TAMs | [226] | ||
| PEGylated silk fibroin NPs | Silk and fibroin | Passive targeting | Reprogramming of TAMs | [207,251] | |
| Hyaluronic acid–PEG blend NPs | p53 plasmid | Passive targeting | Reprogramming of TAMs | [251] | |
| MMP-responsive PLGA NPs | Bisphosphonate-glucomannan | Mannose receptor | Interference with TAMs survival | [237] | |
| Mannose-modified polymeric micelles | siRNA | Mannose receptor | Depletion of TAMs | [229] | |
| PLGA NPs | Radachlorin | Passive targeting | Depletion of TAMs | [252] | |
| Mannose-modified polymeric NPs | siRNAs to modulate NF-κB signaling | Mannose receptor | Reprogramming of TAMs | [253] | |
| PEG and mannose doubly modified trimethyl chitosan NPs | VEGF siRNA/PIGF siRNA | Mannose receptor and passive targeting | Reprogramming TAMs | [127] | |
| Nanovectors made of galactose-functionalized n-butylamine-poly(l-lysine)-b-poly(l-cysteine) polypeptides coated with DCA-grafted sheddable PEG-PLL copolymers | miR155 | C-type lectin (MGL) receptor | Reprogramming TAMs | [242] | |
| Mannose/acid-sensitive PEG-modified PLGA NPs | Doxorubicin (DOX) | Mannose receptor | Depletion of TAMs | [233] | |
| M2pep-coated PLGA NPs | PLX3397 | Passive targeting | Depletion of TAMs | [246] | |
| Biomimetic NPs | CpG, baicalin and antigenic peptide | Passive targeting | Reprogramming TAMs | [239] | |
| Microenvironment-responsive polymeric (P1) NPs | IL-12 | Passive targeting | Reprogramming TAMs | [218] | |
| Mannan-coated PLGA NPs | Didanosine | Mannose receptor | Depletion of TAMs | [206] | |
| Mannose-modified PLGA NPs | ICG, NH4HCO3 and titanium dioxide | Mannose receptor | Reprogramming of TAMs | [230] | |
| miR155-loaded sPEG/GLC (sPEG/GLC/155) NPs | miR155 | Passive targeting | Reprogramming TAMs | [242] | |
| Arginine NPs | Cas9 and gRNA | Passive targeting | Increased phagocytosis of tumor cells by macrophages | [254,255] | |
| PEG-histidine-modified alginate NPs | Oligodeoxynucleotide and galactosylated cationic dextran | Macrophage galactose-type lectin (Mgl) | Reprogramming TAMs | [243] | |
| Cyclodextrin NPs | R848 | Passive targeting | Reprogramming of TAMs | [220] | |
| Lipid-based NPs | Liposomes | C6-ceramide (LipC6) | Passive targeting | Depletion and reprogramming of TAMs | [31] |
| MMP2-sensitive apoptotic body-mimicking NPs and phosphatidylserine-modified NPs | Phosphatidylserine | MMP2-sensitive | Depletion of TAMs | [256] | |
| Cationic liposomes | Curcumin and anti-STAT3 siRNA | Passive targeting | Reprogramming of TAMs | [210] | |
| Sialic acid–cholesterol conjugate-modified liposomes | Epirubicin and sialic acid | Siglec receptors | Depletion of TAMs | [247] | |
| Liposomes | CCR2 siRNA | Passive targeting | Depletion of TAMs | [208] | |
| Liposomes | Hydrazinocurcumin | Legumain | Reprogramming of TAMs | [248] | |
| Clodronate liposomes | Clodrolip | Passive targeting | Depletion of TAMs | [212,213] | |
| Liposomes | Zoledronate | Passive targeting | Depletion of TAMs | [214] | |
| PEGylated liposomes | Alendronate | Passive targeting | Reprogramming of TAMs | [215] | |
| Long-circulating liposomes | Simvastatin | Passive targeting | Interference with TAM survival | [257] | |
| PEGylated liposomes | Anti-CD40 and CpG oligonucleotides | CD40 | Reprogramming of TAMs | [216] | |
| Supramolecular lipid NPs | Dual-kinase (MEK and CSF1R) inhibitor | Passive targeting | Reprogramming of TAMs | [225] | |
| Sorafenib-loaded lipid NPs (SLNPs) | Sorafenib | Passive targeting | Relieve of the immunosuppressive TME | [258] | |
| α-peptide- and M2pep-linked lipid NPs | Anti-CSF-1R siRNA | Scavenger receptor B type 1 (SR-B1) and M2 macrophage binding peptide | Depletion of TAMs | [126] | |
| Clodronate-loaded liposomes | Bindarit and clodronate | Passive targeting | Inhibition of macrophage recruitment | [213] | |
| Inorganic NPs | Mesoporous silica NPs | IL-4 | Passive targeting | Reprogramming of TAMs | [223] |
| Fe2O3 NP | TLR3 agonist, poly (I:C) | Passive targeting | Reprogramming of TAMs | [221] | |
| Fe3O4/PLGA NPs | Anti-CD206 | CD206 | Reprogramming of TAM | [149] | |
| Ca-CO3/Au-NPs | Passive targeting | Reprogramming of TAMs | [227] | ||
| Au@SiO2 NPs (GNPs) | Anti-CD163 | CD163 | Depletion of TAMs | [259] | |
| Stöber silica NPs | Passive targeting | Sequestration in M1 TAMs | [200] | ||
| Peptide/hyaluronic acid/protamine/CaCO3/DNA-NPs | pDNA and IL-12 | CD44 | Reprogramming of TAMs | [260] | |
| Hyaluronic acid-manganese dioxide (MnO2) NPs | Doxorubicin | CD44 | Reprogramming of TAMs | [261] | |
| αvβ3-integrin-targeting (αvβ3-antagonist, a quinalone nonpeptide)-perfluorocarbon NPs | MYC inhibitor | Vitronectin receptor αvβ3 | Depletion and reprogramming of TAMs | [262] | |
| MnO2 NPs | Sorafeni | Passive targeting | Reprogramming of TAMs | [263] | |
| Iron oxide NPs | Trastuzumab, hIgG | Passive targeting and active targeting | Depletion of TAMs | [264] | |
| Zinc oxide (ZnO) NPs | Doxorubicin (Dox) | Passive targeting | Reprogramming of TAMs | [265] | |
| Mesoporous silica-coated white-light emitting NaYbF4:Tm@NaYF4:Yb/Er upconversion NPs | Roussin’s black salt and doxorubicin | Passive targeting | Interference with TAMs survival | [266] | |
| NaYF4: Yb, Er@NaYF4 rare-earth upconversion-NPs conjugated with rose bengal (NPR) | Conjugated photosensitizer | Passive targeting | Reprogramming of TAMs | [249] | |
| Peptide/hyaluronic acid/protamine/CaCO3/DNA-NPs | pDNA and IL-12 | CD44 | Reprogramming of TAMs | [260,265] | |
| Ca-CO3/Au-NPs | Passive targeting | Reprogramming of TAMs | [227] | ||
| Calcium-biphosphonate-PEG NPs | Chelator-free radiolabeling | Passive targeting | Depletion of TAMs | [211] | |
| Porous silicon multistage nanovectors (MSV) | Albumin-bound paclitaxel | Passive targeting | Reprogramming of TAMs | [267] | |
| MUC-1-modified AuNPs | Mucin-1 peptide | Passive targeting | Reprogramming of TAMs | [268] | |
| Carbon NPs | Multi-walled carbon nantotubes | Fluorescent dye | Passive targeting | Reprogramming of TAMs | [222,269] |
| Single-walled carbon nanotubes | Passive targeting | Depletion of TAMs | [270] | ||
| Graphene oxide NPs | FITC and PEG | Passive targeting | Reprogramming of TAMs | [201] | |
| PEG and polyethylenimine (PEI) dual-polymer-functionalized graphene oxide NPs | CpG | Passive targeting | Reprogramming of TAMs | [271] | |
| PEG and polyethylenimine (PEI) dual-polymer-functionalized graphene oxide NPs | CpG | Passive targeting | Reprogramming of TAMs | [271] | |
| Polyhydroxylated fullerenols | Passive targeting | Reprogramming of TAMs | [272] | ||
| Hybrid NPs | Lipid polymer (7C1) NP | CX3CL1 siRNA | Passive targeting | Inhibition of macrophage recruitment | [217] |
| Mannosylated cationic nano hydrogel particles (ManNP) | siRNA | Mannose receptor | Depletion of TAMs | [232] | |
| Mannose-decorated Pluronic®-F127 polymer and tannic acid NPs | F127-TA | Mannose receptor | Targeting TAMs | [234] | |
| Block copolymers with a hydrophobic block polystyrene (PS) and a hydrophilic glycoblock (PR) self-assembled into glyco-NPs with an inert organic core PS and a glycopolymer shell | Passive targeting | Reprogramming of TAMs | [244] | ||
| Legumain-sheddable PEG5k and tuftsin dual-modified NPs | PEG5k and tuftsin | Fc receptor | Depletion of TAMs | [273] | |
| Other | Hydroxyl dendrimer | CSF-1R inhibitor BLZ945 | Passive targeting | Reprogramming of TAMs | [224] |
5. NPs as a Delivery Platform for Gene-Editing Toolsdelivery Platform for Gene Editing Tools to Macrophagesmacrophages
6. Comparative Analysis of TAM-Targeting Strategies Targeting Strategies
6.1. Active Versus Passive Targetingversus Passive Targeting of TAMs by NPs: Advantages and Disadvantages
6.2. Comparison of NP-Based TAM-Targeting Therapies and Current Challenges
7. Conclusions and Future Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Urruticoechea, A.; Alemany, R.; Balart, J.; Villanueva, A.; Vinals, F.; Capella, G. Recent Advances in Cancer Therapy: An Overview. Curr. Pharm. Des. 2010, 16, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Khalil, D.N.; Smith, E.L.; Brentjens, R.J.; Wolchok, J.D. The future of cancer treatment: Immunomodulation, CARs and combination immunotherapy. Nat. Rev. Clin. Oncol. 2016, 13, 273–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, K.D.; Nogueira, L.; Mariotto, A.B.; Rowland, J.H.; Yabroff, K.R.; Alfano, C.M.; Jemal, A.; Kramer, J.L.; Siegel, R.L. Cancer treatment and survivorship statistics, 2019. CA Cancer J. Clin. 2019, 69, 363–385. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Tang, Z.; Gao, S.; Li, C.; Feng, Y.; Zhou, X. Tumor-Associated Macrophages: Recent Insights and Therapies. Front. Oncol. 2020, 10, 188. [Google Scholar] [CrossRef] [PubMed]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef]
- Yang, Y.; Guo, J.; Huang, L. Tackling TAMs for Cancer Immunotherapy: It’s Nano Time. Trends Pharmacol. Sci. 2020, 41, 701–714. [Google Scholar] [CrossRef]
- Reichel, D.; Tripathi, M.; Perez, J.M. Biological Effects of Nanoparticles on Macrophage Polarization in the Tumor Microenvironment. Nanotheranostics 2019, 3, 66–88. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Chen, Q.; Ruan, C.; Chen, X.; Zhang, Y.; He, X.; Zhang, Y.; Lu, Y.; Guo, Q.; Sun, T.; et al. Platinum-Based Nanovectors Engineered with Immuno-Modulating Adjuvant for Inhibiting Tumor growth and Promoting Immunity. Theranostics 2018, 8, 2974–2987. [Google Scholar] [CrossRef]
- Mu, W.; Chu, Q.; Liu, Y.; Zhang, N. A Review on Nano-Based Drug Delivery System for Cancer Chemoimmunotherapy. Nano-Micro Lett. 2020, 12, 1–24. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhang, L.; Wu, H. Nanomaterials for cancer therapies. Nanotechnol. Rev. 2017, 6, 473–496. [Google Scholar] [CrossRef]
- Chen, Y.; Xianyu, Y.; Jiang, X. Surface Modification of Gold Nanoparticles with Small Molecules for Biochemical Analysis. Acc. Chem. Res. 2017, 50, 310–319. [Google Scholar] [CrossRef]
- de S.L. Oliveira, A.L.C.; de Araujo Junior, R.F.; Gomes de Carvalho, T.; Chan, A.B.; Schomann, T.; Tamburini, F.; de Geus-Oei, L.F.; Cruz, L.J. Effect of Oxaliplatin-Loaded Poly (d,l-Lactide-co-Glycolic Acid) (PLGA) Nanoparticles Combined with Retinoic Acid and Cholesterol on Apoptosis, Drug Resistance, and Metastasis Factors of Colorectal Cancer. Pharmaceutics 2020, 12, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, L.J.; Stammes, M.A.; Que, I.; van Beek, E.R.; Knol-Blankevoort, V.T.; Snoeks, T.J.A.; Chan, A.; Kaijzel, E.L.; Lowik, C. Effect of PLGA NP size on efficiency to target traumatic brain injury. J. Control Release 2016, 223, 31–41. [Google Scholar] [CrossRef]
- Cruz, L.J.; Tacken, P.J.; Pots, J.M.; Torensma, R.; Buschow, S.I.; Figdor, C.G. Comparison of antibodies and carbohydrates to target vaccines to human dendritic cells via DC-SIGN. Biomaterials 2012, 33, 4229–4239. [Google Scholar] [CrossRef] [PubMed]
- Rudramurthy, G.R.; Swamy, M.K. Potential applications of engineered nanoparticles in medicine and biology: An update. J. Biol. Inorg. Chem. 2018, 23, 1185–1204. [Google Scholar] [CrossRef]
- Khan, I.; Saeed, K.; Khan, I. Nanoparticles: Properties, applications and toxicities. Arab. J. Chem. 2019, 12, 908–931. [Google Scholar] [CrossRef]
- Rueda, F.; Eich, C.; Cordobilla, B.; Domingo, P.; Acosta, G.; Albericio, F.; Cruz, L.J.; Domingo, J.C. Effect of TLR ligands co-encapsulated with multiepitopic antigen in nanoliposomes targeted to human DCs via Fc receptor for cancer vaccines. Immunobiology 2017, 222, 989–997. [Google Scholar] [CrossRef]
- Cruz, L.J.; Tacken, P.J.; van der Schoot, J.M.S.; Rueda, F.; Torensma, R.; Figdor, C.G. ICAM3-Fc Outperforms Receptor-Specific Antibodies Targeted Nanoparticles to Dendritic Cells for Cross-Presentation. Molecules 2019, 24, 1825. [Google Scholar] [CrossRef] [Green Version]
- Butts, C.; Murray, N.; Maksymiuk, A.; Goss, G.; Marshall, E.; Soulieres, D.; Cormier, Y.; Ellis, P.; Price, A.; Sawhney, R.; et al. Randomized phase IIB trial of BLP25 liposome vaccine in stage IIIB and IV non-small-cell lung cancer. J. Clin. Oncol. 2005, 23, 6674–6681. [Google Scholar] [CrossRef] [Green Version]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the clinic: An update. Bioeng. Transl. Med. 2019, 4, e10143. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wen, Q.; Choi, S. FDA’s Regulatory Science Program for Generic PLA/PLGA-Based Drug Products. Am. Pharm. Rev. 2016, 19, 5–9. [Google Scholar]
- Palanikumar, L.; Al-Hosani, S.; Kalmouni, M.; Nguyen, V.P.; Ali, L.; Pasricha, R.; Barrera, F.N.; Magzoub, M. pH-responsive high stability polymeric nanoparticles for targeted delivery of anticancer therapeutics. Commun. Biol. 2020, 3, 95. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, C.G.; Camps, M.G.M.; Li, T.; Zerrillo, L.; Lowik, C.W.; Ossendorp, F.; Cruz, L.J. Effective chemoimmunotherapy by co-delivery of doxorubicin and immune adjuvants in biodegradable nanoparticles. Theranostics 2019, 9, 6485–6500. [Google Scholar] [CrossRef]
- Mogosanu, G.D.; Grumezescu, A.M.; Bejenaru, C.; Bejenaru, L.E. Polymeric protective agents for nanoparticles in drug delivery and targeting. Int. J. Pharm. 2016, 510, 419–429. [Google Scholar] [CrossRef]
- Reardon, P.J.; Parhizkar, M.; Harker, A.H.; Browning, R.J.; Vassileva, V.; Stride, E.; Pedley, R.B.; Edirisinghe, M.; Knowles, J.C. Electrohydrodynamic fabrication of core-shell PLGA nanoparticles with controlled release of cisplatin for enhanced cancer treatment. Int. J. Nanomed. 2017, 12, 3913–3926. [Google Scholar] [CrossRef] [Green Version]
- Cruz, L.J.; Tacken, P.J.; Fokkink, R.; Figdor, C.G. The influence of PEG chain length and targeting moiety on antibody-mediated delivery of nanoparticle vaccines to human dendritic cells. Biomaterials 2011, 32, 6791–6803. [Google Scholar] [CrossRef] [PubMed]
- Rosalia, R.A.; Cruz, L.J.; van Duikeren, S.; Tromp, A.T.; Silva, A.L.; Jiskoot, W.; de Gruijl, T.; Lowik, C.; Oostendorp, J.; van der Burg, S.H.; et al. CD40-targeted dendritic cell delivery of PLGA-nanoparticle vaccines induce potent anti-tumor responses. Biomaterials 2015, 40, 88–97. [Google Scholar] [CrossRef]
- Kinoshita, R.; Ishima, Y.; Chuang, V.T.G.; Nakamura, H.; Fang, J.; Watanabe, H.; Shimizu, T.; Okuhira, K.; Ishida, T.; Maeda, H.; et al. Improved anticancer effects of albumin-bound paclitaxel nanoparticle via augmentation of EPR effect and albumin-protein interactions using S-nitrosated human serum albumin dimer. Biomaterials 2017, 140, 162–169. [Google Scholar] [CrossRef]
- Bobo, D.; Robinson, K.J.; Islam, J.; Thurecht, K.J.; Corrie, S.R. Nanoparticle-Based Medicines: A Review of FDA-Approved Materials and Clinical Trials to Date. Pharm. Res. 2016, 33, 2373–2387. [Google Scholar] [CrossRef]
- Gao, S.; Yang, D.; Fang, Y.; Lin, X.; Jin, X.; Wang, Q.; Wang, X.; Ke, L.; Shi, K. Engineering Nanoparticles for Targeted Remodeling of the Tumor Microenvironment to Improve Cancer Immunotherapy. Theranostics 2019, 9, 126–151. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Liu, D.; Kimchi, E.T.; Kaifi, J.T.; Qi, X.; Manjunath, Y.; Liu, X.; Deering, T.; Avella, D.M.; Fox, T.; et al. Nanoliposome C6-Ceramide Increases the Anti-tumor Immune Response and Slows Growth of Liver Tumors in Mice. Gastroenterology 2018, 154, 1024–1036.e9. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef]
- Italiani, P.; Boraschi, D. New Insights into Tissue Macrophages: From Their Origin to the Development of Memory. Immune Netw. 2015, 15, 167–176. [Google Scholar] [CrossRef] [Green Version]
- Franklin, R.A.; Liao, W.; Sarkar, A.; Kim, M.V.; Bivona, M.R.; Liu, K.; Pamer, E.G.; Li, M.O. The cellular and molecular origin of tumor-associated macrophages. Science 2014, 344, 921–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortez-Retamozo, V.; Etzrodt, M.; Newton, A.; Rauch, P.J.; Chudnovskiy, A.; Berger, C.; Ryan, R.J.; Iwamoto, Y.; Marinelli, B.; Gorbatov, R.; et al. Origins of tumor-associated macrophages and neutrophils. Proc. Natl. Acad. Sci. USA 2012, 109, 2491–2496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Furth, R.; Cohn, Z.A.; Hirsch, J.G.; Humphrey, J.H.; Spector, W.G.; Langevoort, H.L. The mononuclear phagocyte system: A new classification of macrophages, monocytes, and their precursor cells. Bull. World Health Organ. 1972, 46, 845–852. [Google Scholar]
- van de Laar, L.; Saelens, W.; De Prijck, S.; Martens, L.; Scott, C.L.; Van Isterdael, G.; Hoffmann, E.; Beyaert, R.; Saeys, Y.; Lambrecht, B.N.; et al. Yolk Sac Macrophages, Fetal Liver, and Adult Monocytes Can Colonize an Empty Niche and Develop into Functional Tissue-Resident Macrophages. Immunity 2016, 44, 755–768. [Google Scholar] [CrossRef] [Green Version]
- Hoeffel, G.; Ginhoux, F. Ontogeny of Tissue-Resident Macrophages. Front. Immunol. 2015, 6, 486. [Google Scholar] [CrossRef] [Green Version]
- Salmaninejad, A.; Valilou, S.F.; Soltani, A.; Ahmadi, S.; Abarghan, Y.J.; Rosengren, R.J.; Sahebkar, A. Tumor-associated macrophages: Role in cancer development and therapeutic implications. Cell Oncol. 2019, 42, 591–608. [Google Scholar] [CrossRef] [PubMed]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef]
- Wei, C.; Yang, C.; Wang, S.; Shi, D.; Zhang, C.; Lin, X.; Liu, Q.; Dou, R.; Xiong, B. Crosstalk between cancer cells and tumor associated macrophages is required for mesenchymal circulating tumor cell-mediated colorectal cancer metastasis. Mol. Cancer 2019, 18, 64. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.; Qian, B.Z.; Rowan, C.; Muthana, M.; Keklikoglou, I.; Olson, O.C.; Tazzyman, S.; Danson, S.; Addison, C.; Clemons, M.; et al. Perivascular M2 Macrophages Stimulate Tumor Relapse after Chemotherapy. Cancer Res. 2015, 75, 3479–3491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloc, M.; Uosef, A.; Kubiak, J.Z.; Ghobrial, R.M. Macrophage Proinflammatory Responses to Microorganisms and Transplanted Organs. Int. J. Mol. Sci. 2020, 21, 9669. [Google Scholar] [CrossRef] [PubMed]
- Fernando, M.R.; Reyes, J.L.; Iannuzzi, J.; Leung, G.; McKay, D.M. The pro-inflammatory cytokine, interleukin-6, enhances the polarization of alternatively activated macrophages. PLoS ONE 2014, 9, e94188. [Google Scholar] [CrossRef] [PubMed]
- Perry, C.J.; Munoz-Rojas, A.R.; Meeth, K.M.; Kellman, L.N.; Amezquita, R.A.; Thakral, D.; Du, V.Y.; Wang, J.X.; Damsky, W.; Kuhlmann, A.L.; et al. Myeloid-targeted immunotherapies act in synergy to induce inflammation and antitumor immunity. J. Exp. Med. 2018, 215, 877–893. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Song, Y.; Du, W.; Gong, L.; Chang, H.; Zou, Z. Tumor-associated macrophages: An accomplice in solid tumor progression. J. Biomed. Sci. 2019, 26, 78. [Google Scholar] [CrossRef]
- Belguendouz, H.; Lahmar-Belguendouz, K.; Messaoudene, D.; Djeraba, Z.; Otmani, F.; Hakem, D.; Lahlou-Boukoffa, O.S.; Youinou, P.; Touil-Boukoffa, C. Cytokines Modulate the “Immune-Metabolism” Interactions during Behcet Disease: Effect on Arginine Metabolism. Int. J. Inflam. 2015, 2015, 241738. [Google Scholar] [CrossRef]
- Li, X.; Su, X.; Liu, R.; Pan, Y.; Fang, J.; Cao, L.; Feng, C.; Shang, Q.; Chen, Y.; Shao, C.; et al. HDAC inhibition potentiates anti-tumor activity of macrophages and enhances anti-PD-L1-mediated tumor suppression. Oncogene 2021, 40, 1836–1850. [Google Scholar] [CrossRef]
- Chen, Z.; Malhotra, P.S.; Thomas, G.R.; Ondrey, F.G.; Duffey, D.C.; Smith, C.W.; Enamorado, I.; Yeh, N.T.; Kroog, G.S.; Rudy, S.; et al. Expression of Proinflammatory and Proangiogenic Cytokines in Patients with Head and Neck Cancer. Mol. Oncol. 1999, 5, 1369–1379. [Google Scholar]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Verdeil, G.; Lawrence, T.; Schmitt-Verhulst, A.M.; Auphan-Anezin, N. Targeting STAT3 and STAT5 in Tumor-Associated Immune Cells to Improve Immunotherapy. Cancers 2019, 11, 1832. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.K.; Gangi, L.; Paul, S.; Schioppa, T.; Saccani, A.; Sironi, M.; Bottazzi, B.; Doni, A.; Vincenzo, B.; Pasqualini, F.; et al. A distinct and unique transcriptional program expressed by tumor-associated macrophages (defective NF-kappaB and enhanced IRF-3/STAT1 activation). Blood 2006, 107, 2112–2122. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Herndon, J.M.; Sojka, D.K.; Kim, K.W.; Knolhoff, B.L.; Zuo, C.; Cullinan, D.R.; Luo, J.; Bearden, A.R.; Lavine, K.J.; et al. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity 2017, 47, 323–338.e6. [Google Scholar] [CrossRef]
- Pollard, J.W. The Yolk Sac Feeds Pancreatic Tumors. Immunity 2017, 47, 217–218. [Google Scholar] [CrossRef]
- Giurisato, E.; Lonardi, S.; Telfer, B.; Lussoso, S.; Risa-Ebri, B.; Zhang, J.; Russo, I.; Wang, J.; Santucci, A.; Finegan, K.G.; et al. Extracellular-Regulated Protein Kinase 5-Mediated Control of p21 Expression Promotes Macrophage Proliferation Associated with Tumor Growth and Metastasis. Cancer Res. 2020, 80, 3319–3330. [Google Scholar] [CrossRef] [PubMed]
- Giurisato, E.; Xu, Q.; Lonardi, S.; Telfer, B.; Russo, I.; Pearson, A.; Finegan, K.G.; Wang, W.; Wang, J.; Gray, N.S.; et al. Myeloid ERK5 deficiency suppresses tumor growth by blocking protumor macrophage polarization via STAT3 inhibition. Proc. Natl. Acad. Sci. USA 2018, 115, E2801–E2810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.Z.; Wang, X.; Wang, Y.; Niu, A.; Wang, S.; Zou, C.; Harris, R.C. IL-4/IL-13-mediated polarization of renal macrophages/dendritic cells to an M2a phenotype is essential for recovery from acute kidney injury. Kidney Int. 2017, 91, 375–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, L.; Li, X. MicroRNA-32 targeting PTEN enhances M2 macrophage polarization in the glioma microenvironment and further promotes the progression of glioma. Mol. Cell Biochem. 2019, 460, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Arranz, A.; Doxaki, C.; Vergadi, E.; Martinez de la Torre, Y.; Vaporidi, K.; Lagoudaki, E.D.; Ieronymaki, E.; Androulidaki, A.; Venihaki, M.; Margioris, A.N.; et al. Akt1 and Akt2 protein kinases differentially contribute to macrophage polarization. Proc. Natl. Acad. Sci. USA 2012, 109, 9517–9522. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Zheng, J.; Xu, S.; Fang, Y.; Wu, Y.; Zeng, J.; Shao, A.; Shi, L.; Lu, J.; Mei, S.; et al. Mer regulates microglial/macrophage M1/M2 polarization and alleviates neuroinflammation following traumatic brain injury. J. Neuroinflamm. 2021, 18, 2. [Google Scholar] [CrossRef]
- Jarnicki, A.G.; Lysaght, J.; Todryk, S.; Mills, K.H. Suppression of antitumor immunity by IL-10 and TGF-beta-producing T cells infiltrating the growing tumor: Influence of tumor environment on the induction of CD4+ and CD8+ regulatory T cells. J. Immunol. 2006, 177, 896–904. [Google Scholar] [CrossRef] [Green Version]
- Dennis, K.L.; Blatner, N.R.; Gounari, F.; Khazaie, K. Current status of interleukin-10 and regulatory T-cells in cancer. Curr. Opin. Oncol. 2013, 25, 637–645. [Google Scholar] [CrossRef]
- Llopiz, D.; Ruiz, M.; Infante, S.; Sarobe, P.; Hervas-Stubbs, S.; Alignani, D.; Lasarte, J.J.; Villanueva, L.; Silva, L.; Guruceaga, E. IL-10 expression defines an immunosuppressive dendritic cell population induced by antitumor therapeutic vaccination. Oncotarget 2017, 8, 2659. [Google Scholar] [CrossRef]
- Noe, J.T.; Mitchell, R.A. MIF-Dependent Control of Tumor Immunity. Front. Immunol. 2020, 11, 609948. [Google Scholar] [CrossRef]
- Collison, L.W.; Workman, C.J.; Kuo, T.T.; Boyd, K.; Wang, Y.; Vignali, K.M.; Cross, R.; Sehy, D.; Blumberg, R.S.; Vignali, D.A. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature 2007, 450, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.H.; Keam, B.; Ock, C.Y.; Kim, S.; Han, B.; Kim, J.W.; Lee, K.W.; Jeon, Y.K.; Jung, K.C.; Chung, E.J.; et al. Prognostic value of the association between MHC class I downregulation and PD-L1 upregulation in head and neck squamous cell carcinoma patients. Sci. Rep. 2019, 9, 7680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Yang, X.; Zhang, C.; Wang, Y.; Cheng, T.; Duan, L.; Tong, Z.; Tan, S.; Zhang, H.; Saw, P.E.; et al. Tumor cell-intrinsic PD-1 receptor is a tumor suppressor and mediates resistance to PD-1 blockade therapy. Proc. Natl. Acad. Sci. USA 2020, 117, 6640–6650. [Google Scholar] [CrossRef]
- Smyth, M.J.; Teng, M.W. The Nobel Prize in Physiology and Medicine. J. Aust. N. Z. Soc. Immunol. 2018. [Google Scholar] [CrossRef]
- Chen, X.; Gao, A.; Zhang, F.; Yang, Z.; Wang, S.; Fang, Y.; Li, J.; Wang, J.; Shi, W.; Wang, L.; et al. ILT4 inhibition prevents TAM- and dysfunctional T cell-mediated immunosuppression and enhances the efficacy of anti-PD-L1 therapy in NSCLC with EGFR activation. Theranostics 2021, 11, 3392–3416. [Google Scholar] [CrossRef]
- Anfray, C.; Ummarino, A.; Andon, F.T.; Allavena, P. Current Strategies to Target Tumor-Associated-Macrophages to Improve Anti-Tumor Immune Responses. Cells 2019, 9, 46. [Google Scholar] [CrossRef] [Green Version]
- Baghdadi, M.; Endo, H.; Takano, A.; Ishikawa, K.; Kameda, Y.; Wada, H.; Miyagi, Y.; Yokose, T.; Ito, H.; Nakayama, H.; et al. High co-expression of IL-34 and M-CSF correlates with tumor progression and poor survival in lung cancers. Sci. Rep. 2018, 8, 418. [Google Scholar] [CrossRef]
- Magkouta, S.F.; Vaitsi, P.C.; Pappas, A.G.; Iliopoulou, M.; Kosti, C.N.; Psarra, K.; Kalomenidis, I.T. CSF1/CSF1R Axis Blockade Limits Mesothelioma and Enhances Efficiency of Anti-PDL1 Immunotherapy. Cancers 2021, 13, 2546. [Google Scholar] [CrossRef]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, S.; Borin, T.F.; Piranlioglu, R.; Ara, R.; Lebedyeva, I.; Angara, K.; Achyut, B.R.; Arbab, A.S.; Rashid, M.H. Changes in the tumor microenvironment and outcome for TME-targeting therapy in glioblastoma: A pilot study. PLoS ONE 2021, 16, e0246646. [Google Scholar]
- Denny, W.A.; Flanagan, J.U. Small-molecule CSF1R kinase inhibitors; review of patents 2015-present. Expert Opin. Ther. Pat. 2021, 31, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Dowlati, A.; Harvey, R.D.; Carvajal, R.D.; Hamid, O.; Klempner, S.J.; Kauh, J.S.W.; Peterson, D.A.; Yu, D.; Chapman, S.C.; Szpurka, A.M.; et al. LY3022855, an anti-colony stimulating factor-1 receptor (CSF-1R) monoclonal antibody, in patients with advanced solid tumors refractory to standard therapy: Phase 1 dose-escalation trial. Investig. New Drugs 2021, 39, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Donthireddy, L.; Marvel, D.; Condamine, T.; Wang, F.; Lavilla-Alonso, S.; Hashimoto, A.; Vonteddu, P.; Behera, R.; Goins, M.A.; et al. Cancer-Associated Fibroblasts Neutralize the Anti-tumor Effect of CSF1 Receptor Blockade by Inducing PMN-MDSC Infiltration of Tumors. Cancer Cell 2017, 32, 654–668.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fantuzzi, L.; Tagliamonte, M.; Gauzzi, M.C.; Lopalco, L. Dual CCR5/CCR2 targeting: Opportunities for the cure of complex disorders. Cell Mol. Life Sci. 2019, 76, 4869–4886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nywening, T.M.; Wang-Gillam, A.; Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Cusworth, B.M.; Toriola, A.T.; Nieman, R.K.; Worley, L.A.; Yano, M.; et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: A single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016, 17, 651–662. [Google Scholar] [CrossRef] [Green Version]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridlender, Z.G.; Kapoor, V.; Buchlis, G.; Cheng, G.; Sun, J.; Wang, L.C.; Singhal, S.; Snyder, L.A.; Albelda, S.M. Monocyte chemoattractant protein-1 blockade inhibits lung cancer tumor growth by altering macrophage phenotype and activating CD8+ cells. Am. J. Respir. Cell Mol. Biol. 2011, 44, 230–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, M.; Yu, W.; Chang, C.; Miyamoto, H.; Liu, X.; Jiang, K.; Yeh, S. Estrogen receptor alpha promotes lung cancer cell invasion via increase of and cross-talk with infiltrated macrophages through the CCL2/CCR2/MMP9 and CXCL12/CXCR4 signaling pathways. Mol. Oncol. 2020, 14, 1779–1799. [Google Scholar] [CrossRef] [PubMed]
- Brummer, G.; Fang, W.; Smart, C.; Zinda, B.; Alissa, N.; Berkland, C.; Miller, D.; Cheng, N. CCR2 signaling in breast carcinoma cells promotes tumor growth and invasion by promoting CCL2 and suppressing CD154 effects on the angiogenic and immune microenvironments. Oncogene 2020, 39, 2275–2289. [Google Scholar] [CrossRef] [PubMed]
- Xi, S.; Zheng, X.; Li, X.; Jiang, Y.; Wu, Y.; Gong, J.; Jie, Y.; Li, Z.; Cao, J.; Sha, L.; et al. Activated Hepatic Stellate Cells Induce Infiltration and Formation of CD163(+) Macrophages via CCL2/CCR2 Pathway. Front. Med. 2021, 8, 627927. [Google Scholar] [CrossRef] [PubMed]
- Noel, M.; O’Reilly, E.M.; Wolpin, B.M.; Ryan, D.P.; Bullock, A.J.; Britten, C.D.; Linehan, D.C.; Belt, B.A.; Gamelin, E.C.; Ganguly, B.; et al. Phase 1b study of a small molecule antagonist of human chemokine (C-C motif) receptor 2 (PF-04136309) in combination with nab-paclitaxel/gemcitabine in first-line treatment of metastatic pancreatic ductal adenocarcinoma. Investig. New Drugs 2020, 38, 800–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, Y.; Huang, H.; Guo, M.; Chen, J.; Wu, W.; Li, W.; Xu, X.; Lin, X.; Fu, W.; Yao, Y.; et al. Breast Phyllodes Tumors Recruit and Repolarize Tumor-Associated Macrophages via Secreting CCL5 to Promote Malignant Progression, Which Can Be Inhibited by CCR5 Inhibition Therapy. Clin. Cancer Res. 2019, 25, 3873–3886. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.W.; Wu, H.H.; Liu, S.C.; Wang, P.C.; Ou, W.C.; Chou, W.Y.; Shen, Y.S.; Tang, C.H. CCL5 and CCR5 interaction promotes cell motility in human osteosarcoma. PLoS ONE 2012, 7, e35101. [Google Scholar]
- Xiang, P.; Jin, S.; Yang, Y.; Sheng, J.; He, Q.; Song, Y.; Yu, W.; Hu, S.; Jin, J. Infiltrating CD4+ T cells attenuate chemotherapy sensitivity in prostate cancer via CCL5 signaling. Prostate 2019, 79, 1018–1031. [Google Scholar] [CrossRef]
- Jiao, X.; Velasco-Velazquez, M.A.; Wang, M.; Li, Z.; Rui, H.; Peck, A.R.; Korkola, J.E.; Chen, X.; Xu, S.; DuHadaway, J.B.; et al. CCR5 Governs DNA Damage Repair and Breast Cancer Stem Cell Expansion. Cancer Res. 2018, 78, 1657–1671. [Google Scholar] [CrossRef] [Green Version]
- Long, H.; Xie, R.; Xiang, T.; Zhao, Z.; Lin, S.; Liang, Z.; Chen, Z.; Zhu, B. Autocrine CCL5 signaling promotes invasion and migration of CD133+ ovarian cancer stem-like cells via NF-kappaB-mediated MMP-9 upregulation. Stem Cells 2012, 30, 2309–2319. [Google Scholar] [CrossRef] [PubMed]
- Casagrande, N.; Borghese, C.; Visser, L.; Mongiat, M.; Colombatti, A.; Aldinucci, D. CCR5 antagonism by maraviroc inhibits Hodgkin lymphoma microenvironment interactions and xenograft growth. Haematologica 2019, 104, 564–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frankenberger, C.; Rabe, D.; Bainer, R.; Sankarasharma, D.; Chada, K.; Krausz, T.; Gilad, Y.; Becker, L.; Rosner, M.R. Metastasis Suppressors Regulate the Tumor Microenvironment by Blocking Recruitment of Prometastatic Tumor-Associated Macrophages. Cancer Res. 2015, 75, 4063–4073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knowles, H.J.; Harris, A.L. Macrophages and the hypoxic tumour microenvironment. Front. Biosci. 2007, 12, 4298–4314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schioppa, T.; Uranchimeg, B.; Saccani, A.; Biswas, S.K.; Doni, A.; Rapisarda, A.; Bernasconi, S.; Saccani, S.; Nebuloni, M.; Vago, L.; et al. Regulation of the chemokine receptor CXCR4 by hypoxia. J. Exp. Med. 2003, 198, 1391–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mota, J.M.; Leite, C.A.; Souza, L.E.; Melo, P.H.; Nascimento, D.C.; de-Deus-Wagatsuma, V.M.; Temporal, J.; Figueiredo, F.; Noushmehr, H.; Alves-Filho, J.C.; et al. Post-Sepsis State Induces Tumor-Associated Macrophage Accumulation through CXCR4/CXCL12 and Favors Tumor Progression in Mice. Cancer Immunol. Res. 2016, 4, 312–322. [Google Scholar] [CrossRef] [Green Version]
- Yusen, W.; Xia, W.; Shengjun, Y.; Shaohui, Z.; Hongzhen, Z. The expression and significance of tumor associated macrophages and CXCR4 in non-small cell lung cancer. J. BUON 2018, 23, 398–402. [Google Scholar] [PubMed]
- Ma, J.; Su, H.; Yu, B.; Guo, T.; Gong, Z.; Qi, J.; Zhao, X.; Du, J. CXCL12 gene silencing down-regulates metastatic potential via blockage of MAPK/PI3K/AP-1 signaling pathway in colon cancer. Clin. Transl. Oncol. 2018, 20, 1035–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, A.; Maxwell, R.; Xia, Y.; Cardarelli, P.; Oyasu, M.; Belcaid, Z.; Kim, E.; Hung, A.; Luksik, A.S.; Garzon-Muvdi, T.; et al. Combination anti-CXCR4 and anti-PD-1 immunotherapy provides survival benefit in glioblastoma through immune cell modulation of tumor microenvironment. J. Neurooncol. 2019, 143, 241–249. [Google Scholar] [CrossRef]
- Zarif, J.C.; Baena-Del Valle, J.A.; Hicks, J.L.; Heaphy, C.M.; Vidal, I.; Luo, J.; Lotan, T.L.; Hooper, J.E.; Isaacs, W.B.; Pienta, K.J.; et al. Mannose Receptor-positive Macrophage Infiltration Correlates with Prostate Cancer Onset and Metastatic Castration-resistant Disease. Eur. Urol. Oncol. 2019, 2, 429–436. [Google Scholar] [CrossRef]
- Bockorny, B.; Semenisty, V.; Macarulla, T.; Borazanci, E.; Wolpin, B.M.; Stemmer, S.M.; Golan, T.; Geva, R.; Borad, M.J.; Pedersen, K.S.; et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: The COMBAT trial. Nat. Med. 2020, 26, 878–885. [Google Scholar] [CrossRef]
- Zhang, W.; Huang, Q.; Xiao, W.; Zhao, Y.; Pi, J.; Xu, H.; Zhao, H.; Xu, J.; Evans, C.E.; Jin, H. Advances in Anti-Tumor Treatments Targeting the CD47/SIRPalpha Axis. Front. Immunol. 2020, 11, 18. [Google Scholar] [CrossRef] [Green Version]
- Cioffi, M.; Trabulo, S.; Hidalgo, M.; Costello, E.; Greenhalf, W.; Erkan, M.; Kleeff, J.; Sainz, B., Jr.; Heeschen, C. Inhibition of CD47 Effectively Targets Pancreatic Cancer Stem Cells via Dual Mechanisms. Clin. Cancer Res. 2015, 21, 2325–2337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, D.; Volkmer, J.P.; Willingham, S.B.; Contreras-Trujillo, H.; Fathman, J.W.; Fernhoff, N.B.; Seita, J.; Inlay, M.A.; Weiskopf, K.; Miyanishi, M.; et al. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc. Natl. Acad. Sci. USA 2013, 110, 11103–11108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ring, N.G.; Herndler-Brandstetter, D.; Weiskopf, K.; Shan, L.; Volkmer, J.P.; George, B.M.; Lietzenmayer, M.; McKenna, K.M.; Naik, T.J.; McCarty, A.; et al. Anti-SIRPalpha antibody immunotherapy enhances neutrophil and macrophage antitumor activity. Proc. Natl. Acad. Sci. USA 2017, 114, E10578–E10585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvey, C.M.; Spinler, K.R.; Irianto, J.; Pfeifer, C.R.; Hayes, B.; Xia, Y.; Cho, S.; Dingal, P.; Hsu, J.; Smith, L.; et al. SIRPA-Inhibited, Marrow-Derived Macrophages Engorge, Accumulate, and Differentiate in Antibody-Targeted Regression of Solid Tumors. Curr. Biol. 2017, 27, 2065–2077.e6. [Google Scholar] [CrossRef] [Green Version]
- Petrova, P.S.; Viller, N.N.; Wong, M.; Pang, X.; Lin, G.H.; Dodge, K.; Chai, V.; Chen, H.; Lee, V.; House, V.; et al. TTI-621 (SIRPalphaFc): A CD47-Blocking Innate Immune Checkpoint Inhibitor with Broad Antitumor Activity and Minimal Erythrocyte Binding. Clin. Cancer Res. 2017, 23, 1068–1079. [Google Scholar] [CrossRef] [Green Version]
- Sikic, B.I.; Lakhani, N.; Patnaik, A.; Shah, S.A.; Chandana, S.R.; Rasco, D.; Colevas, A.D.; O’rourke, T.; Narayanan, S.; Papadopoulos, K.; et al. First-in-Human, First-in-Class Phase I Trial of the Anti-CD47 Antibody Hu5F9-G4 in Patients With Advanced Cancers. J. Clin. Oncol. 2019, 37, 946. [Google Scholar] [CrossRef]
- Zhang, J.Q.; Zeng, S.; Vitiello, G.A.; Seifert, A.M.; Medina, B.D.; Beckman, M.J.; Loo, J.K.; Santamaria-Barria, J.; Maltbaek, J.H.; Param, N.J.; et al. Macrophages and CD8(+) T Cells Mediate the Antitumor Efficacy of Combined CD40 Ligation and Imatinib Therapy in Gastrointestinal Stromal Tumors. Cancer Immunol. Res. 2018, 6, 434–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitale, L.A.; Thomas, L.J.; He, L.Z.; O’Neill, T.; Widger, J.; Crocker, A.; Sundarapandiyan, K.; Storey, J.R.; Forsberg, E.M.; Weidlick, J.; et al. Development of CDX-1140, an agonist CD40 antibody for cancer immunotherapy. Cancer Immunol. Immunother. 2019, 68, 233–245. [Google Scholar] [CrossRef]
- Djureinovic, D.; Wang, M.; Kluger, H.M. Agonistic CD40 Antibodies in Cancer Treatment. Cancers 2021, 13, 1302. [Google Scholar] [CrossRef]
- Kaneda, M.M.; Messer, K.S.; Ralainirina, N.; Li, H.; Leem, C.J.; Gorjestani, S.; Woo, G.; Nguyen, A.V.; Figueiredo, C.C.; Foubert, P.; et al. PI3Kgamma is a molecular switch that controls immune suppression. Nature 2016, 539, 437–442. [Google Scholar] [CrossRef] [Green Version]
- Mohan, S.; Vander Broek, R.; Shah, S.; Eytan, D.F.; Pierce, M.L.; Carlson, S.G.; Coupar, J.F.; Zhang, J.; Cheng, H.; Chen, Z.; et al. MEK Inhibitor PD-0325901 Overcomes Resistance to PI3K/mTOR Inhibitor PF-5212384 and Potentiates Antitumor Effects in Human Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2015, 21, 3946–3956. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Somiya, M.; Kuroda, S. Enhancing antibody-dependent cellular phagocytosis by Re-education of tumor-associated macrophages with resiquimod-encapsulated liposomes. Biomaterials 2021, 268, 120601. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, Y. Tumor-associated macrophages: From basic research to clinical application. J. Hematol. Oncol. 2017, 10, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sui, A.; Chen, X.; Demetriades, A.M.; Shen, J.; Cai, Y.; Yao, Y.; Yao, Y.; Zhu, Y.; Shen, X.; Xie, B. Inhibiting NF-kappaB Signaling Activation Reduces Retinal Neovascularization by Promoting a Polarization Shift in Macrophages. Investig. Ophthalmol. Vis. Sci. 2020, 61, 4. [Google Scholar] [CrossRef]
- Cavalcante, R.S.; Ishikawa, U.; Silva, E.S.; Silva Junior, A.A.; Araujo, A.A.; Cruz, L.J.; Chan, A.B.; Araujo Junior, R.F. M2 TAM-associated STAT3/NF-kappaB signalling suppression as major target of immunomodulatory therapy with PLGA-based nanocarriers and anti-PD-L1 in breast cancer. Br. J. Pharmacol. 2021, 178, 2284–2304. [Google Scholar] [CrossRef] [PubMed]
- de Araujo Junior, R.F.; Eich, C.; Jorquera, C.; Schomann, T.; Baldazzi, F.; Chan, A.B.; Cruz, L.J. Ceramide and palmitic acid inhibit macrophage-mediated epithelial-mesenchymal transition in colorectal cancer. Mol. Cell Biochem. 2020, 468, 153–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, Y.; Liu, X. Bioresponsive Nanomedicine: The Next Step of Deadliest Cancers’ Theranostics. Front. Chem. 2020, 8, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobera, M.; Madauss, K.P.; Pohlhaus, D.T.; Wright, Q.G.; Trocha, M.; Schmidt, D.R.; Baloglu, E.; Trump, R.P.; Head, M.S.; Hofmann, G.A.; et al. Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nat. Chem. Biol. 2013, 9, 319–325. [Google Scholar] [CrossRef]
- Guerriero, J.L.; Sotayo, A.; Ponichtera, H.E.; Castrillon, J.A.; Pourzia, A.L.; Schad, S.; Johnson, S.F.; Carrasco, R.D.; Lazo, S.; Bronson, R.T.; et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature 2017, 543, 428–432. [Google Scholar] [CrossRef]
- Frank, A.C.; Raue, R.; Fuhrmann, D.C.; Sirait-Fischer, E.; Reuse, C.; Weigert, A.; Lutjohann, D.; Hiller, K.; Syed, S.N.; Brune, B. Lactate dehydrogenase B regulates macrophage metabolism in the tumor microenvironment. Theranostics 2021, 11, 7570–7588. [Google Scholar] [CrossRef]
- Zheng, P.; Chen, L.; Yuan, X.; Luo, Q.; Liu, Y.; Xie, G.; Ma, Y.; Shen, L. Exosomal transfer of tumor-associated macrophage-derived miR-21 confers cisplatin resistance in gastric cancer cells. J. Exp. Clin. Cancer Res. 2017, 36, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wayne, E.C.; Long, C.; Haney, M.J.; Batrakova, E.V.; Leisner, T.M.; Parise, L.V.; Kabanov, A.V. Targeted Delivery of siRNA Lipoplexes to Cancer Cells Using Macrophage Transient Horizontal Gene Transfer. Adv. Sci. 2019, 6, 1900582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Li, H.; Zhao, S.; Wang, E.; Zhu, J.; Feng, D.; Zhu, Y.; Dou, W.; Fan, Q.; Hu, J.; et al. Epigenetic silencing of miR-144/451a cluster contributes to HCC progression via paracrine HGF/MIF-mediated TAM remodeling. Mol. Cancer 2021, 20, 46. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Qiao, S.; Dai, Y.; Xu, G.; Dai, B.; Lu, L.; Yu, X.; Luo, Q.; Zhang, Z. Molecular-Targeted Immunotherapeutic Strategy for Melanoma via Dual-Targeting Nanoparticles Delivering Small Interfering RNA to Tumor-Associated Macrophages. ACS Nano 2017, 11, 9536–9549. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Tang, C.; Yin, C. Combination antitumor immunotherapy with VEGF and PIGF siRNA via systemic delivery of multi-functionalized nanoparticles to tumor-associated macrophages and breast cancer cells. Biomaterials 2018, 185, 117–132. [Google Scholar] [CrossRef] [PubMed]
- Rodon, J.; Brana, I.; Siu, L.L.; De Jonge, M.J.; Homji, N.; Mills, D.; Di Tomaso, E.; Sarr, C.; Trandafir, L.; Massacesi, C.; et al. Phase I dose-escalation and -expansion study of buparlisib (BKM120), an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2014, 32, 670–681. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Rodon, J.; Burris, H.A.; de Jonge, M.; Verweij, J.; Birle, D.; Demanse, D.; De Buck, S.S.; Ru, Q.C.; Peters, M.; et al. Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2012, 30, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.S.; Lee, K.S.; Chao, T.Y.; Tseng, L.M.; Chitapanarux, I.; Chen, S.C.; Liu, C.T.; Sohn, J.; Kim, J.H.; Chang, Y.C.; et al. A Phase Ib Study of Alpelisib or Buparlisib Combined with Tamoxifen Plus Goserelin in Premenopausal Women with HR-Positive HER2-Negative Advanced Breast Cancer. Clin. Cancer Res. 2021, 27, 408–417. [Google Scholar] [CrossRef]
- Treilleux, I.; Arnedos, M.; Cropet, C.; Wang, Q.; Ferrero, J.M.; Abadie-Lacourtoisie, S.; Levy, C.; Legouffe, E.; Lortholary, A.; Pujade-Lauraine, E.; et al. Translational studies within the TAMRAD randomized GINECO trial: Evidence for mTORC1 activation marker as a predictive factor for everolimus efficacy in advanced breast cancer. Ann. Oncol. 2015, 26, 120–125. [Google Scholar] [CrossRef]
- Wagner, U.; du Bois, A.; Pfisterer, J.; Huober, J.; Loibl, S.; Luck, H.J.; Sehouli, J.; Gropp, M.; Stahle, A.; Schmalfeldt, B.; et al. Gefitinib in combination with tamoxifen in patients with ovarian cancer refractory or resistant to platinum-taxane based therapy--a phase II trial of the AGO Ovarian Cancer Study Group (AGO-OVAR 2.6). Gynecol. Oncol. 2007, 105, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Mayer, E.L.; DeMichele, A.; Rugo, H.S.; Miller, K.; Waks, A.G.; Come, S.E.; Mulvey, T.; Jeselsohn, R.; Overmoyer, B.; Guo, H.; et al. A phase II feasibility study of palbociclib in combination with adjuvant endocrine therapy for hormone receptor-positive invasive breast carcinoma. Ann. Oncol. 2019, 30, 1514–1520. [Google Scholar] [CrossRef] [PubMed]
- Tap, W.D.; Gelderblom, H.; Palmerini, E.; Desai, J.; Bauer, S.; Blay, J.-Y.; Alcindor, T.; Ganjoo, K.; Martín-Broto, J.; Ryan, C.W.; et al. Pexidartinib versus placebo for advanced tenosynovial giant cell tumour (ENLIVEN): A randomised phase 3 trial. Lancet 2019, 394, 478–487. [Google Scholar] [CrossRef]
- Tap, W. ENLIVEN study: Pexidartinib for tenosynovial giant cell tumor (TGCT). Futur. Oncol. 2020, 16, 1875–1878. [Google Scholar] [CrossRef]
- Gelhorn, H.L.; Tong, S.; McQuarrie, K.; Vernon, C.; Hanlon, J.; Maclaine, G.; Lenderking, W.; Ye, X.; Speck, R.M.; Lackman, R.D.; et al. Patient-reported Symptoms of Tenosynovial Giant Cell Tumors. Clin. Ther. 2016, 38, 778–793. [Google Scholar] [CrossRef] [Green Version]
- Lewis, J.H.; Gelderblom, H.; van de Sande, M.; Stacchiotti, S.; Healey, J.H.; Tap, W.D.; Wagner, A.J.; Pousa, A.L.; Druta, M.; Lin, C.C.; et al. Pexidartinib Long-Term Hepatic Safety Profile in Patients with Tenosynovial Giant Cell Tumors. Oncologist 2021, 26, e863–e873. [Google Scholar] [CrossRef]
- Liu, J.; Wang, L.; Zhao, F.; Tseng, S.; Narayanan, C.; Shura, L.; Willingham, S.; Howard, M.; Prohaska, S.; Volkmer, J.; et al. Pre-Clinical Development of a Humanized Anti-CD47 Antibody with Anti-Cancer Therapeutic Potential. PLoS ONE 2015, 10, e0137345. [Google Scholar] [CrossRef] [Green Version]
- Johnson, P.; Challis, R.; Chowdhury, F.; Gao, Y.; Harvey, M.; Geldart, T.; Kerr, P.; Chan, C.; Smith, A.; Steven, N.; et al. Clinical and biological effects of an agonist anti-CD40 antibody: A Cancer Research UK phase I study. Clin. Cancer Res. 2015, 21, 1321–1328. [Google Scholar] [CrossRef] [Green Version]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef] [Green Version]
- Vonderheide, R.H.; Burg, J.M.; Mick, R.; Trosko, J.A.; Li, D.; Shaik, M.N.; Tolcher, A.W.; Hamid, O. Phase I study of the CD40 agonist antibody CP-870,893 combined with carboplatin and paclitaxel in patients with advanced solid tumors. Oncoimmunology 2013, 2, e23033. [Google Scholar] [CrossRef] [Green Version]
- Wolchok, J.D.; Hoos, A.; O’Day, S.; Weber, J.S.; Hamid, O.; Lebbe, C.; Maio, M.; Binder, M.; Bohnsack, O.; Nichol, G.; et al. Guidelines for the evaluation of immune therapy activity in solid tumors: Immune-related response criteria. Clin. Cancer Res. 2009, 15, 7412–7420. [Google Scholar] [CrossRef] [Green Version]
- Vassilios, E.K.; Dardoufas, C.E.; Kouvaris, J.R.; Gennatas, C.S.; Polyzos, A.K.; Gogas, H.J.; Sandilos, P.H.; Uzunoglu, N.K.; Malas, E.G.; Vlahos, L.J. Liposomal Doxorubicin in Conjunction with Reirradiation and Local Hyperthermia Treatment in Recurrent Breast Cancer: A Phase I/II Trial. Clin. Cancer Res. 2002, 8, 374–382. [Google Scholar]
- Ngambenjawong, C.; Gustafson, H.H.; Pun, S.H. Progress in tumor-associated macrophage (TAM)-targeted therapeutics. Adv. Drug Deliv. Rev. 2017, 114, 206–221. [Google Scholar] [CrossRef] [Green Version]
- Vu, M.N.; Rajasekhar, P.; Poole, D.P.; Khor, S.Y.; Truong, N.P.; Nowell, C.J.; Quinn, J.F.; Whittaker, M.; Veldhuis, N.A.; Davis, T.P. Rapid Assessment of Nanoparticle Extravasation in a Microfluidic Tumor Model. ACS Appl. Nano Mater. 2019, 2, 1844–1856. [Google Scholar] [CrossRef] [Green Version]
- Mahlbacher, G.; Curtis, L.T.; Lowengrub, J.; Frieboes, H.B. Mathematical modeling of tumor-associated macrophage interactions with the cancer microenvironment. J. Immunother. Cancer 2018, 6, 10. [Google Scholar] [CrossRef] [Green Version]
- Ovais, M.; Guo, M.; Chen, C. Tailoring Nanomaterials for Targeting Tumor-Associated Macrophages. Adv. Mater. 2019, 31, e1808303. [Google Scholar] [CrossRef] [PubMed]
- Binnemars-Postma, K.; Storm, G.; Prakash, J. Nanomedicine Strategies to Target Tumor-Associated Macrophages. Int. J. Mol. Sci. 2017, 18, 979. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Que, K.T.; Tang, H.M.; Zhang, P.; Fu, Q.M.; Liu, Z.J. Anti-CD206 antibody-conjugated Fe3O4-based PLGA nanoparticles selectively promote tumor-associated macrophages to polarize to the pro-inflammatory subtype. Oncol. Lett. 2020, 20, 298. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Long, L.; Lin, Y.; Peng, C.; Tang, Y.; Zhou, X.; Li, S.; Zhang, C.; Li, X.; Zhou, X. The Double-edged Sword Effect of Macrophage Targeting Delivery System in Different Macrophage Subsets Related Diseases. J. Nanobiotechnol. 2020, 18, 1–14. [Google Scholar] [CrossRef]
- Thomann, J.S.; Heurtault, B.; Weidner, S.; Braye, M.; Beyrath, J.; Fournel, S.; Schuber, F.; Frisch, B. Antitumor activity of liposomal ErbB2/HER2 epitope peptide-based vaccine constructs incorporating TLR agonists and mannose receptor targeting. Biomaterials 2011, 32, 4574–4583. [Google Scholar] [CrossRef]
- Ni, L.; Gayet, I.; Zurawski, S.; Duluc, D.; Flamar, A.L.; Li, X.H.; O’Bar, A.; Clayton, S.; Palucka, A.K.; Zurawski, G.; et al. Concomitant activation and antigen uptake via human dectin-1 results in potent antigen-specific CD8+ T cell responses. J. Immunol. 2010, 185, 3504–3513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leidi, M.; Gotti, E.; Bologna, L.; Miranda, E.; Rimoldi, M.; Sica, A.; Roncalli, M.; Palumbo, G.A.; Introna, M.; Golay, J. M2 macrophages phagocytose rituximab-opsonized leukemic targets more efficiently than m1 cells in vitro. J. Immunol. 2009, 182, 4415–4422. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, D.A.; Vanhecke, D.; Michen, B.; Blank, F.; Gehr, P.; Petri-Fink, A.; Rothen-Rutishauser, B. Different endocytotic uptake mechanisms for nanoparticles in epithelial cells and macrophages. Beilstein J. Nanotechnol. 2014, 5, 1625–1636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- dos Santos, T.; Varela, J.; Lynch, I.; Salvati, A.; Dawson, K.A. Effects of transport inhibitors on the cellular uptake of carboxylated polystyrene nanoparticles in different cell lines. PLoS ONE 2011, 6, e24438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, Y.M.; Hsu, S.J.; Lin, P.C.; Hsu, K.F.; Wu, P.Y.; Su, W.C.; Chang, J.Y.; Shen, M.R. The c.1085A>G Genetic Variant of CSF1R Gene Regulates Tumor Immunity by Altering the Proliferation, Polarization, and Function of Macrophages. Clin. Cancer Res. 2017, 23, 6021–6030. [Google Scholar] [CrossRef] [Green Version]
- Tashiro-Yamaji, J.; Kubota, T.; Yoshida, R. Macrophage MHC receptor 2: A novel receptor on allograft (H-2D(d)K(d))-induced macrophage (H-2D(b)K(b)) recognizing an MHC class I molecule, H-2K(d), in mice. Gene 2006, 384, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Toshchakov, V.; Jones, B.W.; Perera, P.Y.; Thomas, K.; Cody, M.J.; Zhang, S.; Williams, B.R.; Major, J.; Hamilton, T.A.; Fenton, M.J.; et al. TLR4, but not TLR2, mediates IFN-beta-induced STAT1alpha/beta-dependent gene expression in macrophages. Nat. Immunol. 2002, 3, 392–398. [Google Scholar] [CrossRef]
- Roszer, T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediat. Inflamm. 2015, 2015, 816460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, S.; Zhao, X.; Heng, B.C.; Ng, K.W.; Loo, J.S. Cellular uptake of Poly-(d,l-lactide-co-glycolide) (PLGA) nanoparticles synthesized through solvent emulsion evaporation and nanoprecipitation method. Biotechnol. J. 2011, 6, 501–508. [Google Scholar] [CrossRef]
- Augustine, R.; Hasan, A.; Primavera, R.; Wilson, R.J.; Thakor, A.S.; Kevadiya, B.D. Cellular uptake and retention of nanoparticles: Insights on particle properties and interaction with cellular components. Mater. Today Commun. 2020, 25, 101692. [Google Scholar] [CrossRef]
- Cruz, L.J.; Tacken, P.J.; Rueda, F.; Domingo, J.C.; Albericio, F.; Figdor, C.G. Targeting nanoparticles to dendritic cells for immunotherapy. Methods Enzymol. 2012, 509, 143–163. [Google Scholar] [PubMed]
- Sousa de Almeida, M.; Susnik, E.; Drasler, B.; Taladriz-Blanco, P.; Petri-Fink, A.; Rothen-Rutishauser, B. Understanding nanoparticle endocytosis to improve targeting strategies in nanomedicine. Chem. Soc. Rev. 2021, 50, 5397–5434. [Google Scholar] [CrossRef]
- Cruz, L.J.; Tacken, P.J.; Fokkink, R.; Joosten, B.; Stuart, M.C.; Albericio, F.; Torensma, R.; Figdor, C.G. Targeted PLGA nano- but not microparticles specifically deliver antigen to human dendritic cells via DC-SIGN in vitro. J. Control Release 2010, 144, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Kanchan, V.; Panda, A.K. Interactions of antigen-loaded polylactide particles with macrophages and their correlation with the immune response. Biomaterials 2007, 28, 5344–5357. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M.; Boll, W.; Van Oijen, A.; Hariharan, R.; Chandran, K.; Nibert, M.L.; Kirchhausen, T. Endocytosis by random initiation and stabilization of clathrin-coated pits. Cell 2004, 118, 591–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manolova, V.; Flace, A.; Bauer, M.; Schwarz, K.; Saudan, P.; Bachmann, M.F. Nanoparticles target distinct dendritic cell populations according to their size. Eur. J. Immunol. 2008, 38, 1404–1413. [Google Scholar] [CrossRef] [PubMed]
- Alber, F.; Dokudovskaya, S.; Veenhoff, L.M.; Zhang, W.; Kipper, J.; Devos, D.; Suprapto, A.; Karni-Schmidt, O.; Williams, R.; Chait, B.T.; et al. The molecular architecture of the nuclear pore complex. Nature 2007, 450, 695–701. [Google Scholar] [CrossRef]
- Zhu, M.; Nie, G.; Meng, H.; Xia, T.; Nel, A.; Zhao, Y. Physicochemical Properties Determine Nanomaterial Cellular Uptake, Transport, and Fate. Acc. Chem. Res. 2013, 46, 622–631. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.W.; Christison, R.; Bundell, K.; Voyce, C.J.; Brockbank, S.M.; Newham, P.; Lindsay, M.A. Characterisation of cell-penetrating peptide-mediated peptide delivery. Br. J. Pharmacol. 2005, 145, 1093–1102. [Google Scholar] [CrossRef] [PubMed]
- Lutsiak, M.E.; Kwon, G.S.; Samuel, J. Biodegradable nanoparticle delivery of a Th2-biased peptide for induction of Th1 immune responses. J. Pharm. Pharmacol. 2006, 58, 739–747. [Google Scholar] [CrossRef]
- Lundqvist, M.; Stigler, J.; Elia, G.; Lynch, I.; Cedervall, T.; Dawson, K.A. Nanoparticle size and surface properties determine the protein corona with possible implications for biological impacts. Proc. Natl. Acad. Sci. USA 2008, 105, 14265–14270. [Google Scholar] [CrossRef] [Green Version]
- Konduru, N.V.; Molina, R.M.; Swami, A.; Damiani, F.; Pyrgiotakis, G.; Lin, P.; Andreozzi, P.; Donaghey, T.C.; Demokritou, P.; Krol, S.; et al. Protein corona: Implications for nanoparticle interactions with pulmonary cells. Part. Fibre Toxicol. 2017, 14, 42. [Google Scholar] [CrossRef] [Green Version]
- Salvati, A.; Pitek, A.S.; Monopoli, M.P.; Prapainop, K.; Bombelli, F.B.; Hristov, D.R.; Kelly, P.M.; Aberg, C.; Mahon, E.; Dawson, K.A. Transferrin-functionalized nanoparticles lose their targeting capabilities when a biomolecule corona adsorbs on the surface. Nat. Nanotechnol. 2013, 8, 137–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoppstadter, J.; Seif, M.; Dembek, A.; Cavelius, C.; Huwer, H.; Kraegeloh, A.; Kiemer, A.K. M2 polarization enhances silica nanoparticle uptake by macrophages. Front. Pharmacol. 2015, 6, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedeli, C.; Segat, D.; Tavano, R.; Bubacco, L.; De Franceschi, G.; de Laureto, P.P.; Lubian, E.; Selvestrel, F.; Mancin, F.; Papini, E. The functional dissection of the plasma corona of SiO(2)-NPs spots histidine rich glycoprotein as a major player able to hamper nanoparticle capture by macrophages. Nanoscale 2015, 7, 17710–17728. [Google Scholar] [CrossRef] [PubMed]
- Binnemars-Postma, K.A.; Ten Hoopen, H.W.; Storm, G.; Prakash, J. Differential uptake of nanoparticles by human M1 and M2 polarized macrophages: Protein corona as a critical determinant. Nanomedicine 2016, 11, 2889–2902. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, J.E.; Choi, C.H.; Han, H.; Davis, M.E. Polycation-siRNA nanoparticles can disassemble at the kidney glomerular basement membrane. Proc. Natl. Acad. Sci. USA 2012, 109, 3137–3142. [Google Scholar] [CrossRef] [Green Version]
- Longmire, M.; Choyke, P.L.; Kobayashi, H. Clearance properties of nano-sized particles and molecules as imaging agents: Considerations and caveats. Nanomedicine 2008, 3, 703–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabral, H.; Matsumoto, Y.; Mizuno, K.; Chen, Q.; Murakami, M.; Kimura, M.; Terada, Y.; Kano, M.R.; Miyazono, K.; Uesaka, M.; et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat. Nanotechnol. 2011, 6, 815–823. [Google Scholar] [CrossRef]
- Matsumura, Y.; Maeda, H. A New Concept for Macromolecular Therapeutics in Cancer Chemotherapy: Mechanism of Tumoritropic Accumulation of Proteins and the Antitumor Agent Smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Ren, H.; He, Y.; Liang, J.; Cheng, Z.; Zhang, M.; Zhu, Y.; Hong, C.; Qin, J.; Xu, X.; Wang, J. Role of Liposome Size, Surface Charge, and PEGylation on Rheumatoid Arthritis Targeting Therapy. ACS Appl. Mater. Interfaces 2019, 11, 20304–20315. [Google Scholar] [CrossRef]
- Liu, Y.; Tan, J.; Thomas, A.; Ou-Yang, D.; Muzykantov, V.R. The shape of things to come: Importance of design in nanotechnology for drug delivery. Ther. Deliv. 2012, 3, 181–194. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Li, X.; Zhang, Y.; Chen, T.; Xiao, S.; Liang, H. Morphological and Mechanical Determinants of Cellular Uptake of Deformable Nanoparticles. Nanoscale 2018, 10, 11969–11979. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Xiao, S.; Zhu, H.; Wang, L.; Liang, H. Shape-dependent internalization kinetics of nanoparticles by membranes. Soft Matter 2016, 12, 2632–2641. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Sun, L.; Zhang, Y.; Dove, A.P.; O’Reilly, R.K.; Chen, G. Shape Effect of Glyco-Nanoparticles on Macrophage Cellular Uptake and Immune Response. ACS Macro Lett. 2016, 5, 1059–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niikura, K.; Matsunaga, T.; Suzuki, T.; Kobayashi, S.; Yamaguchi, H.; Orba, Y.; Kawaguchi, A.; Hasegawa, H.; Kajino, K.; Ninomiya, T.; et al. Gold Nanoparticles as a Vaccine Platform: Influence of Size and Shape on Immunological Responses in Vitro and in Vivo. ACS Nano 2013, 7, 3926–3938. [Google Scholar] [CrossRef] [PubMed]
- Bartneck, M.; Keul, H.A.; Singh, S.; Czaja, K.; Bornemann, J.; Bockstaller, M.; Moeller, M.; Zwadlo-Klarwasser, G.; Groll, J. Rapid Uptake of Gold Nanorods by Primary Human Blood Phagocytes and Immunomodulatory Effects of Surface Chemistry. ACS Nano 2010, 4, 3073–3086. [Google Scholar] [CrossRef]
- Hinde, E.; Thammasiraphop, K.; Duong, H.T.; Yeow, J.; Karagoz, B.; Boyer, C.; Gooding, J.J.; Gaus, K. Pair correlation microscopy reveals the role of nanoparticle shape in intracellular transport and site of drug release. Nat. Nanotechnol. 2017, 12, 81–89. [Google Scholar] [CrossRef]
- Du, X.J.; Wang, J.L.; Iqbal, S.; Li, H.J.; Cao, Z.T.; Wang, Y.C.; Du, J.Z.; Wang, J. The effect of surface charge on oral absorption of polymeric nanoparticles. Biomater. Sci. 2018, 6, 642–650. [Google Scholar] [CrossRef]
- Hsu, F.F.; Turk, J. Studies on phosphatidylserine by tandem quadrupole and multiple stage quadrupole ion-trap mass spectrometry with electrospray ionization: Structural characterization and the fragmentation processes. J. Am. Soc. Mass. Spectrom. 2005, 16, 1510–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, K.; Li, Y.; Luo, J.; Lee, J.S.; Xiao, W.; Gonik, A.M.; Agarwal, R.G.; Lam, K.S. The effect of surface charge on in vivo biodistribution of PEG-oligocholic acid based micellar nanoparticles. Biomaterials 2011, 32, 3435–3446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brannon-Peppas, L.; Blanchette, J.O. Nanoparticle and targeted systems for cancer therapy. Adv. Drug Deliv. Rev. 2012, 64, 206–212. [Google Scholar] [CrossRef]
- Getts, D.R.; Martin, A.J.; McCarthy, D.P.; Terry, R.L.; Hunter, Z.N.; Yap, W.T.; Getts, M.T.; Pleiss, M.; Luo, X.; King, N.J.; et al. Microparticles bearing encephalitogenic peptides induce T-cell tolerance and ameliorate experimental autoimmune encephalomyelitis. Nat. Biotechnol. 2012, 30, 1217–1224. [Google Scholar] [CrossRef]
- Song, Q.; Wang, X.; Hu, Q.; Huang, M.; Yao, L.; Qi, H.; Qiu, Y.; Jiang, X.; Chen, J.; Chen, H.; et al. Cellular internalization pathway and transcellular transport of pegylated polyester nanoparticles in Caco-2 cells. Int. J. Pharm. 2013, 445, 58–68. [Google Scholar] [CrossRef]
- Peracchia, M.T.; Fattal, E.; Desmaele, D.; Besnard, M.; Noel, J.P.; Gomis, J.M.; Appel, M.; d’Angelo, J.; Couvreur, P. Stealth PEGylated polycyanoacrylate nanoparticles for intravenous administration and splenic targeting. J. Control. Release 1999, 60, 121–128. [Google Scholar] [CrossRef]
- Yu, Q.; Zhao, L.; Guo, C.; Yan, B.; Su, G. Regulating Protein Corona Formation and Dynamic Protein Exchange by Controlling Nanoparticle Hydrophobicity. Front. Bioeng. Biotechnol. 2020, 8, 210. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.W.; Roberts, R.A.; Robbins, G.R.; Perry, J.L.; Kai, M.P.; Chen, K.; Bo, T.; Napier, M.E.; Ting, J.P.; Desimone, J.M.; et al. Nanoparticle clearance is governed by Th1/Th2 immunity and strain background. J. Clin. Investig. 2013, 123, 3061–3073. [Google Scholar] [CrossRef] [Green Version]
- MacParland, S.A.; Tsoi, K.M.; Ouyang, B.; Ma, X.Z.; Manuel, J.; Fawaz, A.; Ostrowski, M.A.; Alman, B.A.; Zilman, A.; Chan, W.C.; et al. Phenotype Determines Nanoparticle Uptake by Human Macrophages from Liver and Blood. ACS Nano 2017, 11, 2428–2443. [Google Scholar] [CrossRef]
- Herd, H.L.; Bartlett, K.T.; Gustafson, J.A.; McGill, L.D.; Ghandehari, H. Macrophage silica nanoparticle response is phenotypically dependent. Biomaterials 2015, 53, 574–582. [Google Scholar] [CrossRef] [Green Version]
- Feito, M.J.; Diez-Orejas, R.; Cicuendez, M.; Casarrubios, L.; Rojo, J.M.; Portoles, M.T. Characterization of M1 and M2 polarization phenotypes in peritoneal macrophages after treatment with graphene oxide nanosheets. Colloids Surf B Biointerfaces 2019, 176, 96–105. [Google Scholar] [CrossRef] [Green Version]
- Pajarinen, J.; Kouri, V.P.; Jamsen, E.; Li, T.F.; Mandelin, J.; Konttinen, Y.T. The response of macrophages to titanium particles is determined by macrophage polarization. Acta Biomater. 2013, 9, 9229–9240. [Google Scholar] [CrossRef] [PubMed]
- Barua, S.; Mitragotri, S. Challenges associated with Penetration of Nanoparticles across Cell and Tissue Barriers: A Review of Current Status and Future Prospects. Nano Today 2014, 9, 223–243. [Google Scholar] [CrossRef] [PubMed]
- Katifelis, H.; Mukha, I.; Bouziotis, P.; Vityuk, N.; Tsoukalas, C.; Lazaris, A.C.; Lyberopoulou, A.; Theodoropoulos, G.E.; Efstathopoulos, E.P.; Gazouli, M. Ag/Au Bimetallic Nanoparticles Inhibit Tumor Growth and Prevent Metastasis in a Mouse Model. Int. J. Nanomed. 2020, 15, 6019–6032. [Google Scholar] [CrossRef] [PubMed]
- Golombek, S.K.; May, J.N.; Theek, B.; Appold, L.; Drude, N.; Kiessling, F.; Lammers, T. Tumor targeting via EPR: Strategies to enhance patient responses. Adv. Drug Deliv. Rev. 2018, 130, 17–38. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Jain, S.; Tiwary, A. Mannan-coated gelatin nanoparticles for sustained and targeted delivery of didanosine: In vitro and in vivo evaluation. Acta Pharm. 2008, 58, 61–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Totten, J.D.; Wongpinyochit, T.; Carrola, J.; Duarte, I.F.; Seib, F.P. PEGylation-Dependent Metabolic Rewiring of Macrophages with Silk Fibroin Nanoparticles. ACS Appl. Mater. Interfaces 2019, 11, 14515–14525. [Google Scholar] [CrossRef] [Green Version]
- Leuschner, F.; Dutta, P.; Gorbatov, R.; Novobrantseva, T.I.; Donahoe, J.S.; Courties, G.; Lee, K.M.; Kim, J.I.; Markmann, J.F.; Marinelli, B.; et al. Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat. Biotechnol. 2011, 29, 1005–1010. [Google Scholar] [CrossRef]
- Shen, S.; Zhang, Y.; Chen, K.G.; Luo, Y.L.; Wang, J. Cationic Polymeric Nanoparticle Delivering CCR2 siRNA to Inflammatory Monocytes for Tumor Microenvironment Modification and Cancer Therapy. Mol. Pharm. 2018, 15, 3642–3653. [Google Scholar] [CrossRef]
- Jose, A.; Labala, S.; Ninave, K.M.; Gade, S.K.; Venuganti, V.V.K. Effective Skin Cancer Treatment by Topical Co-delivery of Curcumin and STAT3 siRNA Using Cationic Liposomes. AAPS PharmSciTech 2018, 19, 166–175. [Google Scholar] [CrossRef]
- Tian, L.; Yi, X.; Dong, Z.; Xu, J.; Liang, C.; Chao, Y.; Wang, Y.; Yang, K.; Liu, Z. Calcium Bisphosphonate Nanoparticles with Chelator-Free Radiolabeling to Deplete Tumor-Associated Macrophages for Enhanced Cancer Radioisotope Therapy. ACS Nano 2018, 12, 11541–11551. [Google Scholar] [CrossRef]
- Zeisberger, S.M.; Odermatt, B.; Marty, C.; Zehnder-Fjällman, A.H.M.; Ballmer-Hofer, K.; Schwendener, R.A. Clodronate-liposome-mediated depletion of tumour-associated macrophages: A new and highly effective antiangiogenic therapy approach. Br. J. Cancer 2006, 95, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Gazzaniga, S.; Bravo, A.I.; Guglielmotti, A.; van Rooijen, N.; Maschi, F.; Vecchi, A.; Mantovani, A.; Mordoh, J.; Wainstok, R. Targeting Tumor-Associated Macrophages and Inhibition of MCP-1 Reduce Angiogenesis and Tumor Growth in a Human Melanoma Xenograft. J. Investig. Dermatol. 2007, 127, 2031–2041. [Google Scholar] [CrossRef] [Green Version]
- Sousa, S.; Auriola, S.; Mönkkönen, J.; Määttä, J. Liposome encapsulated zoledronate favours M1-like behaviour in murine macrophages cultured with soluble factors from breast cancer cells. BMC Cancer 2015, 15, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajan, R.; Sabnani, M.K.; Mavinkurve, V.; Shmeeda, H.; Mansouri, H.; Bonkoungou, S.; Le, A.D.; Wood, L.M.; Gabizon, A.A.; La-Beck, N.M. Liposome-induced immunosuppression and tumor growth is mediated by macrophages and mitigated by liposome-encapsulated alendronate. J. Control. Release Off. J. Control. Release Soc. 2018, 271, 139–148. [Google Scholar] [CrossRef]
- Kwong, B.; Liu, H.; Irvine, D.J. Induction of potent anti-tumor responses while eliminating systemic side effects via liposome-anchored combinatorial immunotherapy. Biomaterials 2011, 32, 5134–5147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, K.; Heishi, T.; Khan, O.F.; Kowalski, P.S.; Incio, J.; Rahbari, N.N.; Chung, E.; Clark, J.W.; Willett, C.G.; Luster, A.D.; et al. Ly6Clo monocytes drive immunosuppression and confer resistance to anti-VEGFR2 cancer therapy. J. Clin. Investig. 2017, 127, 3039–3051. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lin, Y.X.; Qiao, S.L.; An, H.W.; Ma, Y.; Qiao, Z.Y.; Rajapaksha, R.P.; Wang, H. Polymeric nanoparticles promote macrophage reversal from M2 to M1 phenotypes in the tumor microenvironment. Biomaterials 2017, 112, 153–163. [Google Scholar] [CrossRef]
- van Woensel, M.; Mathivet, T.; Wauthoz, N.; Rosière, R.; Garg, A.D.; Agostinis, P.; Mathieu, V.; Kiss, R.; Lefranc, F.; Boon, L.; et al. Sensitization of glioblastoma tumor micro-environment to chemo- and immunotherapy by Galectin-1 intranasal knock-down strategy. Sci. Rep. 2017, 7, 1217. [Google Scholar] [CrossRef]
- Rodell, C.B.; Arlauckas, S.P.; Cuccarese, M.F.; Garris, C.S.; Li, R.; Ahmed, M.S.; Kohler, R.H.; Pittet, M.J.; Weissleder, R. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat. Biomed. Eng. 2018, 2, 578–588. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, Z.; Xue, Y.; Wang, G.; Cheng, Y.; Pan, Y.; Zhao, S.; Hou, Y. Anti-tumor macrophages activated by ferumoxytol combined or surface-functionalized with the TLR3 agonist poly (I: C) promote melanoma regression. Theranostics 2018, 8, 6307–6321. [Google Scholar] [CrossRef] [PubMed]
- VanHandel, M.; Alizadeh, D.; Zhang, L.; Kateb, B.; Bronikowski, M.; Manohara, H.; Badie, B. Selective uptake of multi-walled carbon nanotubes by tumor macrophages in a murine glioma model. J. Neuroimmunol. 2009, 208, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Kwon, D.; Cha, B.G.; Cho, Y.; Min, J.; Park, E.-B.; Kang, S.-J.; Kim, J. Extra-Large Pore Mesoporous Silica Nanoparticles for Directing in Vivo M2 Macrophage Polarization by Delivering IL-4. Nano Lett. 2017, 17, 2747–2756. [Google Scholar] [CrossRef] [PubMed]
- Liaw, K.; Reddy, R.; Sharma, A.; Li, J.; Chang, M.; Sharma, R.; Salazar, S.; Kannan, S.; Kannan, R.M. Targeted systemic dendrimer delivery of CSF-1R inhibitor to tumor-associated macrophages improves outcomes in orthotopic glioblastoma. Bioeng. Transl. Med. 2021, 6, e10205. [Google Scholar] [CrossRef]
- Ramesh, A.; Brouillard, A.; Kumar, S.; Nandi, D.; Kulkarni, A. Dual inhibition of CSF1R and MAPK pathways using supramolecular nanoparticles enhances macrophage immunotherapy. Biomaterials 2020, 227, 119559. [Google Scholar] [CrossRef]
- Fuchs, A.K.; Syrovets, T.; Haas, K.A.; Loos, C.; Musyanovych, A.; Mailander, V.; Landfester, K.; Simmet, T. Carboxyl- and amino-functionalized polystyrene nanoparticles differentially affect the polarization profile of M1 and M2 macrophage subsets. Biomaterials 2016, 85, 78–87. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, Y.; Lu, S.; Yang, L.; Yu, S.; Yang, H. CaCO3-Encapsulated Au Nanoparticles Modulate Macrophages toward M1-like Phenotype. ACS Appl. Bio Mater. 2021, 4, 3214–3223. [Google Scholar] [CrossRef]
- Etzerodt, A.; Tsalkitzi, K.; Maniecki, M.; Damsky, W.; Delfini, M.; Baudoin, E.; Moulin, M.; Bosenberg, M.; Graversen, J.H.; Auphan-Anezin, N.; et al. Specific targeting of CD163(+) TAMs mobilizes inflammatory monocytes and promotes T cell-mediated tumor regression. J. Exp. Med. 2019, 216, 2394–2411. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.S.; Lau, C.M.; Barham, W.J.; Onishko, H.M.; Nelson, C.E.; Li, H.; Smith, C.A.; Yull, F.E.; Duvall, C.L.; Giorgio, T.D. Macrophage-Specific RNA Interference Targeting via “Click”, Mannosylated Polymeric Micelles. Mol. Pharm. 2013, 10, 975–987. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.; Liu, T.; Guo, Z.; Zhuang, R.; Zhang, X.; Chen, X. Reprogramming Tumor-Associated Macrophages by Nanoparticle-Based Reactive Oxygen Species Photogeneration. Nano Lett. 2018, 18, 7330–7342. [Google Scholar] [CrossRef]
- Zhu, S.; Niu, M.; O’Mary, H.; Cui, Z. Targeting of tumor-associated macrophages made possible by PEG-sheddable, mannose-modified nanoparticles. Mol. Pharm. 2013, 10, 3525–3530. [Google Scholar] [CrossRef] [Green Version]
- Leber, N.; Kaps, L.; Yang, A.; Aslam, M.; Giardino, M.; Klefenz, A.; Choteschovsky, N.; Rosigkeit, S.; Mostafa, A.; Nuhn, L.; et al. Alpha-Mannosyl-Functionalized Cationic Nanohydrogel Particles for Targeted Gene Knockdown in Immunosuppressive Macrophages. Macromol. Biosci. 2019, 19, e1900162. [Google Scholar] [CrossRef]
- Niu, M.; Naguib, Y.W.; Aldayel, A.M.; Shi, Y.C.; Hursting, S.D.; Hersh, M.A.; Cui, Z. Biodistribution and in vivo activities of tumor-associated macrophage-targeting nanoparticles incorporated with doxorubicin. Mol. Pharm. 2014, 11, 4425–4436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatami, E.; Mu, Y.; Shields, D.N.; Chauhan, S.C.; Kumar, S.; Cory, T.J.; Yallapu, M. Mannose-decorated hybrid nanoparticles for enhanced macrophage targeting. Biochem. Biophys. Rep. 2019, 17, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.F.; Chao, T.T.; Su, Y.F.; Hsu, C.C.; Chien, C.Y.; Chiu, K.C.; Shiah, S.G.; Lee, C.H.; Liu, S.Y.; Shieh, Y.S. Metformin-treated cancer cells modulate macrophage polarization through AMPK-NF-κB signaling. Oncotarget 2017, 8, 20706–20718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Z.; Zhang, X.; Yong, T.; Bie, N.; Zhan, G.; Li, X.; Liang, Q.; Li, J.; Yu, J.; Huang, G.; et al. Boosting anti-PD-1 therapy with metformin-loaded macrophage-derived microparticles. Nat. Commun. 2021, 12, 440. [Google Scholar] [CrossRef]
- Wang, Y.; Guo, G.; Feng, Y.; Long, H.; Ma, D.-L.; Leung, C.-H.; Dong, L.; Wang, C. A tumour microenvironment-responsive polymeric complex for targeted depletion of tumour-associated macrophages (TAMs). J. Mater. Chem. B 2017, 5, 7307–7318. [Google Scholar] [CrossRef] [PubMed]
- Maalej, M.; Forgione, R.E.; Marchetti, R.; Bulteau, F.; Thépaut, M.; Lanzetta, R.; Laguri, C.; Simorre, J.P.; Fieschi, F.; Molinaro, A.; et al. Human Macrophage Galactose-Type Lectin (MGL) Recognizes the Outer Core of Escherichia coli Lipooligosaccharide. ChemBioChem 2019, 20, 1778–1782. [Google Scholar] [PubMed]
- Han, S.; Wang, W.; Wang, S.; Wang, S.; Ju, R.; Pan, Z.; Yang, T.; Zhang, G.; Wang, H.; Wang, L. Multifunctional biomimetic nanoparticles loading baicalin for polarizing tumor-associated macrophages. Nanoscale 2019, 11, 20206–20220. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Xu, Z.; Ding, T.; Kuang, D.M.; Zheng, L. MicroRNA-155 regulates inflammatory cytokine production in tumor-associated macrophages via targeting C/EBPbeta. Cell Mol. Immunol. 2009, 6, 343–352. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Yin, Y.; Li, N.; Zhu, D.; Zhang, J.; Zhang, C.Y.; Zen, K. Re-polarization of tumor-associated macrophages to pro-inflammatory M1 macrophages by microRNA-155. J. Mol. Cell Biol. 2012, 4, 341–343. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yi, H.; He, H.; Pan, H.; Cai, L.; Ma, Y. Tumor associated macrophage-targeted microRNA delivery with dual-responsive polypeptide nanovectors for anti-cancer therapy. Biomaterials 2017, 134, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Zhang, Z.; Jiang, Y.; Zhang, D.; Chen, J.; Dong, L.; Zhang, J. Targeted delivery of oligonucleotides into tumor-associated macrophages for cancer immunotherapy. J. Control Release 2012, 158, 286–292. [Google Scholar] [CrossRef]
- Su, L.; Zhang, W.; Wu, X.; Zhang, Y.; Chen, X.; Liu, G.; Chen, G.; Jiang, M. Glycocalyx-Mimicking Nanoparticles for Stimulation and Polarization of Macrophages via Specific Interactions. Small 2015, 11, 4191–4200. [Google Scholar] [CrossRef]
- Cieslewicz, M.; Tang, J.; Yu, J.L.; Cao, H.; Zavaljevski, M.; Motoyama, K.; Lieber, A.; Raines, E.W.; Pun, S.H. Targeted delivery of proapoptotic peptides to tumor-associated macrophages improves survival. Proc. Natl. Acad. Sci. USA 2013, 110, 15919–15924. [Google Scholar] [CrossRef] [Green Version]
- Pang, L.; Pei, Y.; Uzunalli, G.; Hyun, H.; Lyle, L.T.; Yeo, Y. Surface Modification of Polymeric Nanoparticles with M2pep Peptide for Drug Delivery to Tumor-Associated Macrophages. Pharm. Res. 2019, 36, 65. [Google Scholar] [CrossRef]
- Zhou, S.; Zhang, T.; Peng, B.; Luo, X.; Liu, X.; Hu, L.; Liu, Y.; Di, D.; Song, Y.; Deng, Y. Targeted delivery of epirubicin to tumor-associated macrophages by sialic acid-cholesterol conjugate modified liposomes with improved antitumor activity. Int. J. Pharm. 2017, 523, 203–216. [Google Scholar] [CrossRef]
- Zhang, X.; Tian, W.; Cai, X.; Wang, X.; Dang, W.; Tang, H.; Cao, H.; Wang, L.; Chen, T. Hydrazinocurcumin Encapsuled nanoparticles “re-educate” tumor-associated macrophages and exhibit anti-tumor effects on breast cancer following STAT3 suppression. PLoS ONE 2013, 8, e65896. [Google Scholar] [CrossRef]
- Chen, C.; Song, M.; Du, Y.; Yu, Y.; Li, C.; Han, Y.; Yan, F.; Shi, Z.; Feng, S. Tumor-Associated-Macrophage-Membrane-Coated Nanoparticles for Improved Photodynamic Immunotherapy. Nano Lett. 2021, 21, 5522–5531. [Google Scholar] [CrossRef]
- Brase, J.C.; Wuttig, D.; Kuner, R.; Sültmann, H. Serum microRNAs as non-invasive biomarkers for cancer. Mol. Cancer 2010, 9, 306. [Google Scholar] [CrossRef] [Green Version]
- Trivedi, M.; Talekar, M.; Shah, P.; Ouyang, Q.; Amiji, M. Modification of tumor cell exosome content by transfection with wt-p53 and microRNA-125b expressing plasmid DNA and its effect on macrophage polarization. Oncogenesis 2016, 5, e250. [Google Scholar] [CrossRef] [Green Version]
- Huis In ‘t Veld, R.V.; Ritsma, L.; Kleinovink, J.W.; Que, I.; Ossendorp, F.; Cruz, L.J. Photodynamic cancer therapy enhances accumulation of nanoparticles in tumor-associated myeloid cells. J. Control Release 2020, 320, 19–31. [Google Scholar] [CrossRef]
- Ortega, R.A.; Barham, W.; Sharman, K.; Tikhomirov, O.; Giorgio, T.D.; Yull, F.E. Manipulating the NF-kappaB pathway in macrophages using mannosylated, siRNA-delivering nanoparticles can induce immunostimulatory and tumor cytotoxic functions. Int. J. Nanomed. 2016, 11, 2163–2177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, M.; Lee, Y.-W.; Hardie, J.; Mout, R.; Yeşilbag Tonga, G.; Farkas, M.E.; Rotello, V.M. CRISPRed Macrophages for Cell-Based Cancer Immunotherapy. Bioconjug. Chem. 2018, 29, 445–450. [Google Scholar] [CrossRef]
- Lee, Y.-W.; Mout, R.; Luther, D.C.; Liu, Y.; Castellanos-García, L.; Burnside, A.S.; Ray, M.; Tonga, G.Y.; Hardie, J.; Nagaraj, H.; et al. In Vivo Editing of Macrophages through Systemic Delivery of CRISPR-Cas9-Ribonucleoprotein-Nanoparticle Nanoassemblies. Adv. Ther. 2019, 2, 1900041. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, J.; Zhang, J.; Marbach, S.; Xu, W.; Zhu, L. Targeting Tumor-Associated Macrophages by MMP2-Sensitive Apoptotic Body-Mimicking Nanoparticles. ACS Appl. Mater. Interfaces 2020, 12, 52402–52414. [Google Scholar] [CrossRef]
- Alupei, M.C.; Licarete, E.; Patras, L.; Banciu, M. Liposomal simvastatin inhibits tumor growth via targeting tumor-associated macrophages-mediated oxidative stress. Cancer Lett. 2015, 356, 946–952. [Google Scholar] [CrossRef]
- Hou, T.; Wang, T.; Mu, W.; Yang, R.; Liang, S.; Zhang, Z.; Fu, S.; Gao, T.; Liu, Y.; Zhang, N. Nanoparticle-Loaded Polarized-Macrophages for Enhanced Tumor Targeting and Cell-Chemotherapy. Nanomicro Lett. 2020, 13, 6. [Google Scholar] [PubMed]
- Kim, M.S.; Lee, J.S.; Kim, J.E.; Kim, J.-W.; Bok, S.; Keum, K.C.; Koh, W.-G.; Koom, W.S. Enhancement of antitumor effect of radiotherapy via combination with Au@SiO2 nanoparticles targeted to tumor-associated macrophages. J. Ind. Eng. Chem. 2020, 84, 349–357. [Google Scholar] [CrossRef]
- He, X.Y.; Liu, B.Y.; Xu, C.; Zhuo, R.X.; Cheng, S.X. A multi-functional macrophage and tumor targeting gene delivery system for the regulation of macrophage polarity and reversal of cancer immunoresistance. Nanoscale 2018, 10, 15578–15587. [Google Scholar] [CrossRef]
- Song, M.; Liu, T.; Shi, C.; Zhang, X.; Chen, X. Bioconjugated Manganese Dioxide Nanoparticles Enhance Chemotherapy Response by Priming Tumor-Associated Macrophages toward M1-like Phenotype and Attenuating Tumor Hypoxia. ACS Nano 2016, 10, 633–647. [Google Scholar] [CrossRef] [Green Version]
- Esser, A.K.; Ross, M.H.; Fontana, F.; Su, X.; Gabay, A.; Fox, G.C.; Xu, Y.; Xiang, J.; Schmieder, A.H.; Yang, X.; et al. Nanotherapy delivery of c-myc inhibitor targets Protumor Macrophages and preserves Antitumor Macrophages in Breast Cancer. Theranostics 2020, 10, 7510–7526. [Google Scholar] [CrossRef]
- Chang, C.C.; Dinh, T.K.; Lee, Y.A.; Wang, F.N.; Sung, Y.C.; Yu, P.L.; Chiu, S.C.; Shih, Y.C.; Wu, C.Y.; Huang, Y.D.; et al. Nanoparticle Delivery of MnO2 and Antiangiogenic Therapy to Overcome Hypoxia-Driven Tumor Escape and Suppress Hepatocellular Carcinoma. ACS Appl. Mater. Interfaces 2020, 12, 44407–44419. [Google Scholar] [CrossRef] [PubMed]
- Korangath, P.; Barnett, J.D.; Sharma, A.; Henderson, E.T.; Stewart, J.; Yu, S.-H.; Kandala, S.K.; Yang, C.-T.; Caserto, J.S.; Hedayati, M.; et al. Nanoparticle interactions with immune cells dominate tumor retention and induce T cell–mediated tumor suppression in models of breast cancer. Sci. Adv. 2020, 6, eaay1601. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Lee, J.S.; Kim, D.; Zhu, L. Exploration of Zinc Oxide Nanoparticles as a Multitarget and Multifunctional Anticancer Nanomedicine. ACS Appl. Mater. Interfaces 2017, 9, 39971–39984. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Tian, G.; Yin, W.; Wang, L.; Zheng, X.; Yan, L.; Li, J.; Su, H.; Chen, C.; Gu, Z.; et al. Controllable Generation of Nitric Oxide by Near-Infrared-Sensitized Upconversion Nanoparticles for Tumor Therapy. Adv. Funct. Mater. 2015, 25, 3049–3056. [Google Scholar] [CrossRef]
- Leonard, F.; Curtis, L.T.; Ware, M.J.; Nosrat, T.; Liu, X.; Yokoi, K.; Frieboes, H.B.; Godin, B. Macrophage Polarization Contributes to the Anti-Tumoral Efficacy of Mesoporous Nanovectors Loaded with Albumin-Bound Paclitaxel. Front. Immunol. 2017, 8, 693. [Google Scholar] [CrossRef] [Green Version]
- Mocan, T.; Matea, C.; Tabaran, F.; Iancu, C.; Orasan, R.; Mocan, L. In Vitro Administration of Gold Nanoparticles Functionalized with MUC-1 Protein Fragment Generates Anticancer Vaccine Response via Macrophage Activation and Polarization Mechanism. J. Cancer 2015, 6, 583–592. [Google Scholar] [CrossRef]
- Dong, J.; Ma, Q. Macrophage polarization and activation at the interface of multi-walled carbon nanotube-induced pulmonary inflammation and fibrosis. Nanotoxicology 2018, 12, 153–168. [Google Scholar] [CrossRef]
- Noonan, D.M.; Principi, E.; Girardello, R.; Bruno, A.; Manni, I.; Gini, E.; Pagani, A.; Grimaldi, A.; Ivaldi, F.; Congiu, T.; et al. Systemic distribution of single-walled carbon nanotubes in a novel model: Alteration of biochemical parameters, metabolic functions, liver accumulation, and inflammation in vivo. Int. J. Nanomed. 2016, 11, 4299–4316. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y.; Ju, E.; Ren, J.; Qu, X. Immunostimulatory oligonucleotides-loaded cationic graphene oxide with photothermally enhanced immunogenicity for photothermal/immune cancer therapy. Biomaterials 2014, 35, 9963–9971. [Google Scholar] [CrossRef]
- Tang, J.; Chen, Z.; Sun, B.; Dong, J.; Liu, J.; Zhou, H.; Wang, L.; Bai, R.; Miao, Q.; Zhao, Y.; et al. Polyhydroxylated fullerenols regulate macrophage for cancer adoptive immunotherapy and greatly inhibit the tumor metastasis. Nanomedicine 2016, 12, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.-S.; Wen, Z.-J.; Wang, J.-H.; Zhu, F.-F.; Guo, F.; Zhou, J.-L.; Xu, J.-J.; Zhong, H.-J. Legumain protease-sheddable PEGylated, tuftsin-modified nanoparticles for selective targeting to tumor-associated macrophages. J. Drug Target. 2021, 1–25. [Google Scholar]
- Duan, L.; Ouyang, K.; Xu, X.; Xu, L.; Wen, C.; Zhou, X.; Qin, Z.; Xu, Z.; Sun, W.; Liang, Y. Nanoparticle Delivery of CRISPR/Cas9 for Genome Editing. Front. Genet. 2021, 12, 673286. [Google Scholar] [CrossRef]
- Li, S.D.; Chen, Y.C.; Hackett, M.J.; Huang, L. Tumor-targeted delivery of siRNA by self-assembled nanoparticles. Mol. Ther. 2008, 16, 163–169. [Google Scholar] [CrossRef]
- Cardoso, M.M.; Peca, N.I.; Roque, C.A. A Antibody-Conjugated Nanoparticles for Therapeutic Applications. Curr. Med. Chem. 2012, 19, 3103–3127. [Google Scholar] [CrossRef]
- Ngoune, R.; Peters, A.; von Elverfeldt, D.; Winkler, K.; Putz, G. Accumulating nanoparticles by EPR: A route of no return. J. Control Release 2016, 238, 58–70. [Google Scholar] [CrossRef] [Green Version]
- Huis In ‘t Veld, R.V.; Da Silva, C.G.; Jager, M.J.; Cruz, L.J.; Ossendorp, F. Combining Photodynamic Therapy with Immunostimulatory Nanoparticles Elicits Effective Anti-Tumor Immune Responses in Preclinical Murine Models. Pharmaceutics 2021, 13, 1470. [Google Scholar] [CrossRef]
- Harrington, K.J.; Mohammadtaghi, S.; Uster, P.S.; Glass, D.; Peters, A.M.; Vile, R.G.; Stewart, J.S.W. Effective Targeting of Solid Tumors in Patients With Locally Advanced Cancers by Radiolabeled Pegylated Liposomes. Clin. Cancer Res. 2021, 7, 243–254. [Google Scholar]
- Venneri, M.A.; De Palma, M.; Ponzoni, M.; Pucci, F.; Scielzo, C.; Zonari, E.; Mazzieri, R.; Doglioni, C.; Naldini, L. Identification of proangiogenic TIE2-expressing monocytes (TEMs) in human peripheral blood and cancer. Blood 2007, 109, 5276–5285. [Google Scholar] [CrossRef] [Green Version]
- Rohan, T.E.; Xue, X.; Lin, H.M.; D’Alfonso, T.M.; Ginter, P.S.; Oktay, M.H.; Robinson, B.D.; Ginsberg, M.; Gertler, F.B.; Glass, A.G.; et al. Tumor microenvironment of metastasis and risk of distant metastasis of breast cancer. J. Natl. Cancer Inst. 2014, 106, 106. [Google Scholar] [CrossRef] [Green Version]
- Másson, M.; Loftsson, T.; Másson, G.; Stefánsson, E. Cyclodextrins as permeation enhancers: Some theoretical evaluations and in vitro testing. J. Control. Release 1999, 59, 107–118. [Google Scholar] [CrossRef]
- Cruz, L.J.; van Dijk, T.; Vepris, O.; Li, T.; Schomann, T.; Baldazzi, F.; Kurita, R.; Nakamura, Y.; Grosveld, F.; Philipsen, S.; et al. PLGA-Nanoparticles for Intracellular Delivery of the CRISPR-Complex to Elevate Fetal Globin Expression in Erythroid Cells. Biomaterials 2021, 268, 120580. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Bharate, G.Y.; Daruwalla, J. Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur. J. Pharm. Biopharm. 2009, 71, 409–419. [Google Scholar] [CrossRef]
- Cruz, L.J.; Rueda, F.; Tacken, P.; Albericio, F.; Torensma, R.; Figdor, C.G. Enhancing immunogenicity and cross-reactivity of HIV-1 antigens by in vivo targeting to dendritic cells. Nanomedicine 2012, 7, 1591–1610. [Google Scholar] [CrossRef] [PubMed]
- Tacken, P.J.; Zeelenberg, I.S.; Cruz, L.J.; van Hout-Kuijer, M.A.; van de Glind, G.; Fokkink, R.G.; Lambeck, A.J.; Figdor, C.G. Targeted delivery of TLR ligands to human and mouse dendritic cells strongly enhances adjuvanticity. Blood 2011, 118, 6836–6844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, C.E.; Harney, A.S.; Pollard, J.W. The Multifaceted Role of Perivascular Macrophages in Tumors. Cancer Cell 2016, 30, 18–25. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Zheng, L.; Chen, W.; Weng, W.; Song, J.; Ji, J. Delivery strategies of cancer immunotherapy: Recent advances and future perspectives. J. Hematol. Oncol. 2019, 12, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shmeeda, H.; Amitay, Y.; Tzemach, D.; Gorin, J.; Gabizon, A. Liposome encapsulation of zoledronic acid results in major changes in tissue distribution and increase in toxicity. J. Control Release 2013, 167, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Condeelis, J.; Pollard, J.W. Macrophages: Obligate partners for tumor cell migration, invasion, and metastasis. Cell 2006, 124, 263–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Tresckow, B.; Morschhauser, F.; Ribrag, V.; Topp, M.S.; Chien, C.; Seetharam, S.; Aquino, R.; Kotoulek, S.; de Boer, C.J.; Engert, A. An Open-Label, Multicenter, Phase I/II Study of JNJ-40346527, a CSF-1R Inhibitor, in Patients with Relapsed or Refractory Hodgkin Lymphoma. Clin. Cancer Res. 2015, 21, 1843–1850. [Google Scholar] [CrossRef] [Green Version]
- Butowski, N.; Colman, H.; De Groot, J.F.; Omuro, A.M.; Nayak, L.; Wen, P.Y.; Cloughesy, T.F.; Marimuthu, A.; Haidar, S.; Perry, A.; et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: An Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro. Oncol. 2016, 18, 557–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva, C.G.; Camps, M.G.M.; Li, T.; Chan, A.B.; Ossendorp, F.; Cruz, L.J. Co-delivery of immunomodulators in biodegradable nanoparticles improves therapeutic efficacy of cancer vaccines. Biomaterials 2019, 220, 119417. [Google Scholar] [CrossRef]
- Basu, A.; Kodumudi, K. Multimodal approaches to improve immunotherapy in breast cancer. Immunotherapy 2020, 12, 161–165. [Google Scholar] [CrossRef]
- Malesci, A.; Bianchi, P.; Celesti, G.; Basso, G.; Marchesi, F.; Grizzi, F.; Di Caro, G.; Cavalleri, T.; Rimassa, L.; Palmqvist, R.; et al. Tumor-associated macrophages and response to 5-fluorouracil adjuvant therapy in stage III colorectal cancer. OncoImmunology 2017, 6, e1342918. [Google Scholar] [CrossRef]
- Fang, Z.; Shen, Y.; Gao, D. Stimulus-responsive nanocarriers for targeted drug delivery. New J. Chem. 2021, 45, 4534–4544. [Google Scholar] [CrossRef]
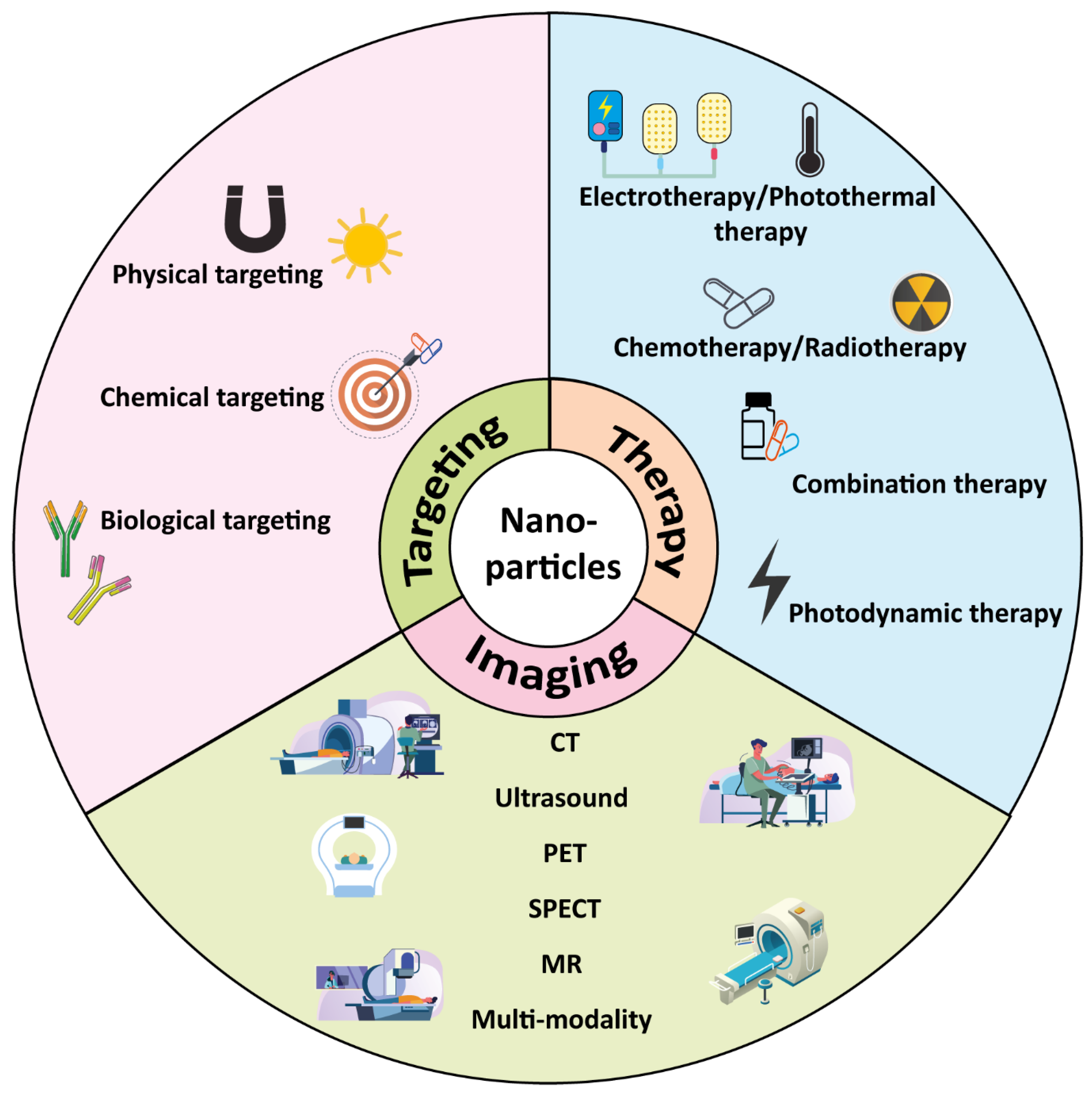
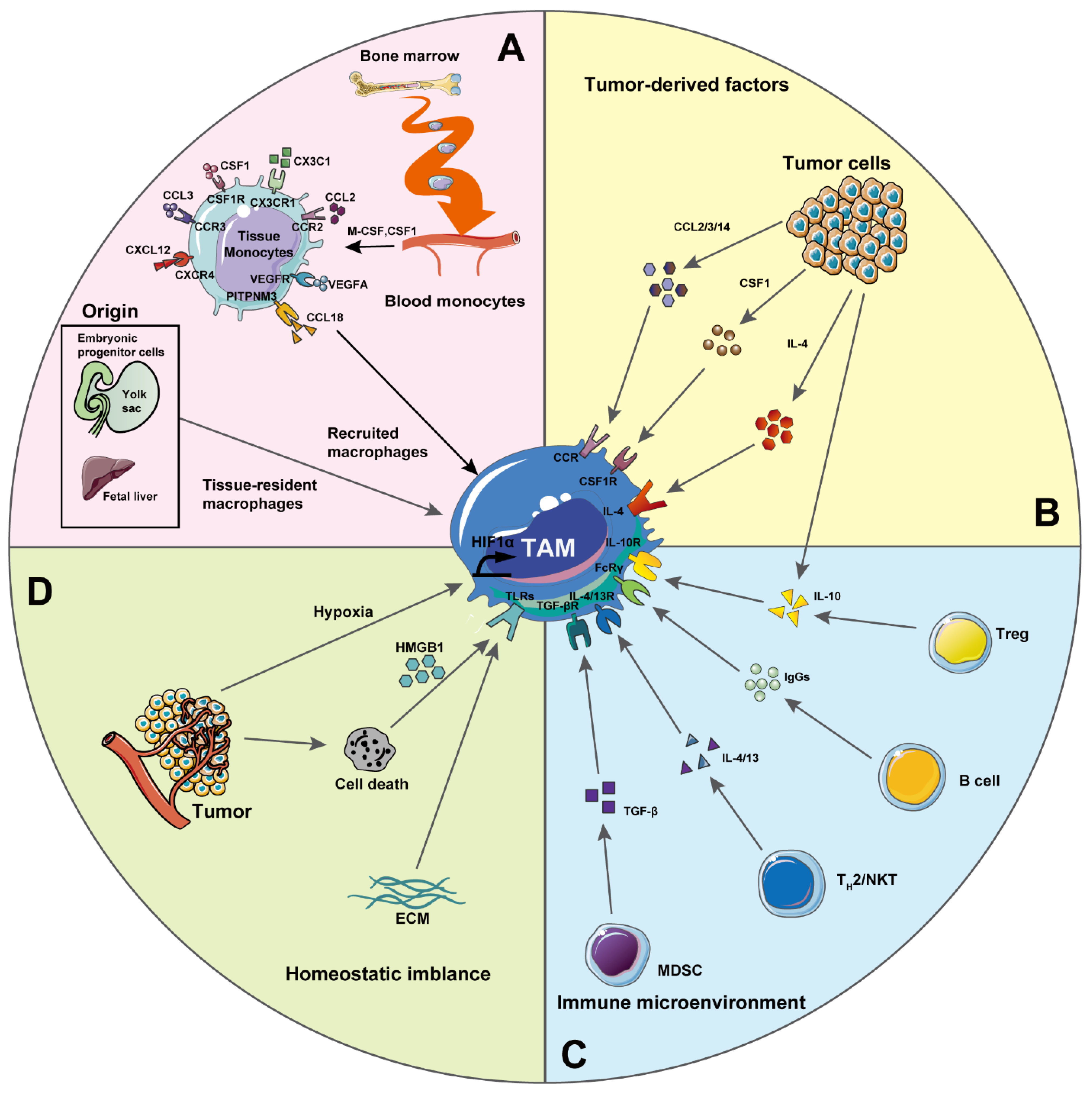
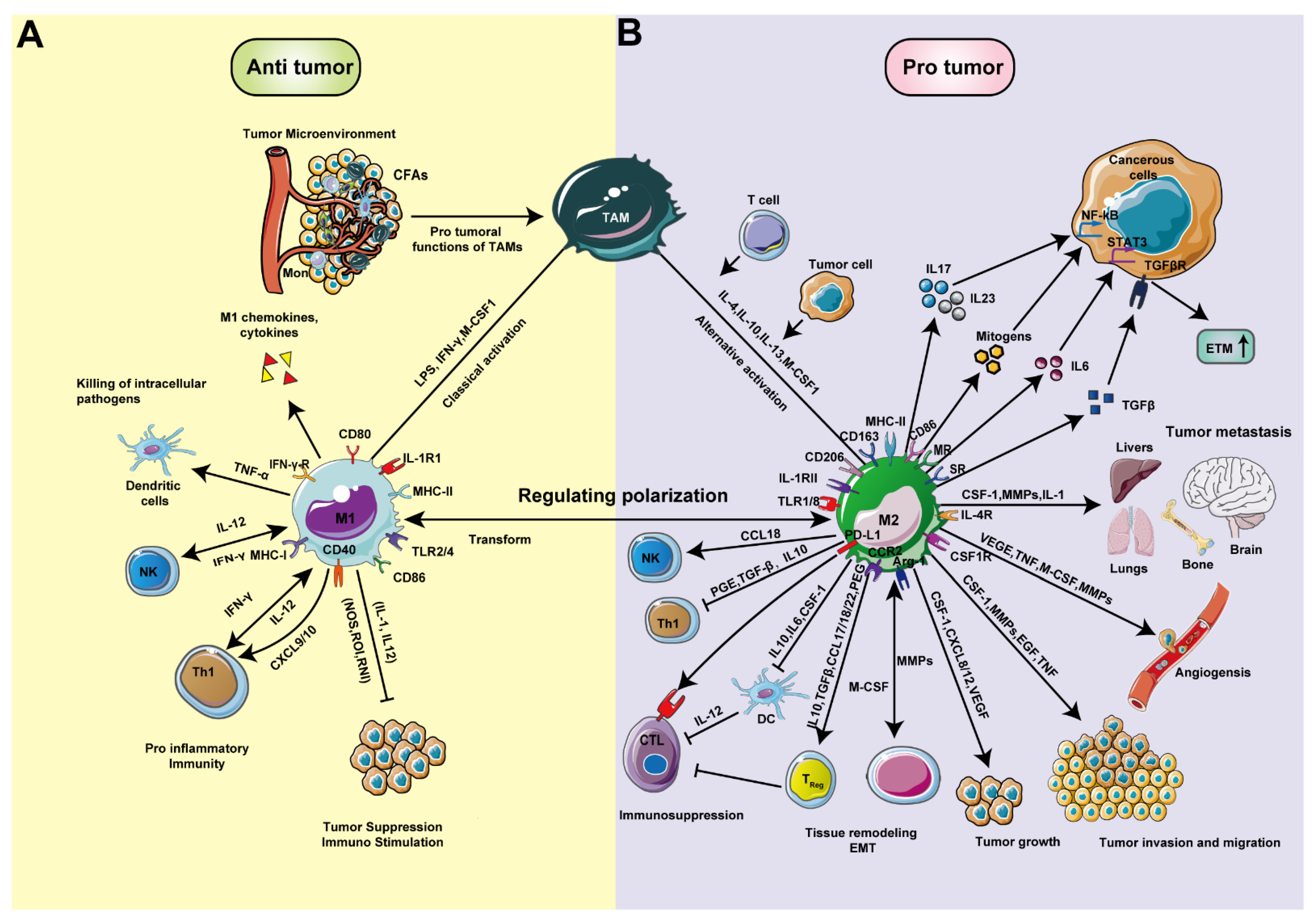
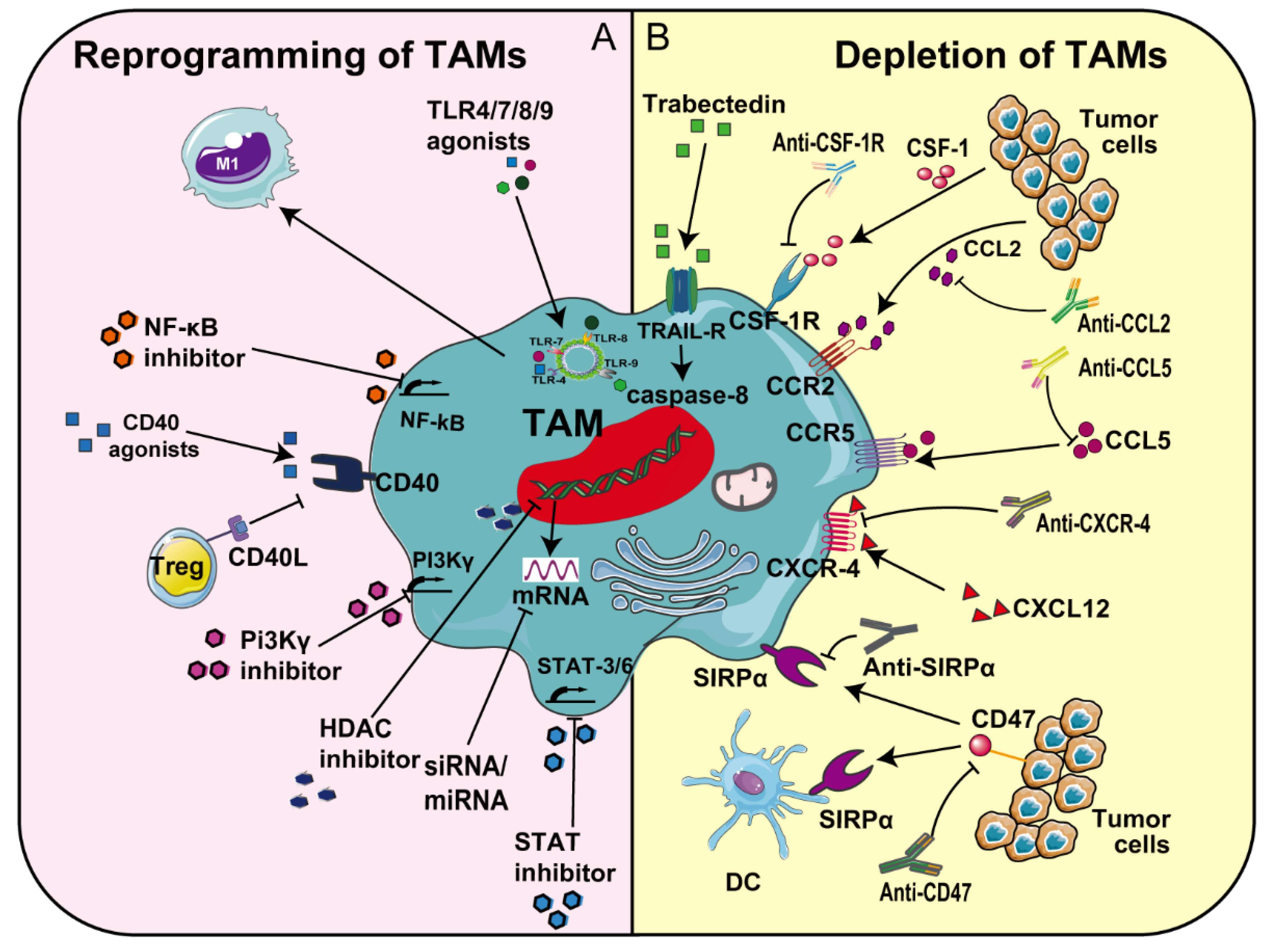
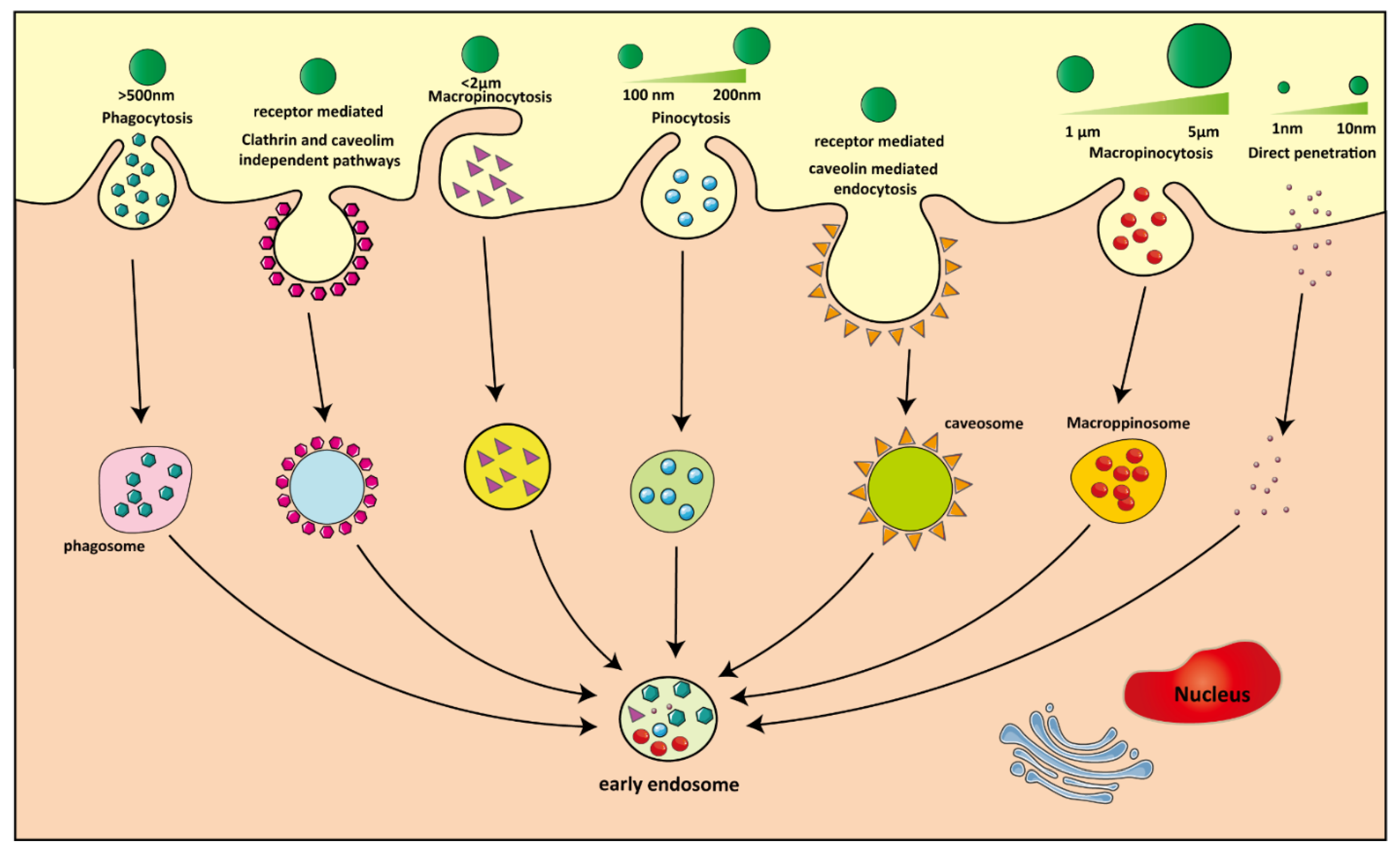
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Y.; de Araújo Júnior, R.F.; Cruz, L.J.; Eich, C. Functionalized Nanoparticles Targeting Tumor-Associated Macrophages as Cancer Therapy. Pharmaceutics 2021, 13, 1670. https://doi.org/10.3390/pharmaceutics13101670
He Y, de Araújo Júnior RF, Cruz LJ, Eich C. Functionalized Nanoparticles Targeting Tumor-Associated Macrophages as Cancer Therapy. Pharmaceutics. 2021; 13(10):1670. https://doi.org/10.3390/pharmaceutics13101670
Chicago/Turabian StyleHe, Yuanyuan, Raimundo Fernandes de Araújo Júnior, Luis J. Cruz, and Christina Eich. 2021. "Functionalized Nanoparticles Targeting Tumor-Associated Macrophages as Cancer Therapy" Pharmaceutics 13, no. 10: 1670. https://doi.org/10.3390/pharmaceutics13101670
APA StyleHe, Y., de Araújo Júnior, R. F., Cruz, L. J., & Eich, C. (2021). Functionalized Nanoparticles Targeting Tumor-Associated Macrophages as Cancer Therapy. Pharmaceutics, 13(10), 1670. https://doi.org/10.3390/pharmaceutics13101670