
Highly Specific Blood-Brain Barrier Transmigrating Single-Domain Antibodies Selected by an In Vivo Phage Display Screening


 ,
,  , , , ,
, , , ,  ,
,  ,
,  ,
,  and
and
Abstract
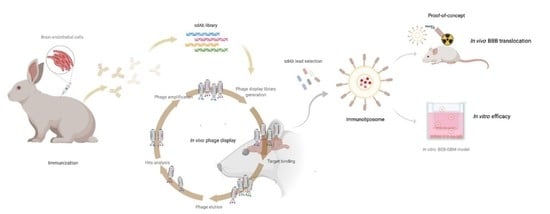
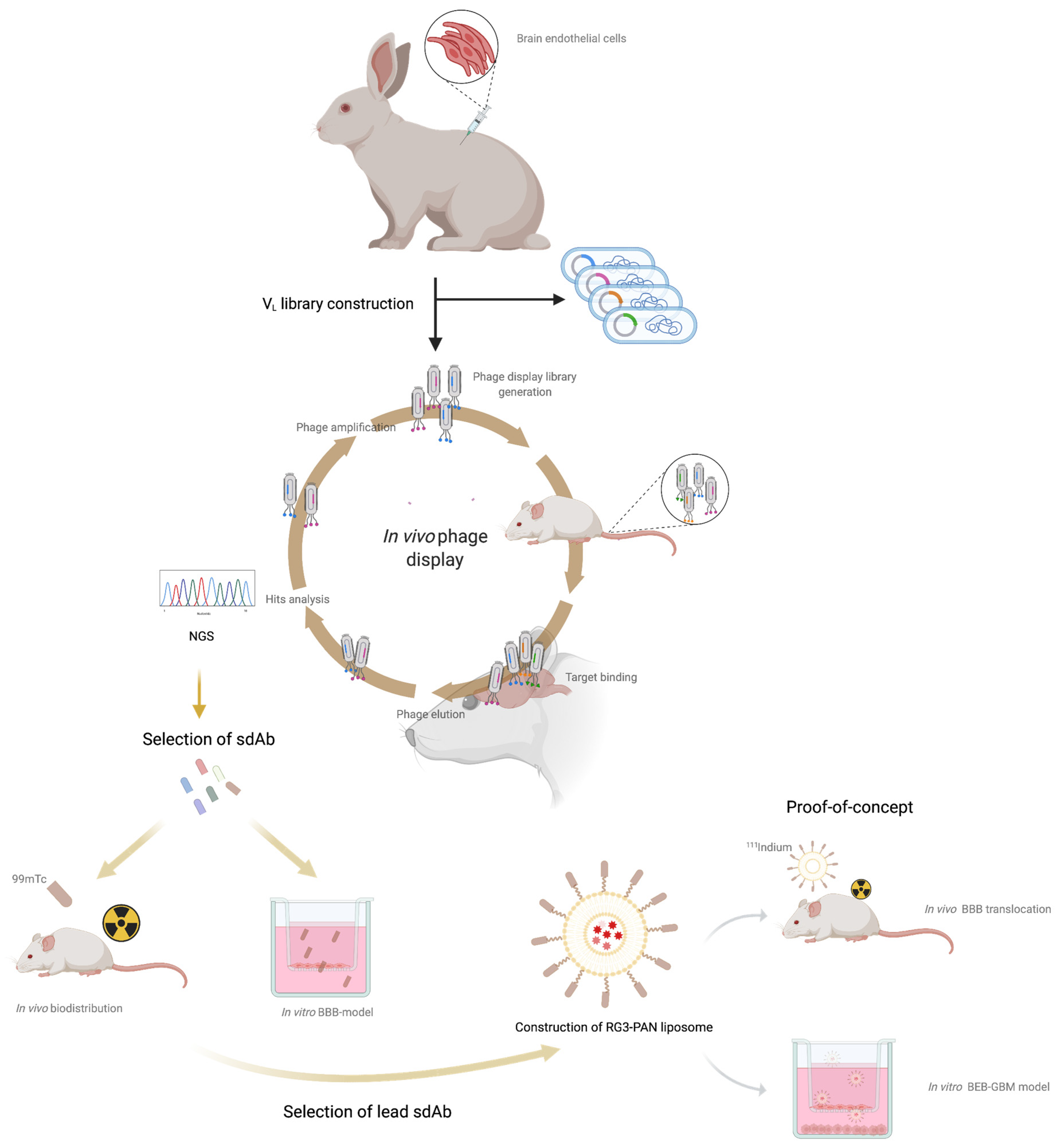
:
1. Introduction
2. Materials and Methods
2.1. Rabbit Immunization
2.2. Characterization of Rabbit Immune Response
2.3. Construction of Single-Domain Antibody Library
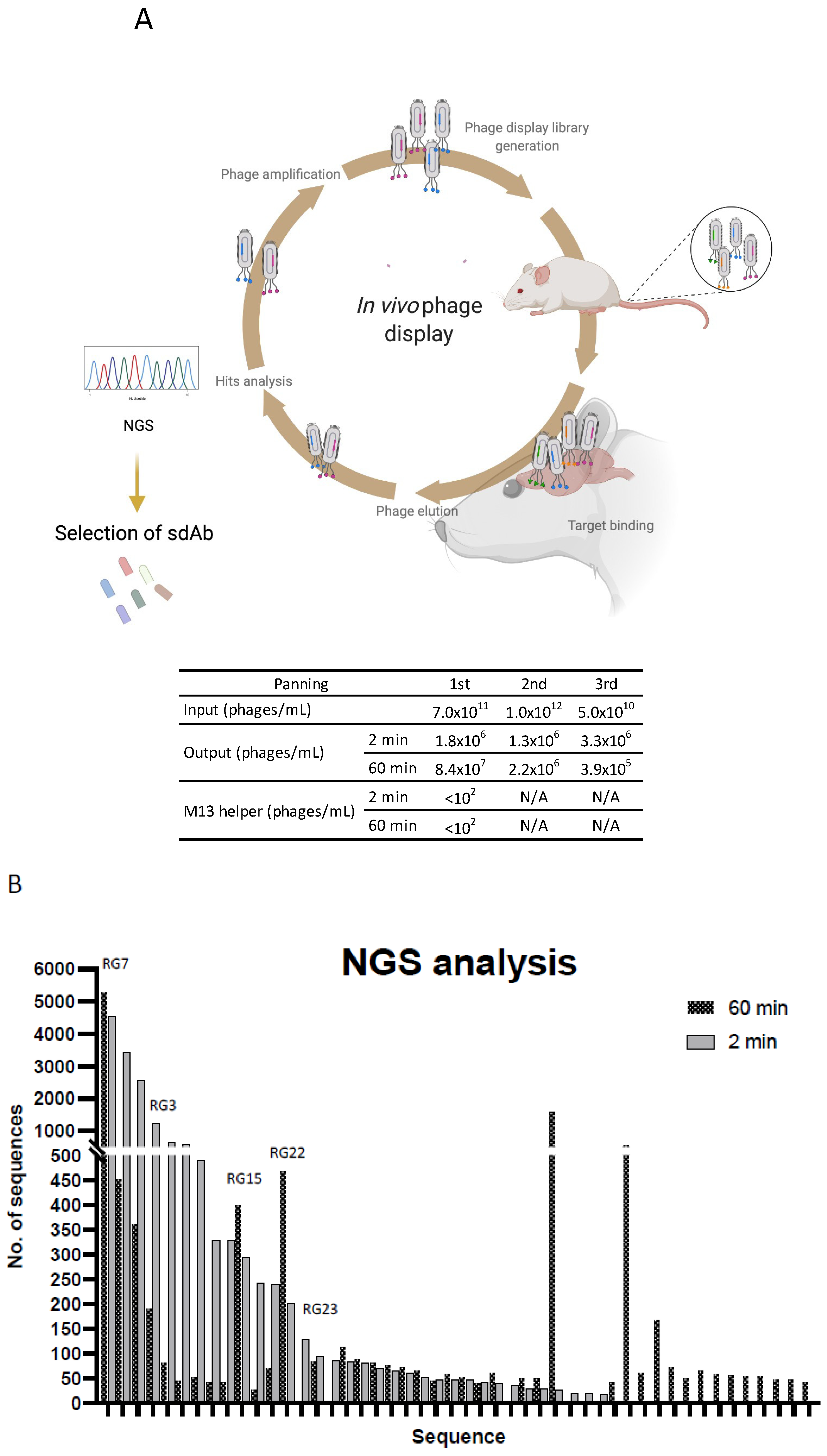
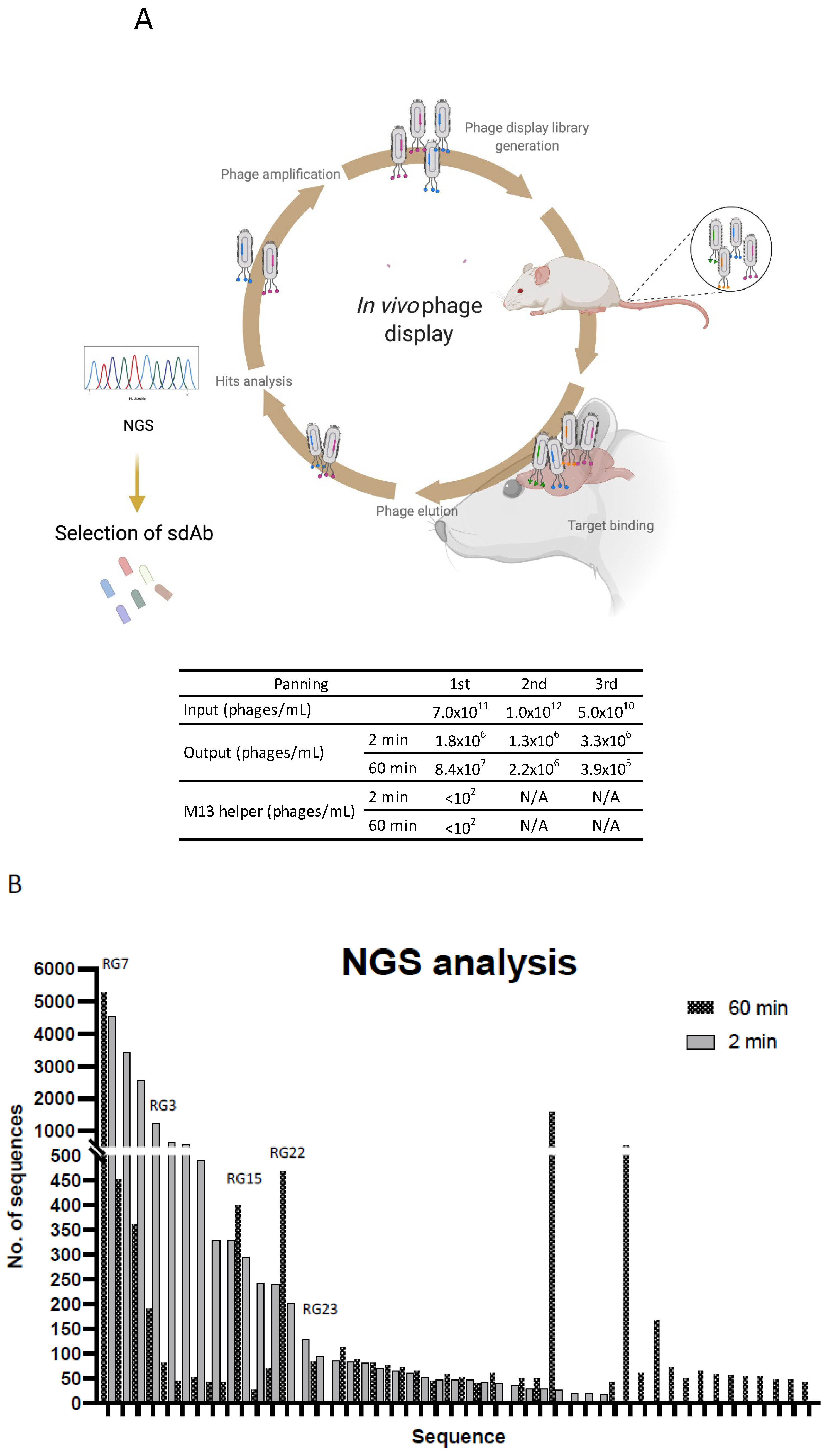
2.4. In Vivo Phage Display
2.5. Analysis of In Vivo Phage Display Enrichment and Next Generation Sequencing
2.6. Expression and Purification of VL sdAbs
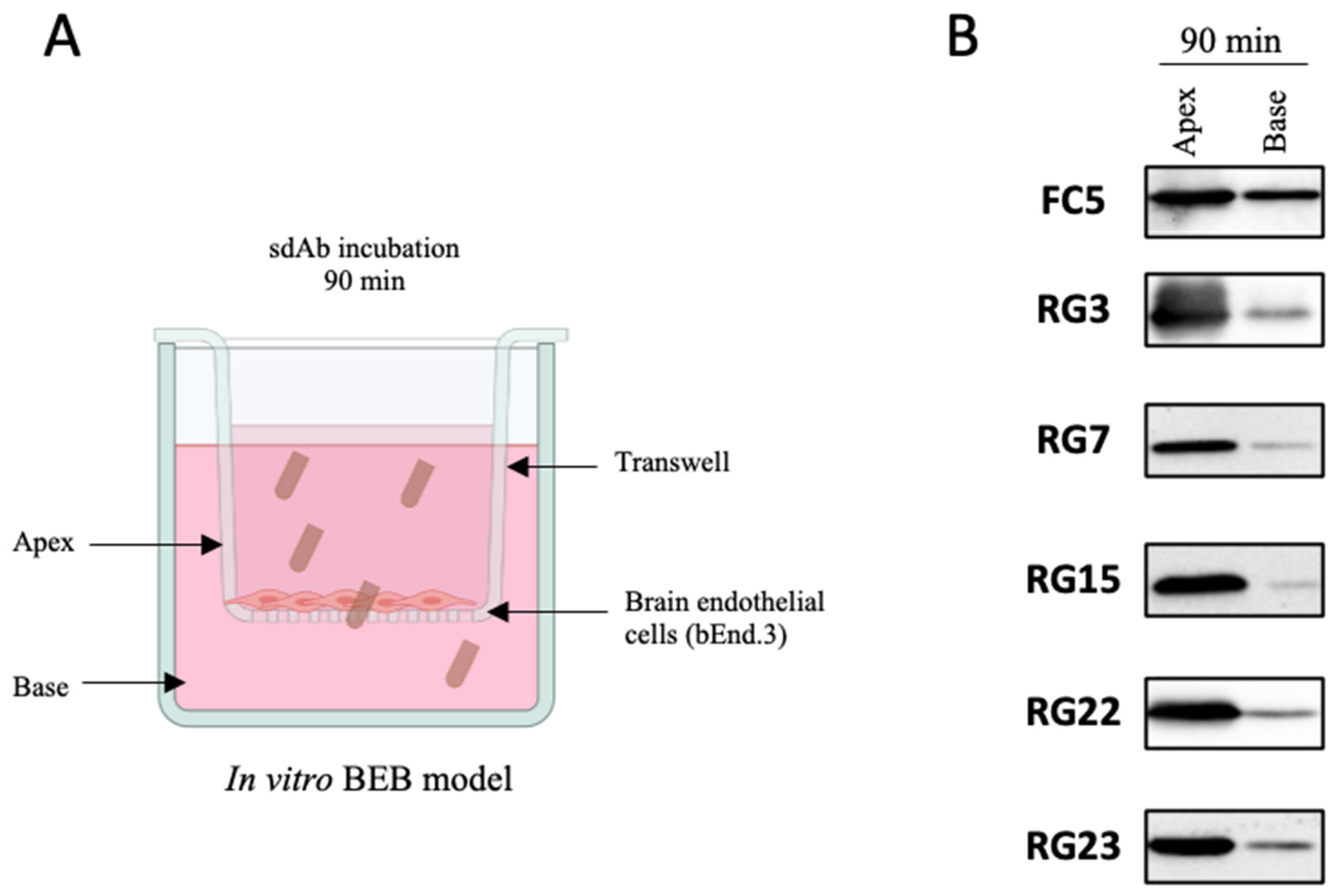
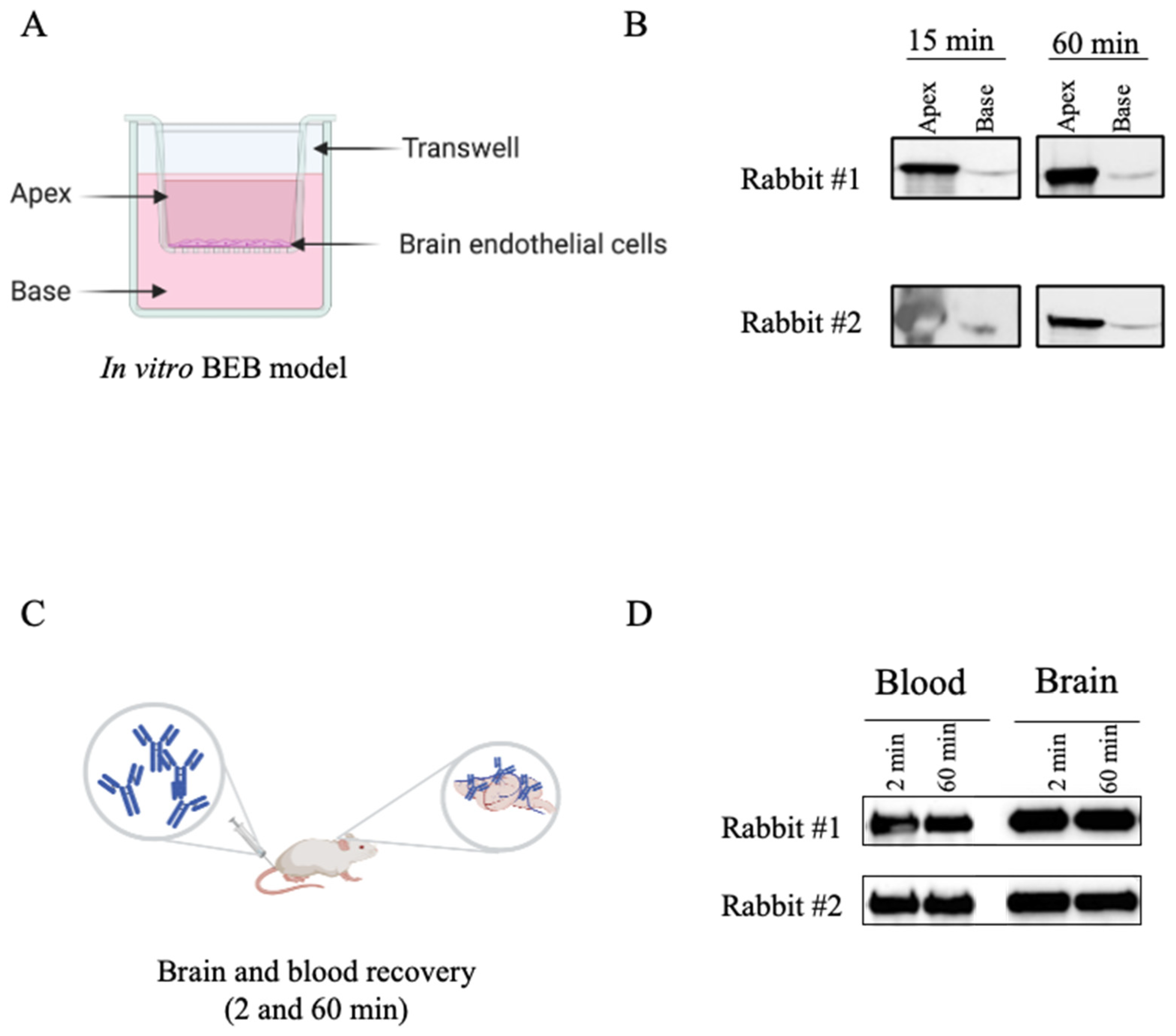
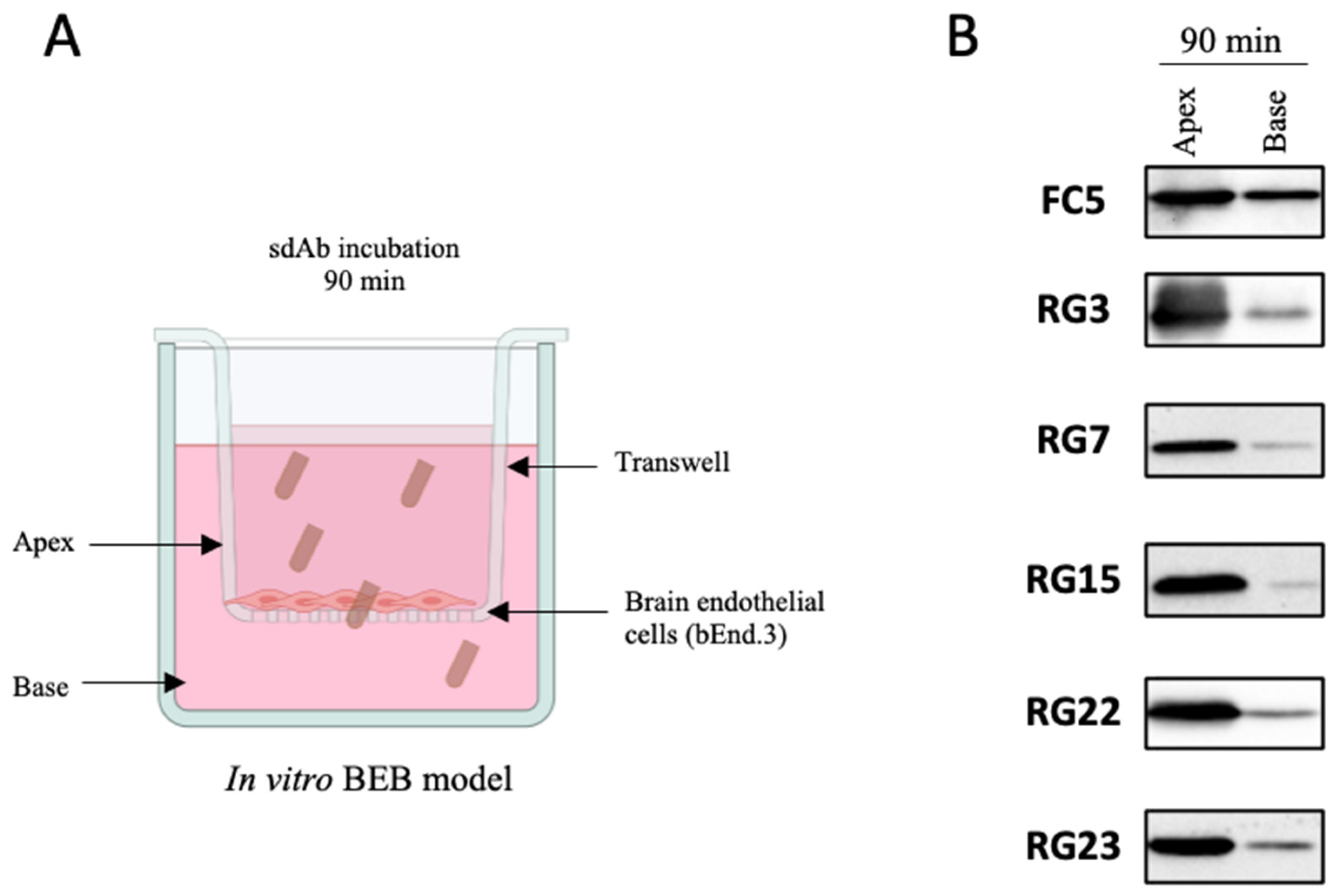
2.7. In Vitro BEB Models
2.8. In Vitro BEB Translocation
2.9. Biodistribution Studies
2.10. Measurement of Brain Antibody Concentrations
2.11. Development of PAN RG3 and FC5 Functionalized Liposomes
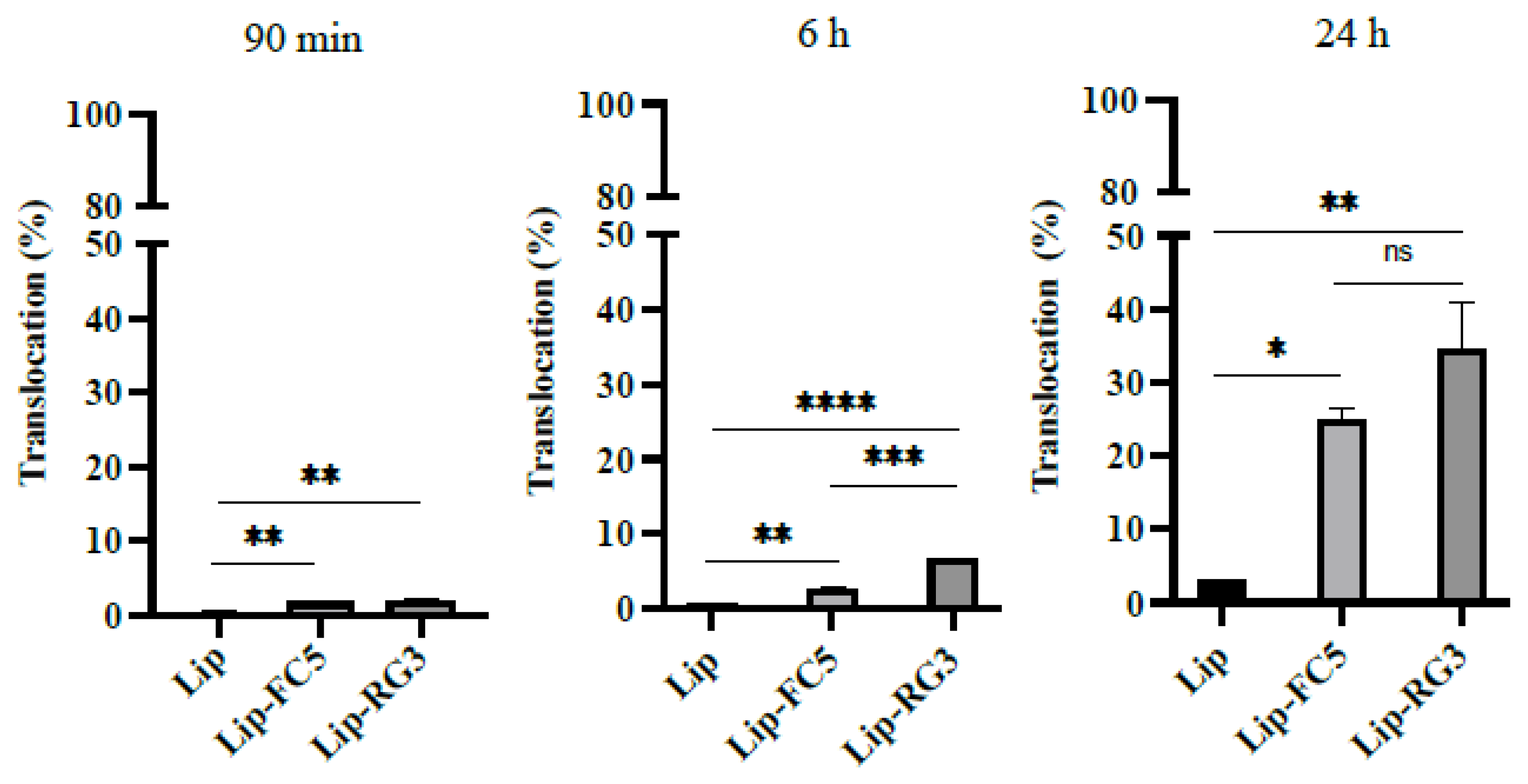
2.12. Translocation Efficiency Assay of Lip-RG3 and Lip-FC5
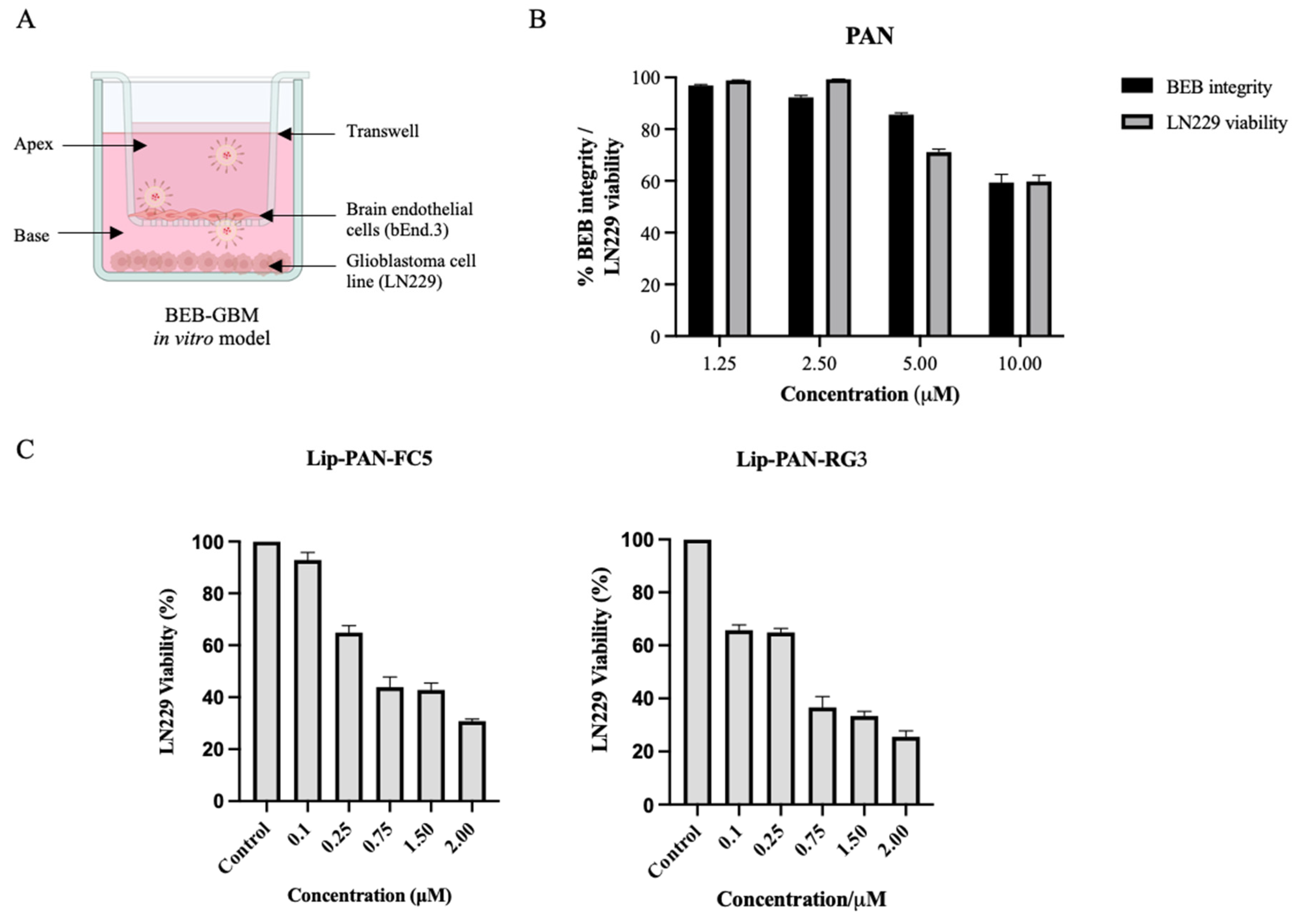
2.13. BEB-Glioblastoma In Vitro Model
2.14. Statistical Analysis
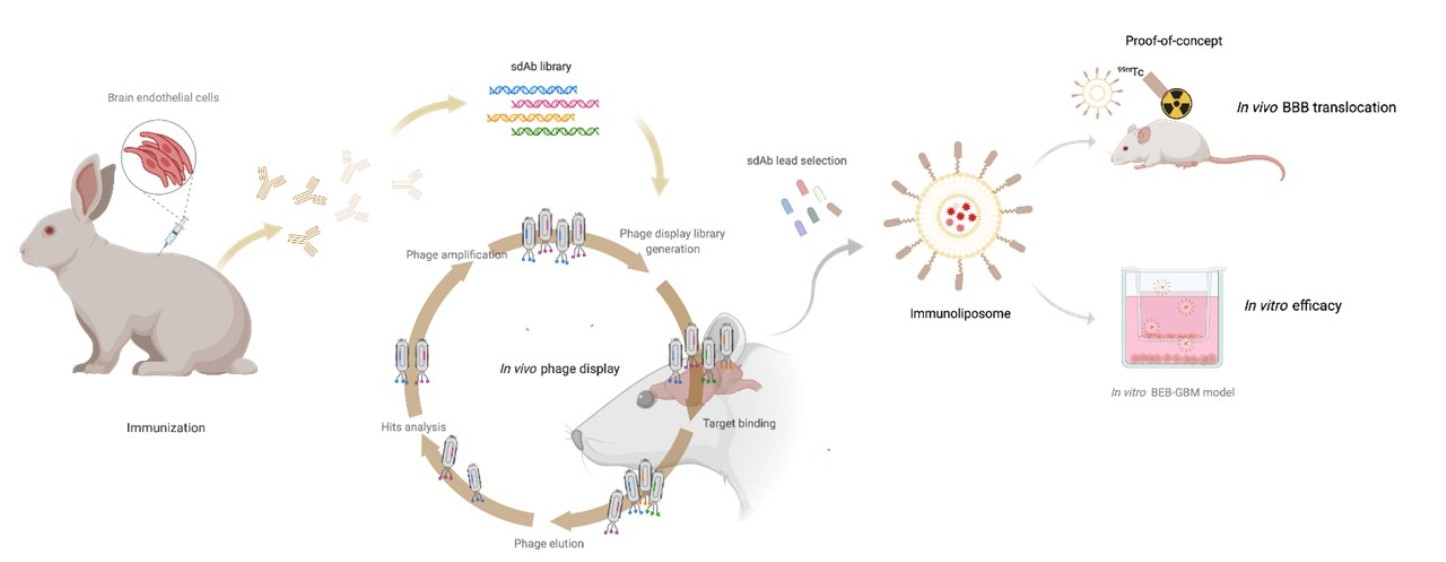
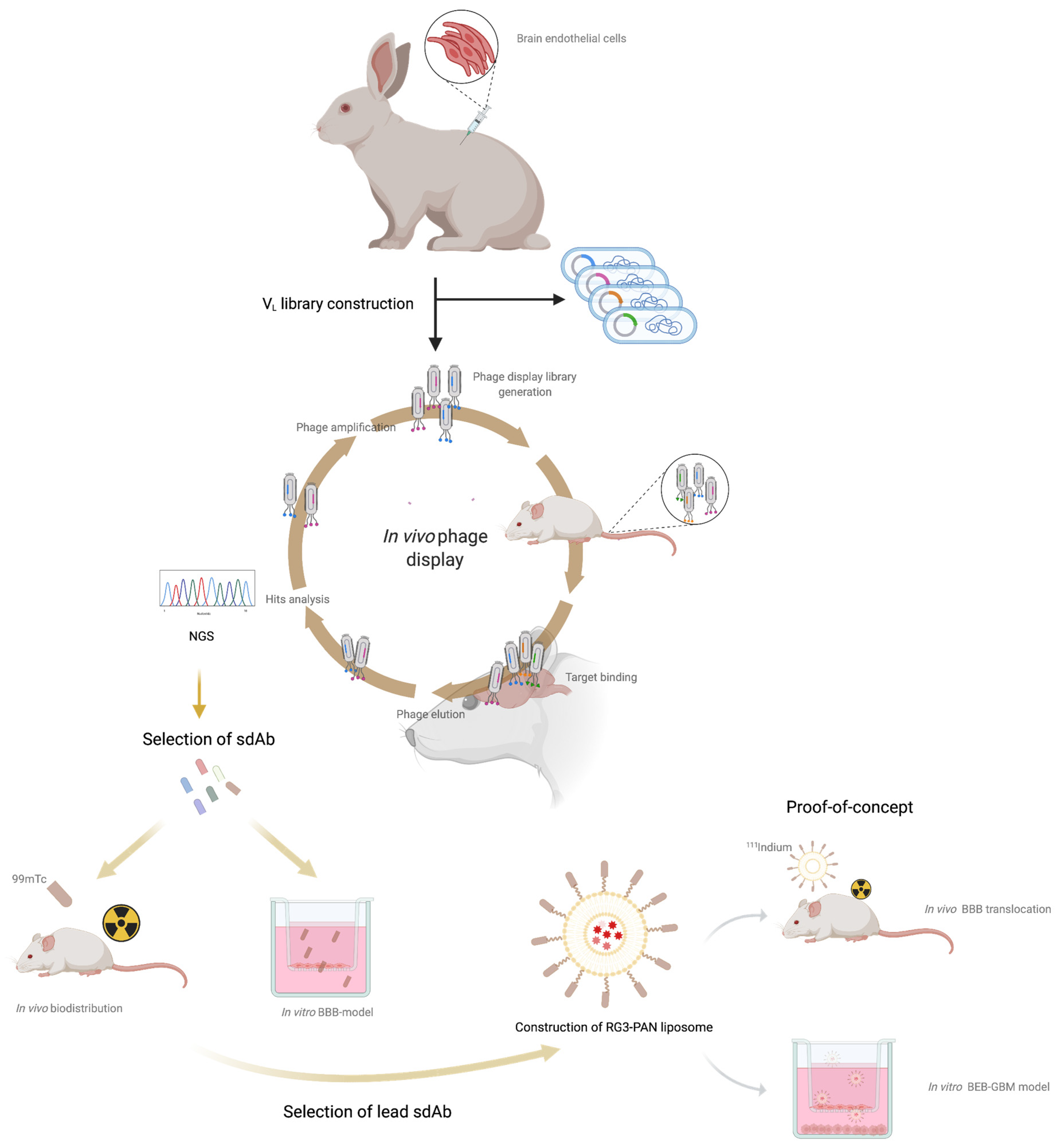
3. Results and Discussion
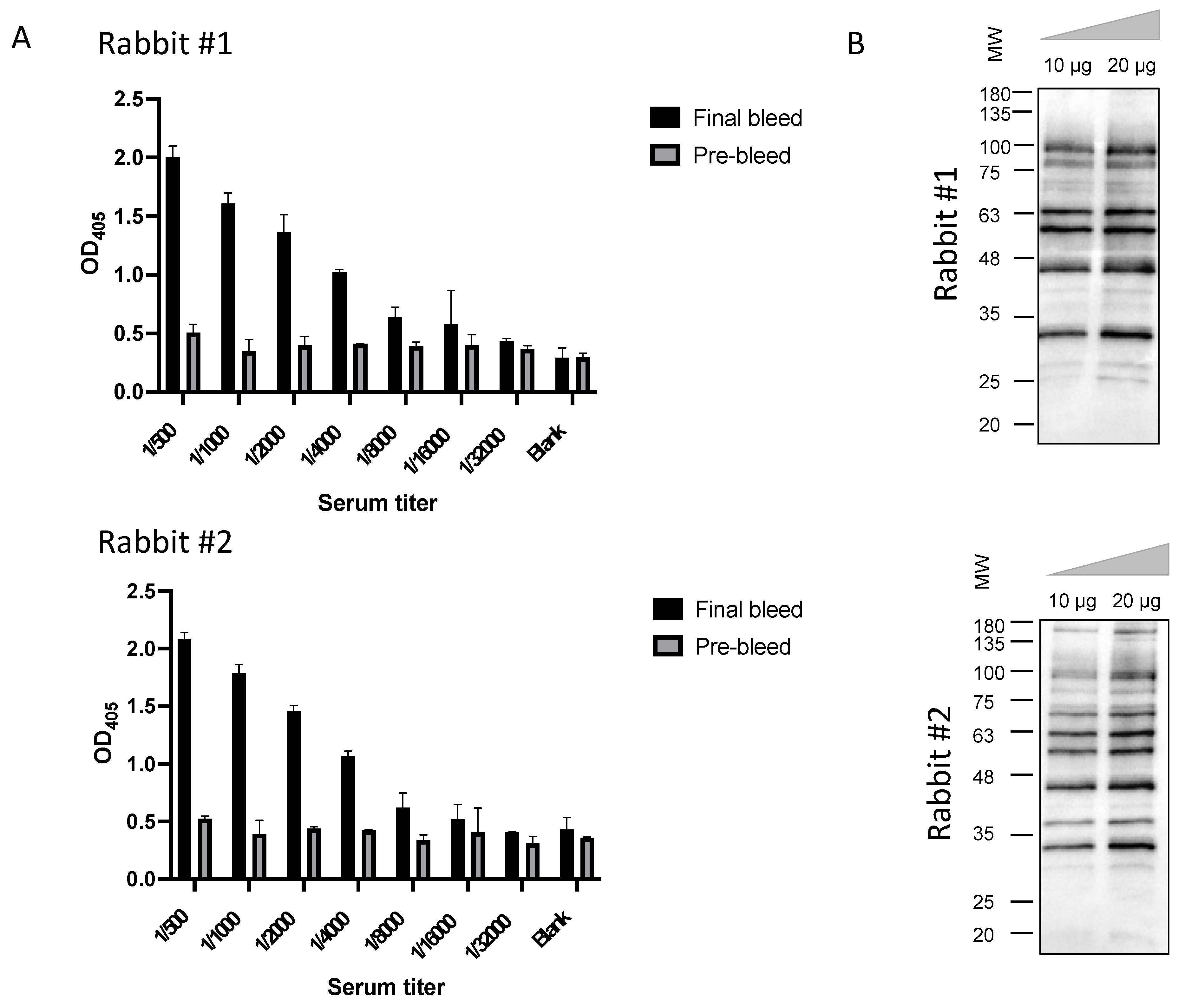
3.1. Construction of the Immunized sdAb Library and Antibody Validation
3.2. In Vivo Phage Display Selection of BBB-Targeting sdAbs
3.3. Screening for Antibodies towards the BBB
3.4. In Vivo Biodistribution of Selected Clones
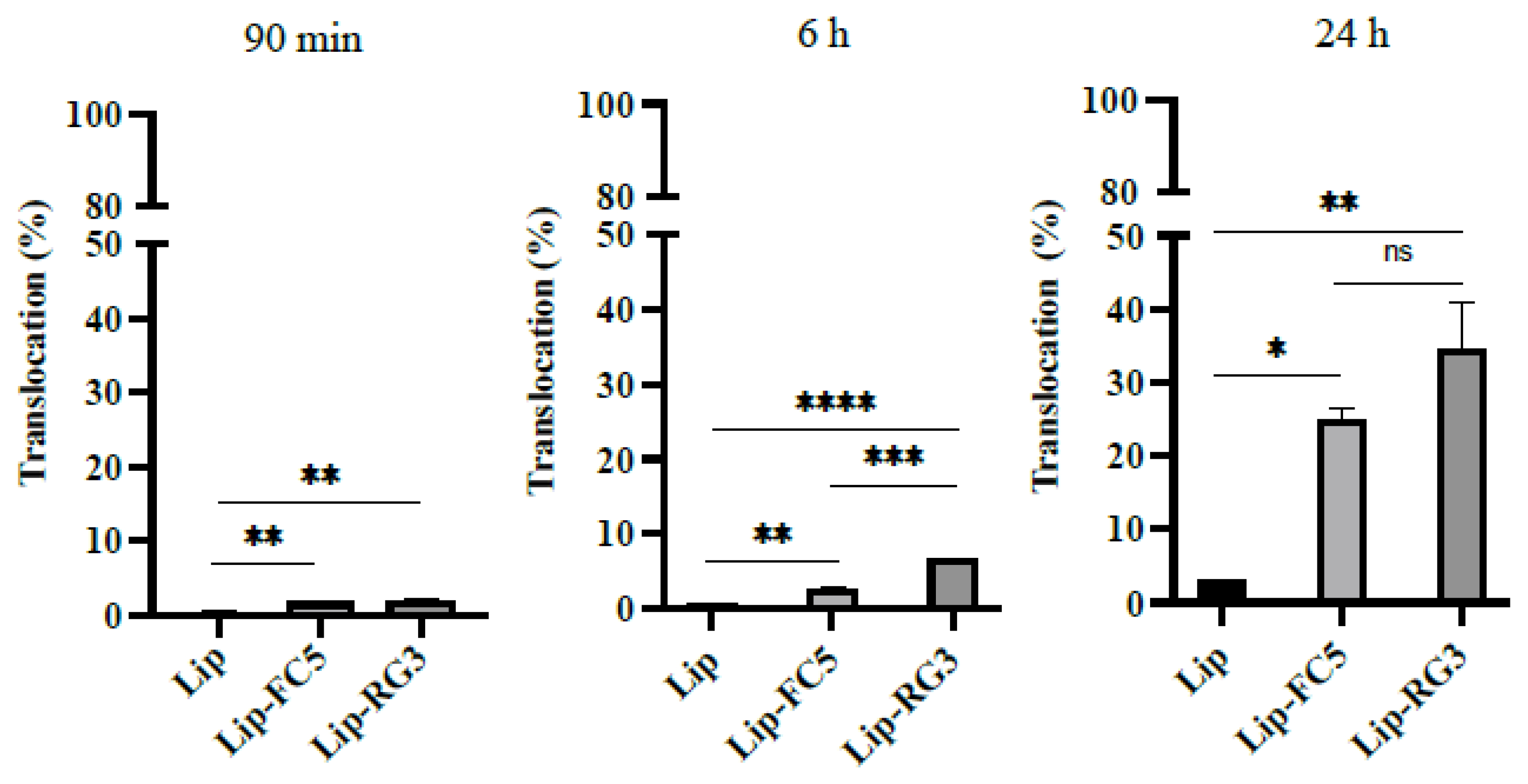
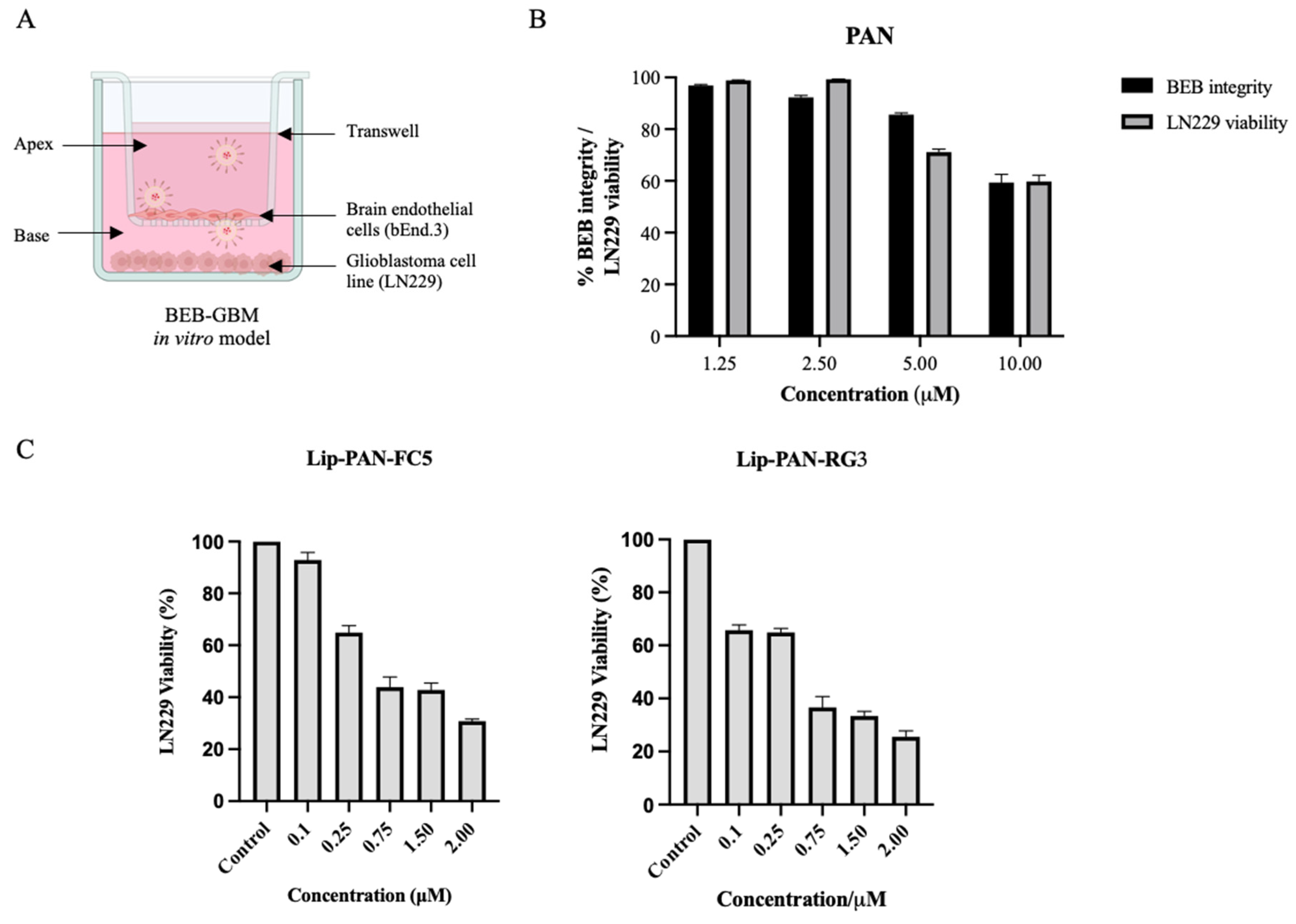
3.5. Validation of the RG3 as a BBB Drug Delivery Vector
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Georgieva, J.V.; Hoekstra, D.; Zuhorn, I.S. Smuggling Drugs into the Brain: An Overview of Ligands Targeting Transcytosis for Drug Delivery across the Blood-Brain Barrier. Pharmaceutics 2014, 6, 557–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardridge, W.M. Delivery of Biologics Across the Blood-Brain Barrier with Molecular Trojan Horse Technology. BioDrugs 2017, 31, 503–519. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and Function of the Blood-Brain Barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Soni, V.; Jain, A.; Khare, P.; Gulbake, A.; Jain, S.K. Potential Approaches for Drug Delivery to the Brain: Past, Present, and Future. Crit. Rev. Ther. Drug Carrier Syst. 2010, 27, 187–236. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.M.; Goyal, B.R.; Bhadada, S.V.; Bhatt, J.S.; Amin, A.F. Getting into the Brain: Approaches to Enhance Brain Drug Delivery. CNS Drugs 2009, 23, 35–58. [Google Scholar] [CrossRef]
- Pardridge, W.M.; Boado, R.J. Reengineering Biopharmaceuticals for Targeted Delivery across the Blood-Brain Barrier. Meth. Enzymol. 2012, 503, 269–292. [Google Scholar] [CrossRef]
- Pardridge, W.M. The Blood-Brain Barrier: Bottleneck in Brain Drug Development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef]
- Pardridge, W.M. Blood-Brain Barrier Delivery. Drug Discov. Today 2007, 12, 54–61. [Google Scholar] [CrossRef]
- Neves, V.; Aires-da-Silva, F.; Corte-Real, S.; Castanho, M.A.R.B. Antibody Approaches To Treat Brain Diseases. Trends Biotechnol. 2016, 34, 36–48. [Google Scholar] [CrossRef]
- Pardridge, W.M. Drug Transport across the Blood–Brain Barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef]
- Boado, R.J.; Zhang, Y.; Zhang, Y.; Pardridge, W.M. Humanization of Anti-Human Insulin Receptor Antibody for Drug Targeting across the Human Blood-Brain Barrier. Biotechnol. Bioeng. 2007, 96, 381–391. [Google Scholar] [CrossRef]
- Pardridge, W.M.; Kang, Y.S.; Buciak, J.L.; Yang, J. Human Insulin Receptor Monoclonal Antibody Undergoes High Affinity Binding to Human Brain Capillaries in vitro and Rapid Transcytosis through the Blood-Brain Barrier in Vivo in the Primate. Pharm. Res. 1995, 12, 807–816. [Google Scholar] [CrossRef]
- Boado, R.J.; Hui, E.K.-W.; Lu, J.Z.; Pardridge, W.M. Glycemic Control and Chronic Dosing of Rhesus Monkeys with a Fusion Protein of Iduronidase and a Monoclonal Antibody against the Human Insulin Receptor. Drug Metab. Dispos. 2012, 40, 2021–2025. [Google Scholar] [CrossRef] [Green Version]
- Boado, R.J.; Ka-Wai Hui, E.; Zhiqiang Lu, J.; Pardridge, W.M. Insulin Receptor Antibody-Iduronate 2-Sulfatase Fusion Protein: Pharmacokinetics, Anti-Drug Antibody, and Safety Pharmacology in Rhesus Monkeys. Biotechnol. Bioeng. 2014, 111, 2317–2325. [Google Scholar] [CrossRef] [Green Version]
- Boado, R.J.; Pardridge, W.M. Brain and Organ Uptake in the Rhesus Monkey in Vivo of Recombinant Iduronidase Compared to an Insulin Receptor Antibody-Iduronidase Fusion Protein. Mol. Pharm. 2017, 14, 1271–1277. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Yang, J.; Pardridge, W.M. Drug Targeting of a Peptide Radiopharmaceutical through the Primate Blood-Brain Barrier in Vivo with a Monoclonal Antibody to the Human Insulin Receptor. J. Clin. Investig. 1997, 100, 1804–1812. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Schlachetzki, F.; Pardridge, W.M. Global Non-Viral Gene Transfer to the Primate Brain Following Intravenous Administration. Mol. Ther. 2003, 7, 11–18. [Google Scholar] [CrossRef]
- Salvati, E.; Re, F.; Sesana, S.; Cambianica, I.; Sancini, G.; Masserini, M.; Gregori, M. Liposomes Functionalized to Overcome the Blood-Brain Barrier and to Target Amyloid-β Peptide: The Chemical Design Affects the Permeability across an in Vitro Model. Int. J. Nanomed. 2013, 8, 1749–1758. [Google Scholar] [CrossRef] [Green Version]
- Xia, C.-F.; Boado, R.J.; Zhang, Y.; Chu, C.; Pardridge, W.M. Intravenous Glial-Derived Neurotrophic Factor Gene Therapy of Experimental Parkinson’s Disease with Trojan Horse Liposomes and a Tyrosine Hydroxylase Promoter. J. Gene Med. 2008, 10, 306–315. [Google Scholar] [CrossRef]
- Watts, R.J.; Dennis, M.S. Bispecific Antibodies for Delivery into the Brain. Curr. Opin. Chem. Biol. 2013, 17, 393–399. [Google Scholar] [CrossRef]
- Yu, Y.J.; Atwal, J.K.; Zhang, Y.; Tong, R.K.; Wildsmith, K.R.; Tan, C.; Bien-Ly, N.; Hersom, M.; Maloney, J.A.; Meilandt, W.J.; et al. Therapeutic Bispecific Antibodies Cross the Blood-Brain Barrier in Nonhuman Primates. Sci. Transl. Med. 2014, 6, 261ra154. [Google Scholar] [CrossRef]
- Stanimirovic, D.; Kemmerich, K.; Haqqani, A.S.; Farrington, G.K. Engineering and Pharmacology of Blood-Brain Barrier-Permeable Bispecific Antibodies. Adv. Pharmacol. 2014, 71, 301–335. [Google Scholar] [CrossRef] [PubMed]
- Niewoehner, J.; Bohrmann, B.; Collin, L.; Urich, E.; Sade, H.; Maier, P.; Rueger, P.; Stracke, J.O.; Lau, W.; Tissot, A.C.; et al. Increased Brain Penetration and Potency of a Therapeutic Antibody Using a Monovalent Molecular Shuttle. Neuron 2014, 81, 49–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrington, G.K.; Caram-Salas, N.; Haqqani, A.S.; Brunette, E.; Eldredge, J.; Pepinsky, B.; Antognetti, G.; Baumann, E.; Ding, W.; Garber, E.; et al. A Novel Platform for Engineering Blood-Brain Barrier-Crossing Bispecific Biologics. FASEB J. 2014, 28, 4764–4778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coloma, M.J.; Lee, H.J.; Kurihara, A.; Landaw, E.M.; Boado, R.J.; Morrison, S.L.; Pardridge, W.M. Transport across the Primate Blood-Brain Barrier of a Genetically Engineered Chimeric Monoclonal Antibody to the Human Insulin Receptor. Pharm. Res. 2000, 17, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Engelhardt, B.; Lesley, J.; Bickel, U.; Pardridge, W.M. Targeting Rat Anti-Mouse Transferrin Receptor Monoclonal Antibodies through Blood-Brain Barrier in Mouse. J. Pharmacol. Exp. Ther. 2000, 292, 1048–1052. [Google Scholar]
- Ohshima-Hosoyama, S.; Simmons, H.A.; Goecks, N.; Joers, V.; Swanson, C.R.; Bondarenko, V.; Velotta, R.; Brunner, K.; Wood, L.D.; Hruban, R.H.; et al. A Monoclonal Antibody-GDNF Fusion Protein Is Not Neuroprotective and Is Associated with Proliferative Pancreatic Lesions in Parkinsonian Monkeys. PLoS ONE 2012, 7, e39036. [Google Scholar] [CrossRef]
- Couch, J.A.; Yu, Y.J.; Zhang, Y.; Tarrant, J.M.; Fuji, R.N.; Meilandt, W.J.; Solanoy, H.; Tong, R.K.; Hoyte, K.; Luk, W.; et al. Addressing Safety Liabilities of TfR Bispecific Antibodies That Cross the Blood-Brain Barrier. Sci. Transl. Med. 2013, 5, 1–12. [Google Scholar] [CrossRef]
- Jefferies, W.A.; Brandon, M.R.; Hunt, S.V.; Williams, A.F.; Gatter, K.C.; Mason, D.Y. Transferrin Receptor on Endothelium of Brain Capillaries. Nature 1984, 312, 162–163. [Google Scholar] [CrossRef]
- Aguiar, S.; Dias, J.; Manuel, A.M.; Russo, R.; Gois, P.M.P.; da Silva, F.A.; Goncalves, J. Chapter Five—Chimeric Small Antibody Fragments as Strategy to Deliver Therapeutic Payloads. In Advances in Protein Chemistry and Structural Biology; Academic Press: Cambridge, MA, USA, 2018; Volume 112, pp. 143–182. ISBN 1876-1623. [Google Scholar]
- Cantante, C.; Lourenço, S.; Morais, M.; Leandro, J.; Gano, L.; Silva, N.; Leandro, P.; Serrano, M.; Henriques, A.O.; Andre, A.; et al. Albumin-Binding Domain from Streptococcus Zooepidemicus Protein Zag as a Novel Strategy to Improve the Half-Life of Therapeutic Proteins. J. Biotechnol. 2017, 253, 23–33. [Google Scholar] [CrossRef]
- Muruganandam, A.; Tanha, J.; Narang, S.; Stanimirovic, D. Selection of Phage-Displayed Llama Single-Domain Antibodies That Transmigrate across Human Blood-Brain Barrier Endothelium. FASEB J. 2002, 16, 240–242. [Google Scholar] [CrossRef] [PubMed]
- Abulrob, A.; Sprong, H.; Van Bergen en Henegouwen, P.; Stanimirovic, D. The Blood-Brain Barrier Transmigrating Single Domain Antibody: Mechanisms of Transport and Antigenic Epitopes in Human Brain Endothelial Cells. J. Neurochem. 2005, 95, 1201–1214. [Google Scholar] [CrossRef] [PubMed]
- Webster, C.I.; Caram-Salas, N.; Haqqani, A.S.; Thom, G.; Brown, L.; Rennie, K.; Yogi, A.; Costain, W.; Brunette, E.; Stanimirovic, D.B. Brain Penetration, Target Engagement, and Disposition of the Blood–Brain Barrier-Crossing Bispecific Antibody Antagonist of Metabotropic Glutamate Receptor Type 1. FASEB J. 2016, 30, 1927–1940. [Google Scholar] [CrossRef] [Green Version]
- Zuchero, Y.J.Y.; Chen, X.; Bien-Ly, N.; Bumbaca, D.; Tong, R.K.; Gao, X.; Zhang, S.; Hoyte, K.; Luk, W.; Huntley, M.A.; et al. Discovery of Novel Blood-Brain Barrier Targets to Enhance Brain Uptake of Therapeutic Antibodies. Neuron 2016, 89, 70–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaillard, P.J.; van der Sandt, I.C.; Voorwinden, L.H.; Vu, D.; Nielsen, J.L.; de Boer, A.G.; Breimer, D.D. Astrocytes Increase the Functional Expression of P-Glycoprotein in an in Vitro Model of the Blood-Brain Barrier. Pharm. Res. 2000, 17, 1198–1205. [Google Scholar] [CrossRef]
- Janzer, R.C.; Raff, M.C. Astrocytes Induce Blood-Brain Barrier Properties in Endothelial Cells. Nature 1987, 325, 253–257. [Google Scholar] [CrossRef]
- Guntner, A.S.; Peyrl, A.; Mayr, L.; Englinger, B.; Berger, W.; Slavc, I.; Buchberger, W.; Gojo, J. Cerebrospinal Fluid Penetration of Targeted Therapeutics in Pediatric Brain Tumor Patients. Acta Neuropathol. Commun. 2020, 8, 1–13. [Google Scholar] [CrossRef]
- da Silva, F.A.; Li, M.; Rato, S.; Maia, S.; Malhó, R.; Warren, K.; Harrich, D.; Craigie, R.; Barbas, C.; Goncalves, J. Recombinant Rabbit Single-Chain Antibodies Bind to the Catalytic and C-Terminal Domains of HIV-1 Integrase Protein and Strongly Inhibit HIV-1 Replication. Biotechnol. Appl. Biochem. 2012, 59, 353–366. [Google Scholar] [CrossRef] [Green Version]
- Calado, R.; Duarte, J.; Borrego, P.; Marcelino, J.M.; Bártolo, I.; Martin, F.; Figueiredo, I.; Almeida, S.; Graça, L.; Vítor, J.; et al. A Prime-Boost Immunization Strategy with Vaccinia Virus Expressing Novel Gp120 Envelope Glycoprotein from a CRF02_AG Isolate Elicits Cross-Clade Tier 2 HIV-1 Neutralizing Antibodies. Vaccines 2020, 8, 171. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, F.N.C.A.; Goncalves, J.M.B. Engineered Rabbit Antibody Variable Domais and Uses Thereof. International Patent Application. No. PCT/PT2008/000018. WO2008136694A9, 13 November 2008. [Google Scholar]
- Barbas, F.C., III; Dennis, R.B.; Jamie, K.S.; Gregg, J.S. Phage Display: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001. [Google Scholar]
- Barbas, C.F.; Kang, A.S.; Lerner, R.A.; Benkovic, S.J. Assembly of Combinatorial Antibody Libraries on Phage Surfaces: The Gene III Site. Proc. Natl. Acad. Sci. USA 1991, 88, 7978–7982. [Google Scholar] [CrossRef] [Green Version]
- Kabat, E.A.; Wu, T.T.; Perry, H.M.; Foeller, C.; Gottesman, K.S. Sequences of Proteins of Immunological Interest; DIANE Publishing: Collingdale, PA, USA, 1992; ISBN 978-0-941375-65-8. [Google Scholar]
- Neves, V.; Aires-da-Silva, F.; Morais, M.; Gano, L.; Ribeiro, E.; Pinto, A.; Aguiar, S.; Gaspar, D.; Fernandes, C.; Correia, J.D.G.; et al. Novel Peptides Derived from Dengue Virus Capsid Protein Translocate Reversibly the Blood–Brain Barrier through a Receptor-Free Mechanism. ACS Chem. Biol. 2017, 12, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, M.M.; Radomska, A.; Gobbo, O.L.; Bakowsky, U.; Radomski, M.W.; Ehrhardt, C. Targeted Delivery of Transferrin-Conjugated Liposomes to an Orthotopic Model of Lung Cancer in Nude Rats. J. Aerosol. Med. Pulm. Drug Deliv. 2012, 25, 310–318. [Google Scholar] [CrossRef]
- Gaspar, M.M.; Boerman, O.C.; Laverman, P.; Corvo, M.L.; Storm, G.; Cruz, M.E.M. Enzymosomes with Surface-Exposed Superoxide Dismutase: In Vivo Behaviour and Therapeutic Activity in a Model of Adjuvant Arthritis. J. Control Release 2007, 117, 186–195. [Google Scholar] [CrossRef]
- Rader, C.; Ritter, G.; Nathan, S.; Elia, M.; Gout, I.; Jungbluth, A.A.; Cohen, L.S.; Welt, S.; Old, L.J.; Barbas, C.F. The Rabbit Antibody Repertoire as a Novel Source for the Generation of Therapeutic Human Antibodies. J. Biol. Chem. 2000, 275, 13668–13676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, J.; Peng, H.; Rader, C. From Rabbit Antibody Repertoires to Rabbit Monoclonal Antibodies. Exp. Mol. Med. 2017, 49, e305. [Google Scholar] [CrossRef] [PubMed]
- Feng, R.; Wang, R.; Hong, J.; Dower, C.M.; Croix, B.S.; Ho, M. Isolation of Rabbit Single Domain Antibodies to B7-H3 via Protein Immunization and Phage Display. Antib. Ther. 2020, 3, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Pasqualini, R.; Ruoslahti, E. Organ Targeting in Vivo Using Phage Display Peptide Libraries. Nature 1996, 380, 364–366. [Google Scholar] [CrossRef]
- Stutz, C.C.; Georgieva, J.V.; Shusta, E.V. Coupling Brain Perfusion Screens and Next Generation Sequencing to Identify Blood-Brain Barrier Binding Antibodies. AIChE J. 2018, 64, 4229–4236. [Google Scholar] [CrossRef]
- Dias, J.N.R.; Aguiar, S.I.; Pereira, D.M.; André, A.S.; Gano, L.; Correia, J.D.G.; Carrapiço, B.; Rütgen, B.; Malhó, R.; Peleteiro, C.; et al. The Histone Deacetylase Inhibitor Panobinostat Is a Potent Antitumor Agent in Canine Diffuse Large B-Cell Lymphoma. Oncotarget 2018, 9, 28586–28598. [Google Scholar] [CrossRef]
- Eleutherakis-Papaiakovou, E.; Kanellias, N.; Kastritis, E.; Gavriatopoulou, M.; Terpos, E.; Dimopoulos, M.A. Efficacy of Panobinostat for the Treatment of Multiple Myeloma. Available online: https://www.hindawi.com/journals/jo/2020/7131802/ (accessed on 20 November 2020).
- Bagcchi, S. Panobinostat Active against Diffuse Intrinsic Pontine Glioma. Lancet Oncol. 2015, 16, e267. [Google Scholar] [CrossRef]
- Chen, R.; Zhang, M.; Zhou, Y.; Guo, W.; Yi, M.; Zhang, Z.; Ding, Y.; Wang, Y. The Application of Histone Deacetylases Inhibitors in Glioblastoma. J. Exp. Clin. Cancer Res. 2020, 39, 1–18. [Google Scholar] [CrossRef]
- Jose, G.; Lu, Y.-J.; Hung, J.-T.; Yu, A.L.; Chen, J.-P. Co-Delivery of CPT-11 and Panobinostat with Anti-GD2 Antibody Conjugated Immunoliposomes for Targeted Combination Chemotherapy. Cancers 2020, 12, 3211. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, L.; McCully, C.L.; Peer, C.; Cruz, R.; Figg, W. Hg-36 Plasma and Cerebrospinal Fluid (CSF) Pharmacokinetics of Panobinostat Following Oral Administration to Nonhuman Primates. Neuro Oncol. 2016, 18, iii55. [Google Scholar] [CrossRef] [Green Version]
- Hersh, D.S.; Wadajkar, A.S.; Roberts, N.; Perez, J.G.; Connolly, N.P.; Frenkel, V.; Winkles, J.A.; Woodworth, G.F.; Kim, A.J. Evolving Drug Delivery Strategies to Overcome the Blood Brain Barrier. Curr. Pharm. Des. 2016, 22, 1177–1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mojarad-Jabali, S.; Farshbaf, M.; Walker, P.R.; Hemmati, S.; Fatahi, Y.; Zakeri-Milani, P.; Sarfraz, M.; Valizadeh, H. An Update on Actively Targeted Liposomes in Advanced Drug Delivery to Glioma. Int. J. Pharm. 2021, 602, 120645. [Google Scholar] [CrossRef]
- Li, X.; Tsibouklis, J.; Weng, T.; Zhang, B.; Yin, G.; Feng, G.; Cui, Y.; Savina, I.N.; Mikhalovska, L.I.; Sandeman, S.R.; et al. Nano Carriers for Drug Transport across the Blood-Brain Barrier. J. Drug Target. 2017, 25, 17–28. [Google Scholar] [CrossRef] [Green Version]
- Lakkadwala, S.; dos Santos Rodrigues, B.; Sun, C.; Singh, J. Biodistribution of TAT or QLPVM Coupled to Receptor Targeted Liposomes for Delivery of Anticancer Therapeutics to Brain in Vitro and in Vivo. Nanomed. Nanotechnol. Biol. Med. 2020, 23, 102112. [Google Scholar] [CrossRef]
- Mu, L.-M.; Bu, Y.-Z.; Liu, L.; Xie, H.-J.; Ju, R.-J.; Wu, J.-S.; Zeng, F.; Zhao, Y.; Zhang, J.-Y.; Lu, W.-L. Lipid Vesicles Containing Transferrin Receptor Binding Peptide TfR-T12 and Octa-Arginine Conjugate Stearyl-R8 Efficiently Treat Brain Glioma along with Glioma Stem Cells. Sci. Rep. 2017, 7, 3487. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organ | RG3-99mTc | RG7-99mTc | RG15-99mTc | RG22-99mTc | RG23-99mTc | FC5-99mTc | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 min | 60 min | 2 min | 60 min | 2 min | 60 min | 2 min | 60 min | 2 min | 60 min | 2 min | 60 min | |
| Blood | 10.1 ± 2.6 | 1.4 ± 0.1 | 11.2 ± 2.4 | 1.5 ± 0.6 | 12.5 ± 2.4 | 3.0 ± 0.1 | 13.2 ± 2.5 | 0.8 ± 0.1 | 7.8 ± 0.2 | 1.1 ± 0.8 | 11.0 ± 2.4 | 1.0 ± 0.20 |
| Liver | 5.0 ± 0.9 | 1.9 ± 0.3 | 3.8 ± 0.8 | 1.0 ± 0.4 | 6.0 ± 1.0 | 3.6 ± 0.7 | 4.6 ± 0.7 | 1.7 ± 0.2 | 5.3 ± 0.2 | 1.9 ± 0.4 | 10.0 ± 1.5 | 12.40 ± 0.4 |
| Intestine | 1.8 ± 0.6 | 0.7 ± 0.1 | 1.8 ± 0.2 | 0.21 ± 0.08 | 1.8 ± 0.4 | 0.4 ± 0.1 | 2.0 ± 0.2 | 0.17 ± 0.03 | 1.6 ± 0.7 | 0.4 ± 0.1 | 0.9 ± 0.4 | 0.79 ± 0.04 |
| Spleen | 2.2 ± 0.6 | 0.6 ± 0.1 | 3.6 ± 0.6 | 0.5 ± 0.3 | 3.5 ± 1.2 | 4.8 ± 1.7 | 2.6 ± 0.6 | 0.54 ± 0.03 | 2.45 ± 0.04 | 0.8 ± 0.1 | 4.2 ± 0.8 | 4.2 ± 0.80 |
| Heart | 3.2 ± 1.2 | 0.4 ± 0.1 | 4.7 ± 1.2 | 0.45 ± 0.08 | 4.3 ± 0.8 | 1.3 ± 0.2 | 3.2 ± 0.5 | 0.29 ± 0.03 | 4.5 ± 0.4 | 0.5 ± 0.1 | 3.9 ± 0.5 | 0.76 ± 0.03 |
| Lung | 7.5 ± 2.7 | 1.1 ± 0.2 | 9.8 ± 2.4 | 1.5 ± 0.7 | 9.9 ± 1.2 | 2.4 ± 0.4 | 6.7 ± 0.6 | 2.1 ± 0.4 | 6.0 ± 0.9 | 1.7 ± 0.3 | 12.6 ± 0.6 | 38.20 ± 6.20 |
| Kidney | 74.6 ± 9.9 | 170.6 ± 19.1 | 73.6 ± 7.5 | 93.1 ± 6.9 | 54.6 ± 7.5 | 52.1 ± 9.0 | 55.5 ± 0.9 | 49.3 ± 8.3 | 27.2 ± 4.1 | 74.3 ± 10.7 | 59.8 ± 2.4 | 96.40 ± 6.80 |
| Muscle | 2.0 ± 1.0 | 0.4 ± 0.2 | 1.9 ± 0.2 | 0.29 ± 0.07 | 1.5 ± 0.4 | 0.6 ± 0.1 | 1.4 ± 0.1 | 0.3 ± 0.1 | 1.5 ± 0.2 | 0.34 ± 0.05 | 1.1 ± 0.1 | 0.39 ± 0.04 |
| Bone | 2.7 ± 0.2 | 0.81 ± 0.05 | 2.3 ± 0.4 | 0.33 ± 0.05 | 2.3 ± 0.8 | 1.0 ± 0.1 | 2.4 ± 0.4 | 0.5 ± 0.1 | 1.5 ± 0.4 | 0.4 ± 0.1 | 1.7 ± 0.2 | 0.80 ± 0.3 |
| Stomach | 1.0 ± 0.4 | 0.97 ± 0.04 | 1.7 ± 0.6 | 0.9 ± 0.4 | 0.7 ± 0.3 | 1.2 ± 0.1 | 0.7 ± 0.3 | 0.26 ± 0.07 | 1.1 ± 0.5 | 0.9 ± 0.4 | 1.2 ± 0.2 | 0.80 ± 0.3 |
| Brain | 0.82 ± 0.05 | 0.06 ± 0.02 | 0.46 ± 0.1 | 0.4 ± 0.1 | 0.61 ± 0.1 | 0.08 ± 0.06 | 0.60 ± 0.15 | 0.03 ± 0.01 | 0.49 ± 0.02 | 0.04 ± 0.02 | 0.52 ± 0.18 | 0.07 ± 0.02 |
| Excretion (%I.A.) | - | 19.6 ± 5.4 | - | 21.1 ± 9.0 | - | 27.5 ± 5.1 | - | 70.3 ± 3.5 | - | 27.5 ± 5.1 | - | 9.60 ± 1.40 |
| Organ | 111In-Lip-RG3 | 111In-Lip-(Control) | |||
|---|---|---|---|---|---|
| 2 min | 60 min | 24 h | 60 min | 24 h | |
| Blood | 33.0 ± 7.4 | 26.2 ± 8.1 | 8.9 ± 1.7 | 12.7 ± 3.8 | 3.5 ± 0.6 |
| Liver | 8.1 ± 2.8 | 10.7 ± 2.1 | 11.7 ± 1.6 | 1.1 ± 0.6 | 5.9 ± 0.5 |
| Intestine | 0.4 ± 0.1 | 0.9 ± 0.4 | 2.3 ± 0.1 | 0.55 ± 0.06 | 0.98 ± 0.07 |
| Spleen | 4.3 ± 0.8 | 19.4 ± 3.2 | 18.2 ± 0.1 | 1.7 ± 0.4 | 12.5 ± 4.2 |
| Heart | 4.2 ± 0.8 | 4.4 ± 1.2 | 3.0 ± 1.3 | 0.7 ± 0.2 | 1.3 ± 0.3 |
| Lung | 13.3 ± 3.9 | 10.2 ± 1.0 | 3.9 ± 1.6 | 1.0 ± 0.5 | 1.6 ± 0.6 |
| Kidney | 7.0 ± 1.7 | 6.6 ± 1.0 | 6.4 ± 0.7 | 1.3 ± 0.6 | 2.7 ± 0.3 |
| Muscle | 0.8 ± 0.2 | 0.62 ± 0.07 | 0.9 ± 0.2 | 0.4 ± 0.1 | 1.3 ± 0.9 |
| Bone | 1.8 ± 0.1 | 1.6 ± 0.2 | 1.1 ± 0.9 | 0.6 ± 0.3 | 0.7 ± 0.3 |
| Stomach | 0.8 ± 0.2 | 0.8 ± 0.6 | 0.8 ± 0.2 | 1.7 ± 0.9 | 1.1 ± 0.2 |
| Brain | 1.0 ± 0.3 | 1.0 ± 0.4 | 0.4 ± 0.2 | 0.13 ± 0.03 | 0.16 ± 0.07 |
| Carcass (%I.A.) | 45.2 ± 1.8 | 43.6 ± 4.8 | 38.3 ± 3.6 | 18.0 ± 0.9 | 26.0 ± 2.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aguiar, S.I.; Dias, J.N.R.; André, A.S.; Silva, M.L.; Martins, D.; Carrapiço, B.; Castanho, M.; Carriço, J.; Cavaco, M.; Gaspar, M.M.; et al. Highly Specific Blood-Brain Barrier Transmigrating Single-Domain Antibodies Selected by an In Vivo Phage Display Screening. Pharmaceutics 2021, 13, 1598. https://doi.org/10.3390/pharmaceutics13101598
Aguiar SI, Dias JNR, André AS, Silva ML, Martins D, Carrapiço B, Castanho M, Carriço J, Cavaco M, Gaspar MM, et al. Highly Specific Blood-Brain Barrier Transmigrating Single-Domain Antibodies Selected by an In Vivo Phage Display Screening. Pharmaceutics. 2021; 13(10):1598. https://doi.org/10.3390/pharmaceutics13101598
Chicago/Turabian StyleAguiar, Sandra Isabel, Joana N. R. Dias, Ana Santos André, Marta Lisete Silva, Diana Martins, Belmira Carrapiço, Miguel Castanho, João Carriço, Marco Cavaco, Maria Manuela Gaspar, and et al. 2021. "Highly Specific Blood-Brain Barrier Transmigrating Single-Domain Antibodies Selected by an In Vivo Phage Display Screening" Pharmaceutics 13, no. 10: 1598. https://doi.org/10.3390/pharmaceutics13101598
APA StyleAguiar, S. I., Dias, J. N. R., André, A. S., Silva, M. L., Martins, D., Carrapiço, B., Castanho, M., Carriço, J., Cavaco, M., Gaspar, M. M., Nobre, R. J., Pereira de Almeida, L., Oliveira, S., Gano, L., Correia, J. D. G., Barbas, C., 3rd, Gonçalves, J., Neves, V., & Aires-da-Silva, F. (2021). Highly Specific Blood-Brain Barrier Transmigrating Single-Domain Antibodies Selected by an In Vivo Phage Display Screening. Pharmaceutics, 13(10), 1598. https://doi.org/10.3390/pharmaceutics13101598