
Variants in COMT, CYP3A5, CYP2B6, and ABCG2 Alter Quetiapine Pharmacokinetics

 ,
,  , , , , ,
, , , , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design and Population
2.2. Pharmacokinetics and Safety
2.3. Genotyping, Haplotyping, and Phenotyping
2.4. Mass Spectrometry
2.5. Statistical Analysis
3. Results
3.1. Safety
3.2. Mass Spectrometry Analysis
4. Discussion
Study Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- DeVane, C.L.; Nemeroff, C.B. Clinical Pharmacokinetics of Quetiapine: An Atypical Antipsychotic. Clin. Pharm. 2001, 40, 509–522. [Google Scholar] [CrossRef]
- Maan, J.S.; Ershadi, M.; Khan, I.; Saadabadi, A. Quetiapine; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Narala, A.; Veerabrahma, K. Preparation, Characterization and Evaluation of Quetiapine Fumarate Solid Lipid Nanoparticles to Improve the Oral Bioavailability. J. Pharm. 2013, 2013, 265741. [Google Scholar] [CrossRef]
- Li, K.-Y.; Li, X.; Cheng, Z.-N.; Li, H.-D. Metabolic Mechanism of Quetiapine in Vivo with Clinical Therapeutic Dose. Methods Find. Exp. Clin. Pharmacol. 2005, 27, 83–86. [Google Scholar] [CrossRef]
- Bakken, G.V.; Rudberg, I.; Christensen, H.; Molden, E.; Refsum, H.; Hermann, M. Metabolism of Quetiapine by CYP3A4 and CYP3A5 in Presence or Absence of Cytochrome B5. Drug Metab. Dispos. 2009, 37, 254–258. [Google Scholar] [CrossRef]
- López-Muñoz, F.; Alamo, C. Active Metabolites as Antidepressant Drugs: The Role of Norquetiapine in the Mechanism of Action of Quetiapine in the Treatment of Mood Disorders. Front. Psychiatry 2013, 4, 102. [Google Scholar] [CrossRef] [PubMed]
- Boulton, D.W.; DeVane, C.L.; Liston, H.L.; Markowitz, J.S. In Vitro P-Glycoprotein Affinity for Atypical and Conventional Antipsychotics. Life Sci. 2002, 71, 163–169. [Google Scholar] [CrossRef]
- Lin, J.H.; Yamazaki, M. Role of P-Glycoprotein in Pharmacokinetics: Clinical Implications. Clin. Pharmacokinet. 2003, 42, 59–98. [Google Scholar] [CrossRef] [PubMed]
- El-Saifi, N.; Moyle, W.; Jones, C.; Tuffaha, H. Quetiapine Safety in Older Adults: A Systematic Literature Review. J. Clin. Pharm. Ther. 2016, 41, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Carr, C.N.; Lopchuk, S.; Beckman, M.E.; Baugh, T.B. Evaluation of the Use of Low-Dose Quetiapine and the Risk of Metabolic Consequences: A Retrospective Review. Ment. Health Clin. 2016, 6, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Cabaleiro, T.; López-Rodríguez, R.; Román, M.; Ochoa, D.; Novalbos, J.; Borobia, A.; Carcas, A.; Abad-Santos, F. Pharmacogenetics of Quetiapine in Healthy Volunteers: Association with Pharmacokinetics, Pharmacodynamics, and Adverse Effects. Int. Clin. Psychopharmacol. 2015, 30, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Wu, X.; Li, M.; Huang, H.; Minica, C.; Yi, Z.; Wang, G.; Shen, L.; Xing, Q.; Shi, Y.; et al. Association Studies of Genomic Variants with Treatment Response to Risperidone, Clozapine, Quetiapine and Chlorpromazine in the Chinese Han Population. Pharm. J. 2016, 16, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Vijayananthan, A.; Nawawi, O. The Importance of Good Clinical Practice Guidelines and Its Role in Clinical Trials. Biomed. Imaging Interv. J. 2008, 4, e5. [Google Scholar] [CrossRef] [PubMed]
- World Medical Association. World Medical Association Declaration of Helsinki: Ethical Principles for Medical Research Involving Human Subjects. JAMA 2013, 310, 2191–2194. [Google Scholar] [CrossRef]
- Aguirre, C.; García, M. Causality assessment in reports on adverse drug reactions. Algorithm of Spanish pharmacovigilance system. Med. Clin. 2016, 147, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Scott, S.A.; Sangkuhl, K.; Stein, C.M.; Hulot, J.-S.; Mega, J.L.; Roden, D.M.; Klein, T.E.; Sabatine, M.S.; Johnson, J.A.; Shuldiner, A.R. Clinical Pharmacogenetics Implementation Consortium Guidelines for CYP2C19 Genotype and Clopidogrel Therapy: 2013 Update. Clin. Pharmacol. Ther. 2013, 94, 317–323. [Google Scholar] [CrossRef]
- Desta, Z.; Gammal, R.S.; Gong, L.; Whirl-Carrillo, M.; Gaur, A.H.; Sukasem, C.; Hockings, J.; Myers, A.; Swart, M.; Tyndale, R.F.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2B6 and Efavirenz-Containing Antiretroviral Therapy. Clin. Pharmacol. Ther. 2019, 106, 726–733. [Google Scholar] [CrossRef]
- Theken, K.N.; Lee, C.R.; Gong, L.; Caudle, K.E.; Formea, C.M.; Gaedigk, A.; Klein, T.E.; Agúndez, J.A.G.; Grosser, T. Clinical Pharmacogenetics Implementation Consortium Guideline (CPIC) for CYP2C9 and Nonsteroidal Anti-Inflammatory Drugs. Clin. Pharmacol. Ther. 2020, 108, 191–200. [Google Scholar] [CrossRef]
- Crews, K.R.; Gaedigk, A.; Dunnenberger, H.M.; Leeder, J.S.; Klein, T.E.; Caudle, K.E.; Haidar, C.E.; Shen, D.D.; Callaghan, J.T.; Sadhasivam, S.; et al. Clinical Pharmacogenetics Implementation Consortium Guidelines for Cytochrome P450 2D6 Genotype and Codeine Therapy: 2014 Update. Clin. Pharmacol. Ther. 2014, 95, 376–382. [Google Scholar] [CrossRef]
- Birdwell, K.; Decker, B.; Barbarino, J.; Peterson, J.; Stein, C.; Sadee, W.; Wang, D.; Vinks, A.; He, Y.; Swen, J.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guidelines for CYP3A5 Genotype and Tacrolimus Dosing. Clin. Pharmacol. Ther. 2015, 98, 19–24. [Google Scholar] [CrossRef]
- Gammal, R.; Court, M.; Haidar, C.; Iwuchukwu, O.; Gaur, A.; Alvarellos, M.; Guillemette, C.; Lennox, J.; Whirl-Carrillo, M.; Brummel, S.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for UGT1A1 and Atazanavir Prescribing. Clin. Pharmacol. Ther. 2016, 99, 363–369. [Google Scholar] [CrossRef]
- Ramsey, L.B.; Johnson, S.G.; Caudle, K.E.; Haidar, C.E.; Voora, D.; Wilke, R.A.; Maxwell, W.D.; McLeod, H.L.; Krauss, R.M.; Roden, D.M.; et al. The Clinical Pharmacogenetics Implementation Consortium Guideline for SLCO1B1 and Simvastatin-Induced Myopathy: 2014 Update. Clin. Pharmacol. Ther. 2014, 96, 423–428. [Google Scholar] [CrossRef]
- Koller, D.; Zubiaur, P.; Saiz-Rodríguez, M.; Abad-Santos, F.; Wojnicz, A. Simultaneous Determination of Six Antipsychotics, Two of Their Metabolites and Caffeine in Human Plasma by LC-MS/MS Using a Phospholipid-Removal Microelution-Solid Phase Extraction Method for Sample Preparation. Talanta 2019, 198, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Van der Weide, K.; van der Weide, J. The Influence of the CYP3A4*22 Polymorphism on Serum Concentration of Quetiapine in Psychiatric Patients. J. Clin. Psychopharmacol. 2014, 34, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Bakken, G.V.; Molden, E.; Hermann, M. Impact of Genetic Variability in CYP2D6, CYP3A5, and ABCB1 on Serum Concentrations of Quetiapine and N-Desalkylquetiapine in Psychiatric Patients. Ther. Drug Monit. 2015, 37, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Royal Dutch Pharmacists Association, Dutch Pharmacogenetics Working Group, The Netherlands, last update: 07.06.2021. Available online: https://www.knmp.nl/downloads/farmacogenetica-engels-recommendation-tekst.pdf (accessed on 10 September 2021).
- Scheggia, D.; Sannino, S.; Scattoni, M.L.; Papaleo, F. COMT as a Drug Target for Cognitive Functions and Dysfunctions. CNS Neurol. Disord. Drug Targets 2012, 11, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.-Z.; Liu, B.-C.; Zhang, J.; Wang, L.; Li, X.-W.; Wang, Y.; Ji, J.; Yang, F.-P.; Wan, C.-L.; Xu, Y.-F.; et al. Association between a COMT Polymorphism and Clinical Response to Risperidone Treatment: A Pharmacogenetic Study. Psychiatr. Genet. 2012, 22, 298–299. [Google Scholar] [CrossRef] [PubMed]
- Nikolac Perkovic, M.; Sagud, M.; Zivkovic, M.; Uzun, S.; Nedic Erjavec, G.; Kozumplik, O.; Svob Strac, D.; Mimica, N.; Mihaljevic Peles, A.; Pivac, N. Catechol-O-Methyltransferase Rs4680 and Rs4818 Haplotype Association with Treatment Response to Olanzapine in Patients with Schizophrenia. Sci. Rep. 2020, 10, 10049. [Google Scholar] [CrossRef] [PubMed]
- Segura, M.; Roura, L.; de la Torre, R.; Joglar, J. Synthesis of the Major Metabolites of Paroxetine. Bioorg. Chem. 2003, 31, 248–258. [Google Scholar] [CrossRef]
- Fisher, D.S.; Handley, S.A.; Taylor, D.; Flanagan, R.J. Measurement of Quetiapine and Four Quetiapine Metabolites in Human Plasma by LC-MS/MS. Biomed. Chromatogr. 2012, 26, 1125–1132. [Google Scholar] [CrossRef]
- Woodward, N.D.; Jayathilake, K.; Meltzer, H.Y. COMT Val108/158met Genotype, Cognitive Function, and Cognitive Improvement with Clozapine in Schizophrenia. Schizophr. Res. 2007, 90, 86–96. [Google Scholar] [CrossRef]
- Goghari, V.M.; Sponheim, S.R. Differential Association of the COMT Val158Met Polymorphism with Clinical Phenotypes in Schizophrenia and Bipolar Disorder. Schizophr. Res. 2008, 103, 186–191. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, K.-A.; Joo, H.-J.; Lee, H.-M.; Park, J.-Y. Influence of ABCB1 and CYP3A5 Genetic Polymorphisms on the Pharmacokinetics of Quetiapine in Healthy Volunteers. Pharmacogenet. Genom. 2014, 24, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Uehlinger, C.; Crettol, S.; Chassot, P.; Brocard, M.; Koeb, L.; Brawand-Amey, M.; Eap, C.B. Increased (R)-Methadone Plasma Concentrations by Quetiapine in Cytochrome P450s and ABCB1 Genotyped Patients. J. Clin. Psychopharmacol. 2007, 27, 273–278. [Google Scholar] [CrossRef]
- Lee, H.-K.; Hu, M.; Lui, S.S.; Ho, C.-S.; Wong, C.-K.; Tomlinson, B. Effects of Polymorphisms in ABCG2, SLCO1B1, SLC10A1 and CYP2C9/19 on Plasma Concentrations of Rosuvastatin and Lipid Response in Chinese Patients. Pharmacogenomics 2013, 14, 1283–1294. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Ding, Y.; Wang, X.; Xin, W.; Du, W.; Chen, W.; Zhang, X.; Li, P. Effects of ABCG2 and SLCO1B1 Gene Variants on Inflammation Markers in Patients with Hypercholesterolemia and Diabetes Mellitus Treated with Rosuvastatin. Eur. J. Clin. Pharmacol. 2020, 76, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Saiz-Rodríguez, M.; Belmonte, C.; Román, M.; Ochoa, D.; Jiang-Zheng, C.; Koller, D.; Mejía, G.; Zubiaur, P.; Wojnicz, A.; Abad-Santos, F. Effect of ABCB1 C3435T Polymorphism on Pharmacokinetics of Antipsychotics and Antidepressants. Basic Clin. Pharmacol. Toxicol. 2018, 123, 474–485. [Google Scholar] [CrossRef]
- Lee, S.; Morris, A.; Kim, S.; Li, F.; Baumgartner, L. Impact of Quetiapine Therapy on QTc Prolongation in Critically Ill Patients. Ann. Pharmacother. 2019, 53, 705–710. [Google Scholar] [CrossRef]
- Harrigan, E.P.; Miceli, J.J.; Anziano, R.; Watsky, E.; Reeves, K.R.; Cutler, N.R.; Sramek, J.; Shiovitz, T.; Middle, M. A Randomized Evaluation of the Effects of Six Antipsychotic Agents on QTc, in the Absence and Presence of Metabolic Inhibition. J. Clin. Psychopharmacol. 2004, 24, 62–69. [Google Scholar] [CrossRef]
- Peridy, E.; Hamel, J.-F.; Rolland, A.-L.; Gohier, B.; Boels, D. Quetiapine Poisoning and Factors Influencing Severity. J. Clin. Psychopharmacol. 2019, 39, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Herink, M.C.; Irwin, A.N.; Zumach, G.M. FDA Breakthrough Therapy Designation: Evaluating the Quality of the Evidence behind the Drug Approvals. Pharmacotherapy 2018, 38, 967–980. [Google Scholar] [CrossRef] [PubMed]



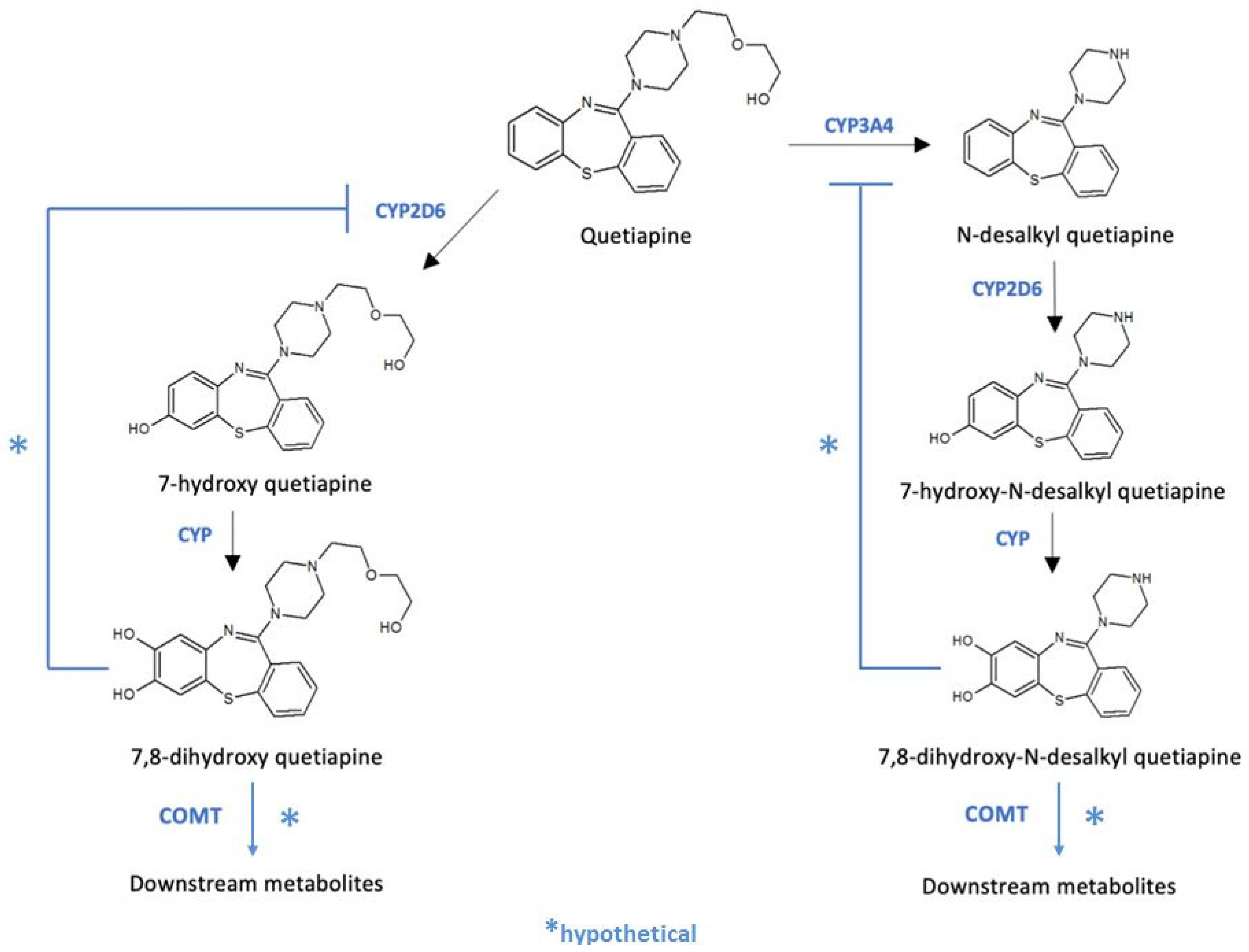{kind=link}
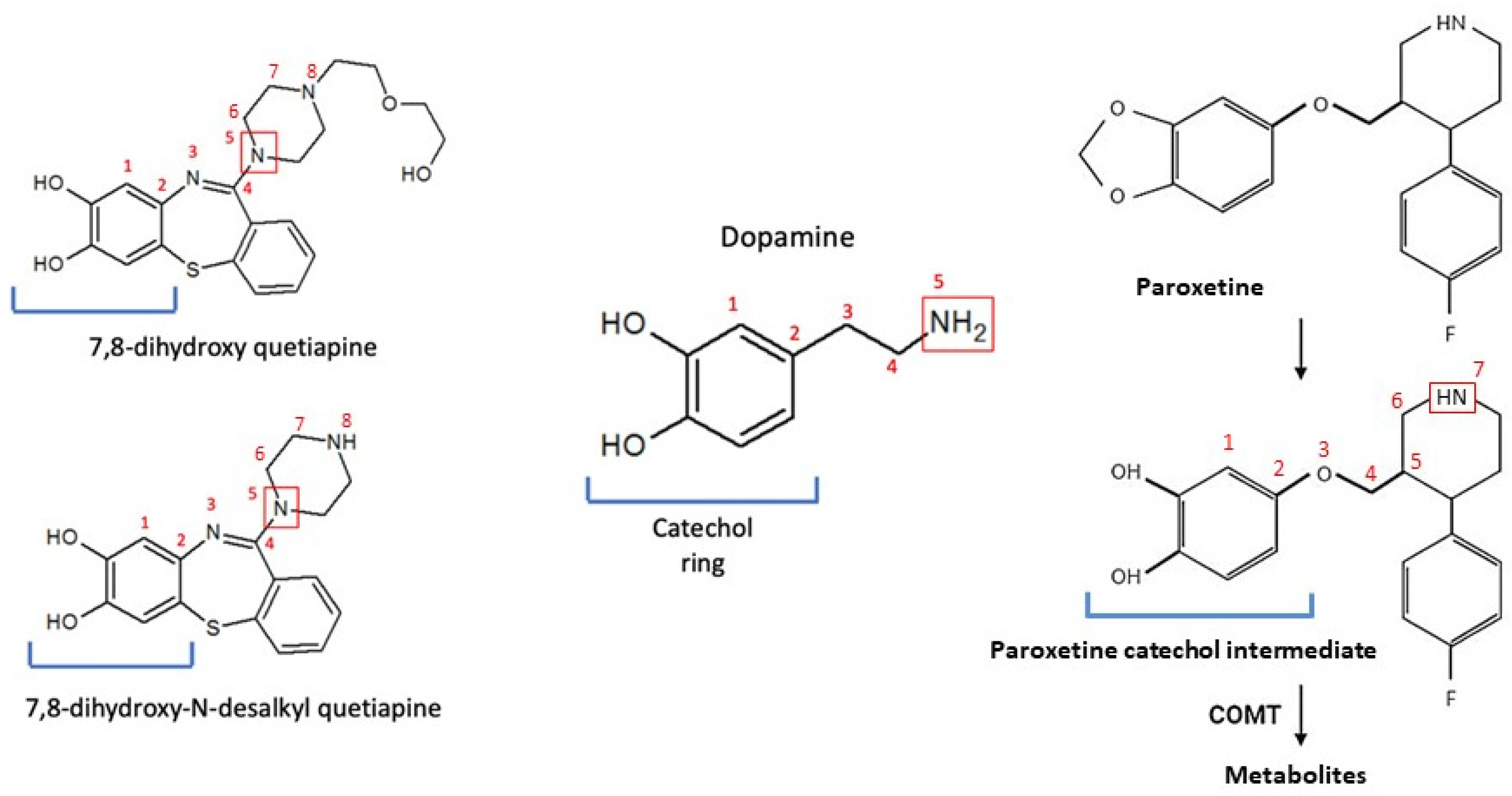{kind=link}
| Gene | Allele | Variant | Gene | Allele | Variant | Gene | Allele | Variant |
|---|---|---|---|---|---|---|---|---|
| ABCB1 | C3435T | rs1045642 | CYP2C8 | * 2 | rs11572103 | CYP3A4 | * 3 | rs4986910 |
| G2677 T/A | rs2032582 | * 3 | rs10509681 | * 2 | rs55785340 | |||
| G2677 T/A | rs2032582 | * 3 | rs11572080 | * 6 | rs4646438 | |||
| C1236T | rs1128503 | * 4 | rs1058930 | * 18 | rs28371759 | |||
| ABCG2 | rs2231142 | CYP2C9 | * 2 | rs1799853 | * 22 | rs35599367 | ||
| ABCC2 | rs2273697 | * 3 | rs1057910 | CYP3A5 | * 3 | rs776746 | ||
| COMT | rs4680 | * 5 | rs28371686 | * 6 | rs10264272 | |||
| rs13306278 | * 8 | rs9332094 | * 7 | rs41303343 | ||||
| CYP1A2 | * 1C | rs2069514 | * 8 | rs7900194 | SCL28A3 | rs7853758 | ||
| * 1F | rs762551 | * 11 | rs28371685 | SLC22A1 | * 2 | rs72552763 | ||
| * 1B | rs2470890 | CYP2D6 | * 3 | rs35742686 | * 3 | rs12208357 | ||
| CYP2A6 | * 9 | rs28399433 | * 4 | rs3892097 | * 5 | rs34059508 | ||
| CYP2B6 | * 9 | rs3745274 | * 6 | rs5030655 | SLCO1B1 | * 5 | rs4149056 | |
| * 5 | rs3211371 | * 7 | rs5030867 | * 1b | rs2306283 | |||
| rs4803419 | * 8 | rs5030865 | rs4149015 | |||||
| * 4 | rs2279343 | * 9 | rs5030656 | * 2 | rs56101265 | |||
| * 22 | rs34223104 | * 10 | rs1065852 | * 3 | rs56061388 | |||
| * 18 | rs28399499 | * 10 | rs1135840 | * 6 | rs55901008 | |||
| CYP2C19 | * 2 | rs4244285 | * 12 | rs5030862 | * 9 | rs59502379 | ||
| * 3 | rs4986893 | * 14 | rs5030865 | * 10 | rs56199088 | |||
| * 4 | rs28399504 | * 15 | rs774671100 | rs11045879 | ||||
| * 6 | rs72552267 | * 17 | rs28371706 | UGT1A1 | * 6 | rs4148323 | ||
| * 5 | rs56337013 | * 19 | rs72549353 | * 80 | rs887829 | |||
| * 7 | rs72558186 | * 29 | rs59421388 | UGT1A4 | rs2011425 | |||
| * 8 | rs41291556 | * 41 | rs28371725 | UGT2B15 | rs1902023 | |||
| * 9 | rs17884712 | * 56B | rs72549347 | |||||
| * 17 | rs12248560 | * 59 | rs79292917 | |||||
| * 35 | rs12769205 |
| Variable | N | Age | Weight (kg) | Height (cm) | BMI |
|---|---|---|---|---|---|
| Total | 49 | 31.7 (9.1) | 69.2 (13.1) | 167.9 (11.2) | 24.4 (2.7) |
| Sex | |||||
| Male | 28 | 31.2 (9.1) | 75.2 (12.0) | 175.0 (9.0) | 24.5 (2.8) |
| Female | 21 | 32.3 (9.8) | 61.1 (9.8) * | 158.5 (5.4) * | 24.2 (2.9) |
| Race | |||||
| Caucasian | 9 | 32.6 (12.6) | 70.1 (16.0) | 172.0 (14.6) | 23.4 (2.9) |
| Latino-American | 40 | 31.5 (8.6) | 69.0 (12.3) | 167.0 (10.3) | 24.6 (2.7) |
| Variable | N | AUC0–¥/DW | Cmax/DW | tmax (h) | t1/2 (h) | Vd/Fw (L/kg) | Cl/Fw (L/h·kg) |
|---|---|---|---|---|---|---|---|
| (kg·ng·h/mL mg) | (kg·ng/mL mg) | ||||||
| Total | 49 | 746.6 (378.7) | 201.61 (90.57) | 1.34 (1.00) | 4.98 (1.18) | 12.04 (6.49) | 1.78 (1.04) |
| Sex: | |||||||
| Male | 28 | 733.09 (379.70) | 210.92 (96.83) | 1.34 (1.00) | 5.01 (0.93) | 12.16 (6.07) | 1.75 (0.90) |
| Female | 21 | 764.52 (385.94) | 189.20 (82.15) | 1.33 (0.95) | 4.94 (1.41) | 11.89 (7.16) | 1.83 (1.23) |
| Race: | |||||||
| Caucasian | 9 | 677.87 (409.88) | 212.51 (124.11) | 1.63 (1.22) | 4.57 (1.30) | 14.39 (11.05) | 2.31 (1.71) |
| Latino-American | 40 | 762.02 (375.11) | 199.16 (83.08) | 1.27 (0.91) | 5.07 (1.11) | 11.52 (5.02) | 1.66 (0.81) |
| Clinical trial: | |||||||
| 1 | 19 | 699.87 (306.78) | 203.71 (92.80) | 1.61 (1.29) | 4.57 (1.19) | 10.81 (4.69) | 1.79 (1.03) |
| 2 | 30 | 776.13 (420.26) | 200.28 (90.70) | 1.17 (0.67) | 5.24 (1.06) * | 12.83 (7.37) | 1.77 (1.07) |
| Genotype or Phenotype | n | AUC0–∞/DW | Cmax/DW | tmax (h) | t1/2 (h) | Vd/Fw (L/kg) | Cl/Fw (L/h·kg) |
|---|---|---|---|---|---|---|---|
| (kg·ng·h/mL·mg) | (kg·ng/mL·mg) | ||||||
| Total | 49 | 746.56 (378.69) | 201.61 (90.57) | 1.34 (0.97) | 4.98 (1.14) | 12.05 (6.49) | 1.78 (1.04) |
| ABCG2 rs2231142: | |||||||
| G/G | 35 | 686.66 (322.63) | 191.4 (86.82) | 1.34 (1.01) | 5.04 (1.10) | 13.23 (7.11) | 1.9 (1.09) |
| G/T | 12 | 855.47 (392.98) | 215.57 (95.05) | 1.41 (0.94) | 5.01 (1.08) | 9.65 (2.96) | 1.48 (0.85) |
| T/T | 2 | 1141.53 (1032.36) | 296.58 (118.52) | 0.79 (0.41) | 3.67 (2.25) | 5.72 (2.22) $ | 1.49 (1.33) |
| COMT rs13306278: | |||||||
| C/C | 42 | 683.92 (345.07) | 190.26 (91.57) | 1.28 (0.95) | 4.89 (1.09) | 12.74 (6.66) | 1.91 (1.06) |
| C/T | 7 | 1122.43 (375.92) * | 269.71 (44.91)* | 1.67 (1.13) | 5.52 (1.41) | 7.87 (3.11) *, $ | 1.01 (0.4) *, $ |
| CYP2B6 phenotype: | |||||||
| RM | 8 | 698.71 (354.07) | 199.1 (99.02) | 1.73 (1.52) | 5.44 (1.08) | 13.45 (5.81) | 1.81 (0.93) |
| NM | 13 | 701.77 (315.35) | 203.28 (87.63) | 1.26 (1.01) | 4.72 (0.53) | 12.6 (8.09) | 1.83 (1.12) |
| IM | 23 | 751.89 (399) | 204.64 (96.92) | 1.18 (0.68) | 4.66 (1.19) | 10.7 (4.57) | 1.76 (1.03) |
| PM | 5 | 915.08 (529.9) | 187.36 (79.06) | 1.63 (1.08) | 6.38 (1.18) **, $ | 14.54 (10.55) | 1.7 (1.39) |
| CYP3A5 phenotype: | |||||||
| NM + IM | 16 | 619.19 (347.49) | 170.99 (81.41) | 1.2 (0.88) | 4.5 (1.4) | 12.77 (6.8) | 2.17 (1.22) |
| PM | 33 | 808.32 (382.71) | 216.46 (92.2) | 1.4 (1.02) | 5.21 (0.94) *, $ | 11.7 (6.41) | 1.59 (0.91) |
| UGT1A1 phenotype: | |||||||
| NM | 19 | 837.76 (482.78) | 208.06 (103.27) | 1.54 (1.04) | 4.92 (1.16) | 11.26 (6.66) | 1.78 (1.31) |
| IM | 22 | 745.33 (296.5) | 203.76 (84) | 1.37 (1.03) | 5.07 (1.17) | 11.36 (5.15) | 1.61 (0.73) |
| PM | 8 | 533.36 (213.91) | 180.39 (83.78) | 0.76 (0.21) *** | 4.88 (1.16) | 15.81 (8.69) | 2.25 (1.04) |
| Drop in MS Abundance between 2 h and 10 h (%) | |||||||
|---|---|---|---|---|---|---|---|
| Volunteer | COMT rs13306278 | CYP2D6 | Que. 384.3 | NorQue. 296.1 | 7-OH-Que/Que-SO. 400.2 | 7.8-diOH-Que *. 416.4 | 7.8-diOH-N-desal-Que *. 328.3 |
| A | C/C | IM | −8.30 | −11.90 | −2.80 | −2.60 | −4.60 |
| B | C/C | NM | −7.50 | −1.60 | −10.10 | 7.80 | 5.00 |
| C | C/T | NM | −21.70 | −16.10 | −31.80 | −11.50 | −12.70 |
| D | C/T | IM | −7.60 | −12.50 | −7.20 | −6.10 | −3.20 |
| E | C/C | UM | −12.30 | −25.70 | −22.60 | −4.50 | −17.20 |
| F | C/T | NM | −10.90 | −16.20 | −12.10 | −0.30 | −7.80 |
| Mean | −11.38 | −14.00 | −14.43 | −2.87 | −6.75 | ||
| SD | 5.41 | 7.83 | 10.77 | 6.45 | 7.76 | ||
| Mean COMT rs13306278 C/T | −13.40 | −14.93 | −17.03 | −5.97 | −7.90 | ||
| Mean COMT rs13306278 C/C | −9.37 | −13.07 | −11.83 | 0.23 | −5.60 | ||
| Mean CYP2D6 UM-NM | −13.10 | −14.90 | −19.15 | −2.13 | −8.18 | ||
| Mean CYP2D6 IM | −7.95 | −12.20 | −5.00 | −4.35 | −3.90 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zubiaur, P.; Fernández-Campos, P.; Navares-Gómez, M.; Soria-Chacartegui, P.; Villapalos-García, G.; Román, M.; Mejía-Abril, G.; Ochoa, D.; Abad-Santos, F. Variants in COMT, CYP3A5, CYP2B6, and ABCG2 Alter Quetiapine Pharmacokinetics. Pharmaceutics 2021, 13, 1573. https://doi.org/10.3390/pharmaceutics13101573
Zubiaur P, Fernández-Campos P, Navares-Gómez M, Soria-Chacartegui P, Villapalos-García G, Román M, Mejía-Abril G, Ochoa D, Abad-Santos F. Variants in COMT, CYP3A5, CYP2B6, and ABCG2 Alter Quetiapine Pharmacokinetics. Pharmaceutics. 2021; 13(10):1573. https://doi.org/10.3390/pharmaceutics13101573
Chicago/Turabian StyleZubiaur, Pablo, Paula Fernández-Campos, Marcos Navares-Gómez, Paula Soria-Chacartegui, Gonzalo Villapalos-García, Manuel Román, Gina Mejía-Abril, Dolores Ochoa, and Francisco Abad-Santos. 2021. "Variants in COMT, CYP3A5, CYP2B6, and ABCG2 Alter Quetiapine Pharmacokinetics" Pharmaceutics 13, no. 10: 1573. https://doi.org/10.3390/pharmaceutics13101573
APA StyleZubiaur, P., Fernández-Campos, P., Navares-Gómez, M., Soria-Chacartegui, P., Villapalos-García, G., Román, M., Mejía-Abril, G., Ochoa, D., & Abad-Santos, F. (2021). Variants in COMT, CYP3A5, CYP2B6, and ABCG2 Alter Quetiapine Pharmacokinetics. Pharmaceutics, 13(10), 1573. https://doi.org/10.3390/pharmaceutics13101573