
Tuning the Cytotoxicity of Bis-Phosphino-Amines Ruthenium(II) Para-Cymene Complexes for Clinical Development in Breast Cancer

 , ,
, ,  ,
,  , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. General Procedure
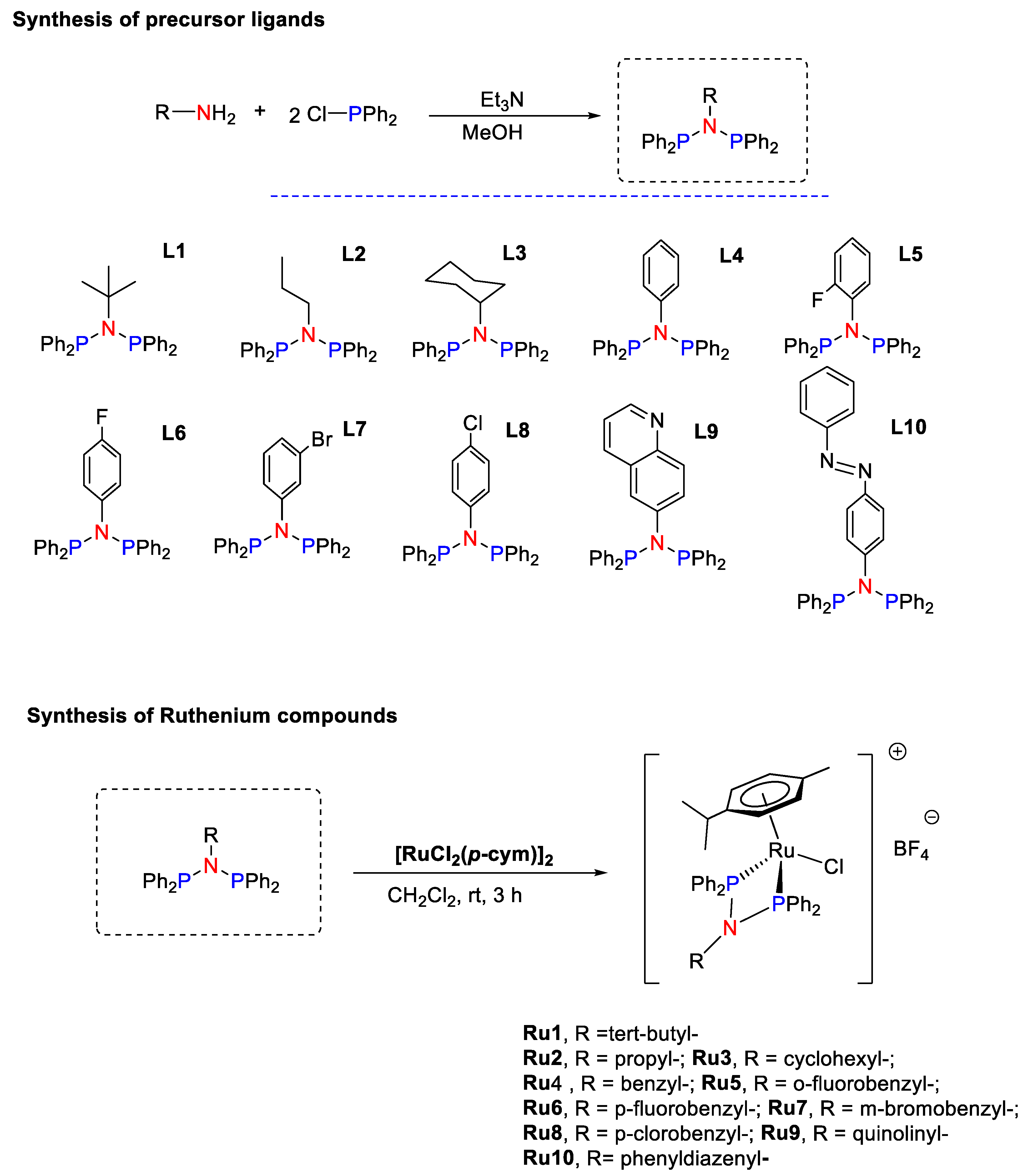
2.2. Synthesis and Characterization of Ligands L1–L10
2.3. Synthesis and Characterization of Ruthenium Compounds Ru1–Ru10
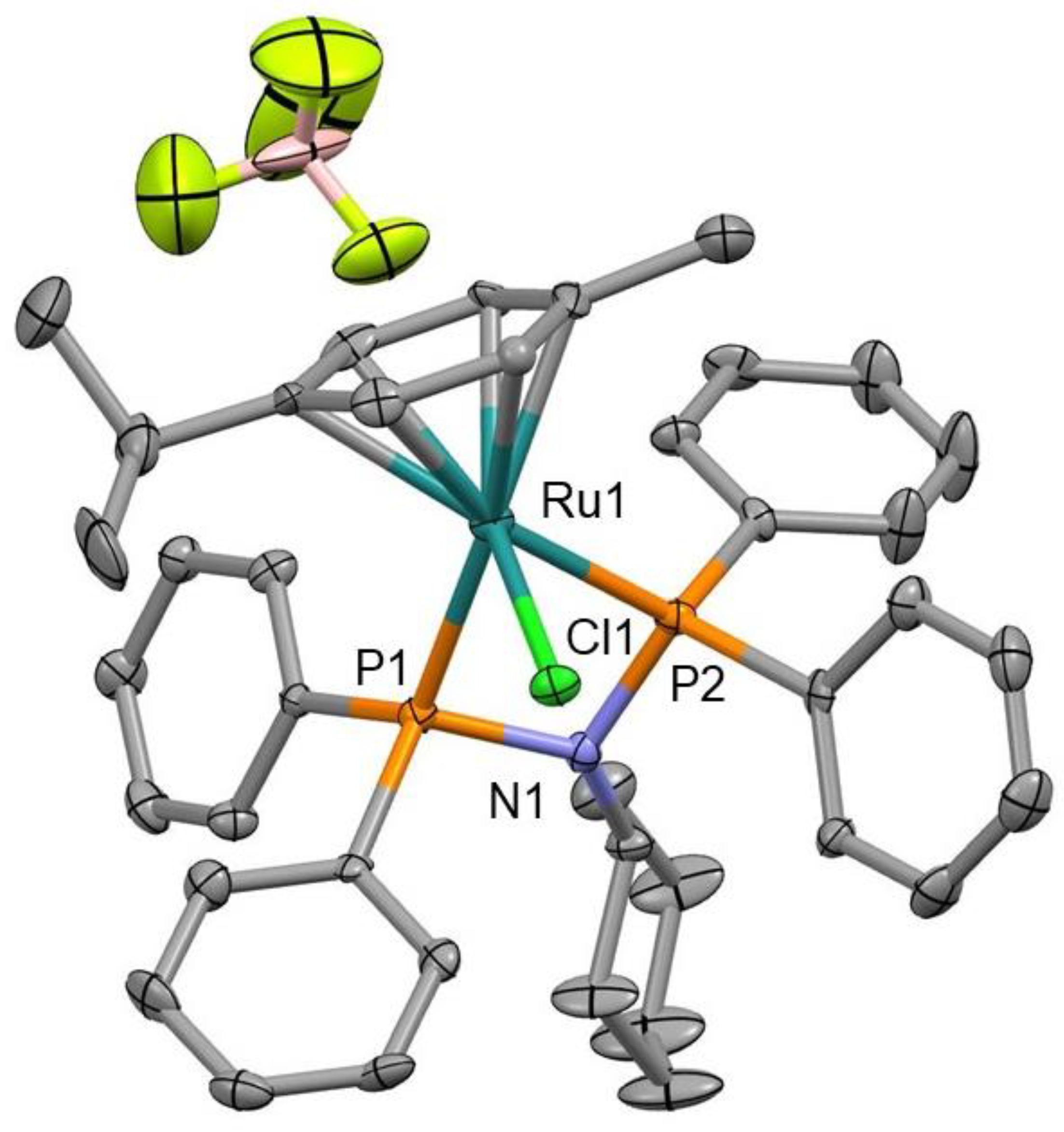
2.4. X-ray Crystallography
2.5. HSA Binding Studies by Steady-State Fluorescence Spectroscopy
2.6. Biological Assays
2.7. Statistical Analysis
3. Results
3.1. Synthesis and Characterization of Precursor Ligands and Ruthenium Compounds
3.2. Stability of the Ruthenium Complexes in Solution
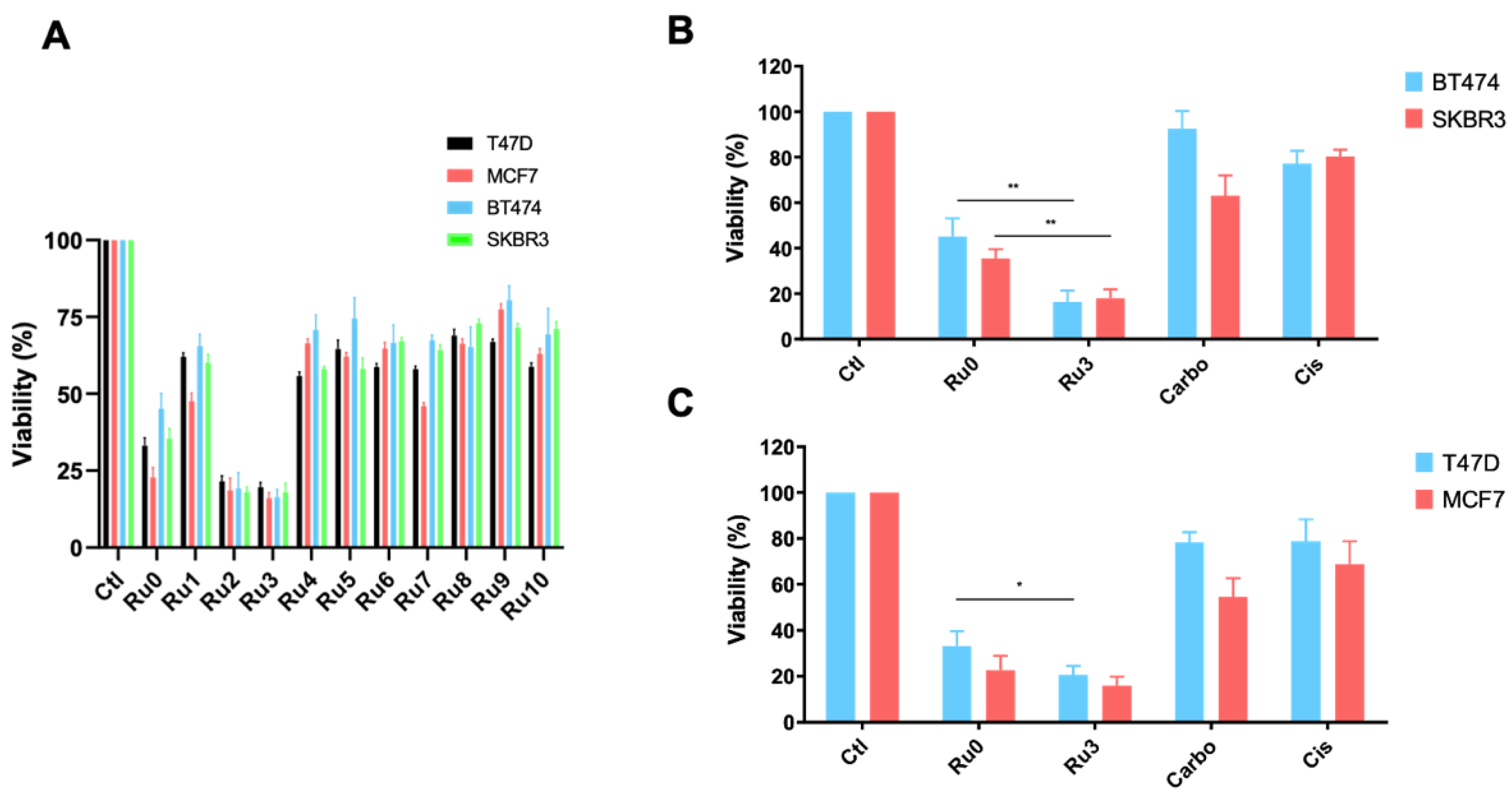
3.3. Antiproliferative Efficacy of New Synthetized Ruthenium Compounds Compared to Ru0 and Platins
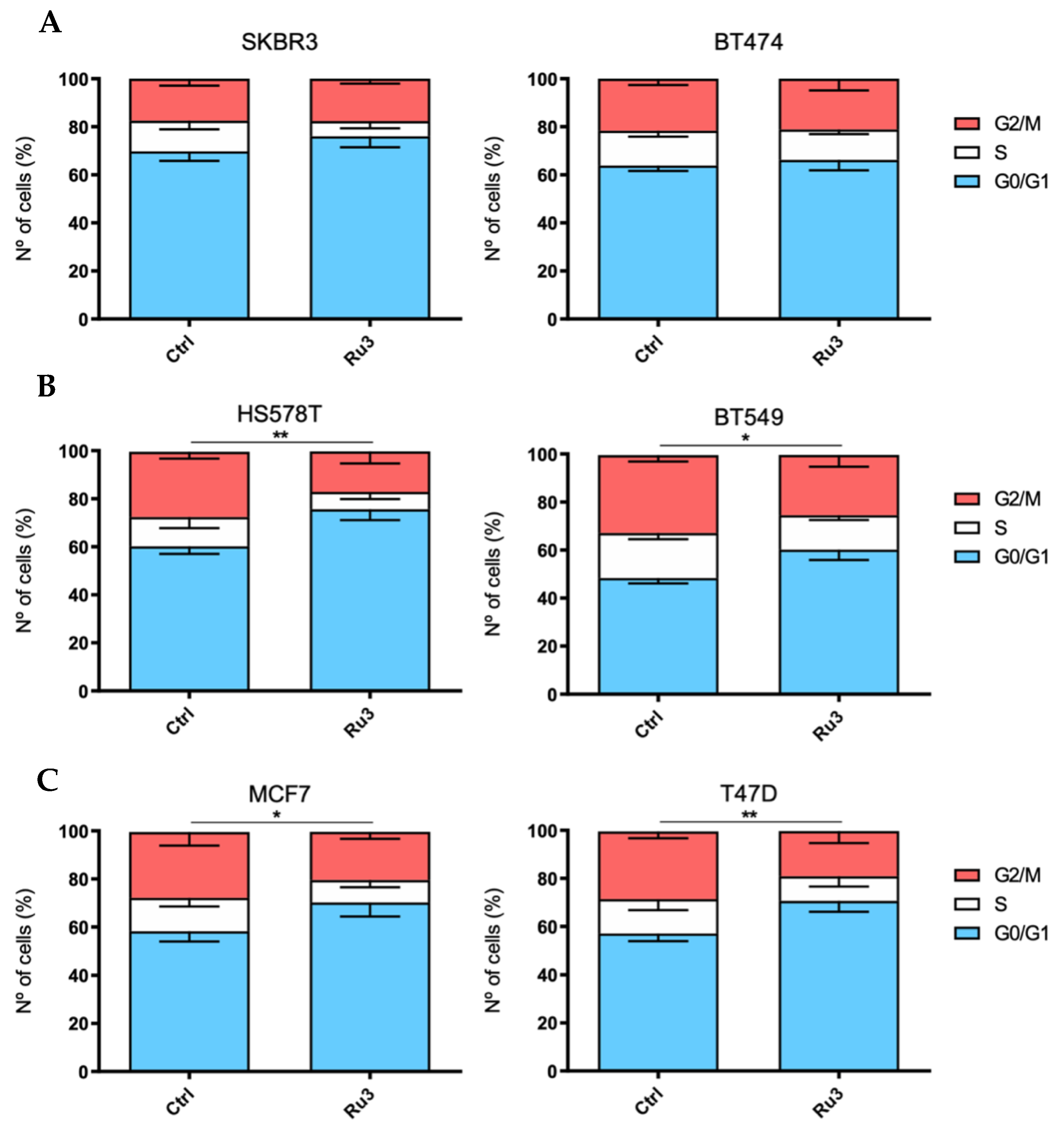
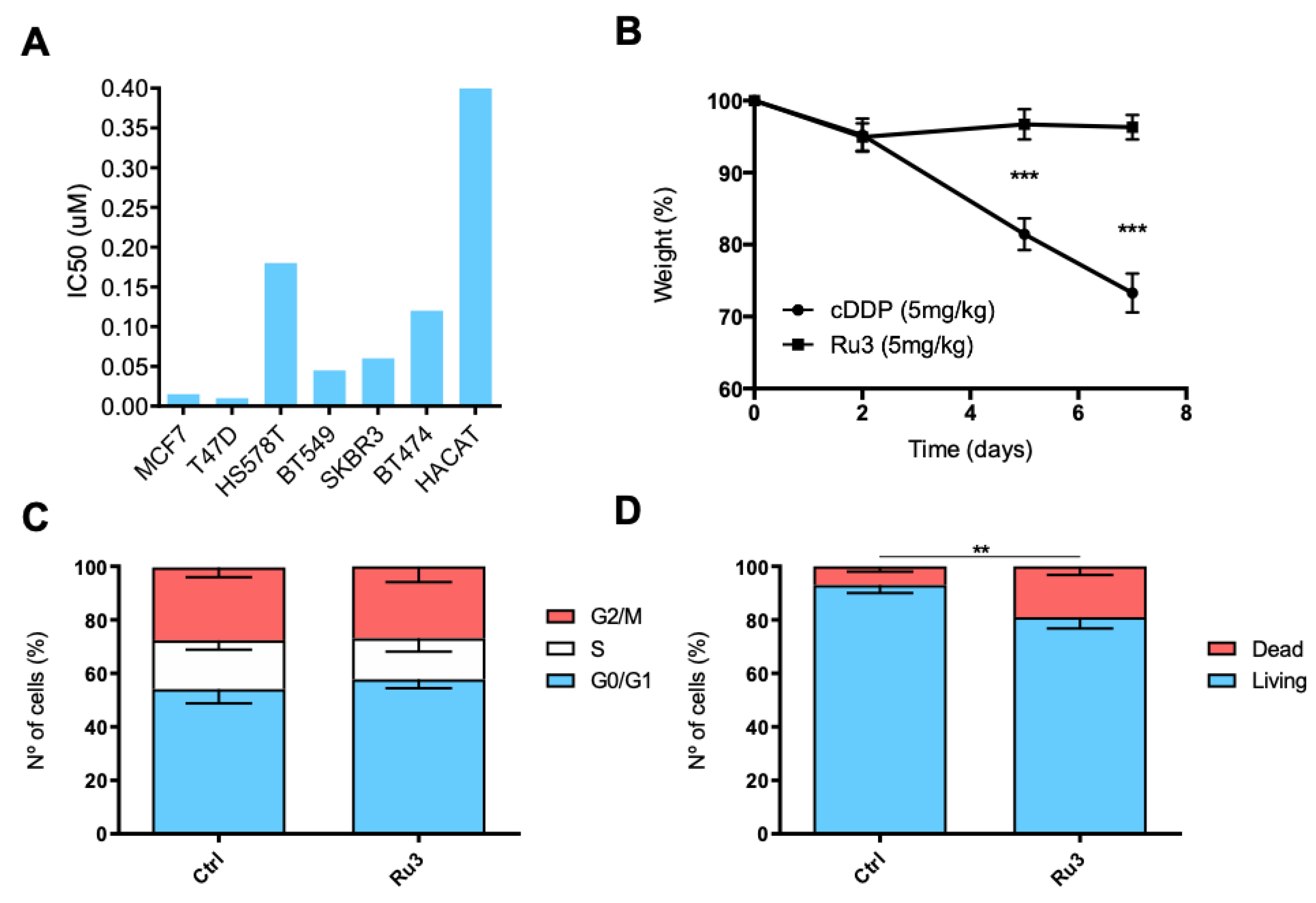
3.4. Ru3 Exerts a G2/M Arrest in Breast Cancer Cell Lines
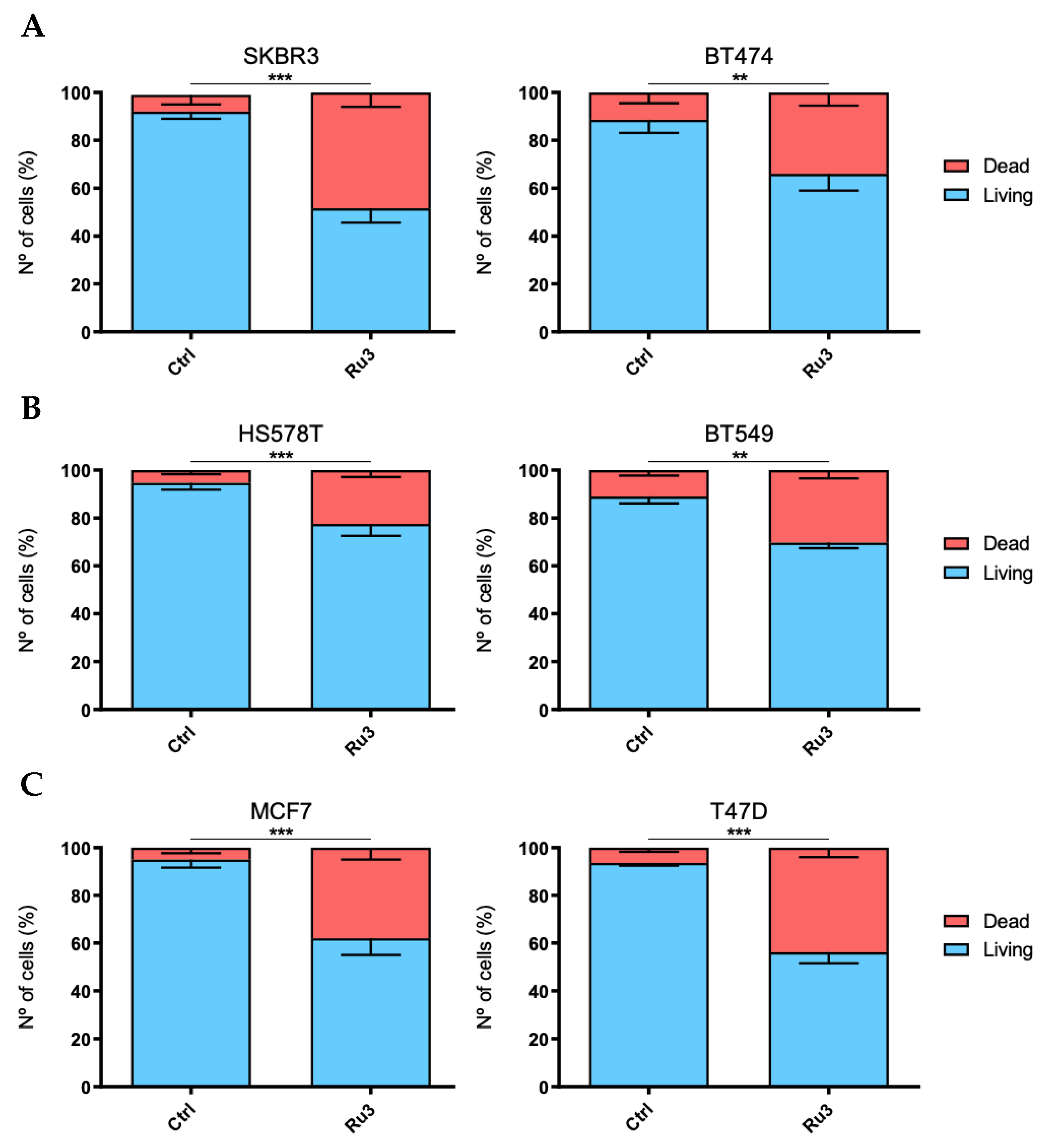
3.5. Ru3 Exerts Apoptotic-Based Cell Death in TNBC
3.6. Ru3 Is Less Toxic Than Cisplatin Using In Vitro and in In Vivo Studies
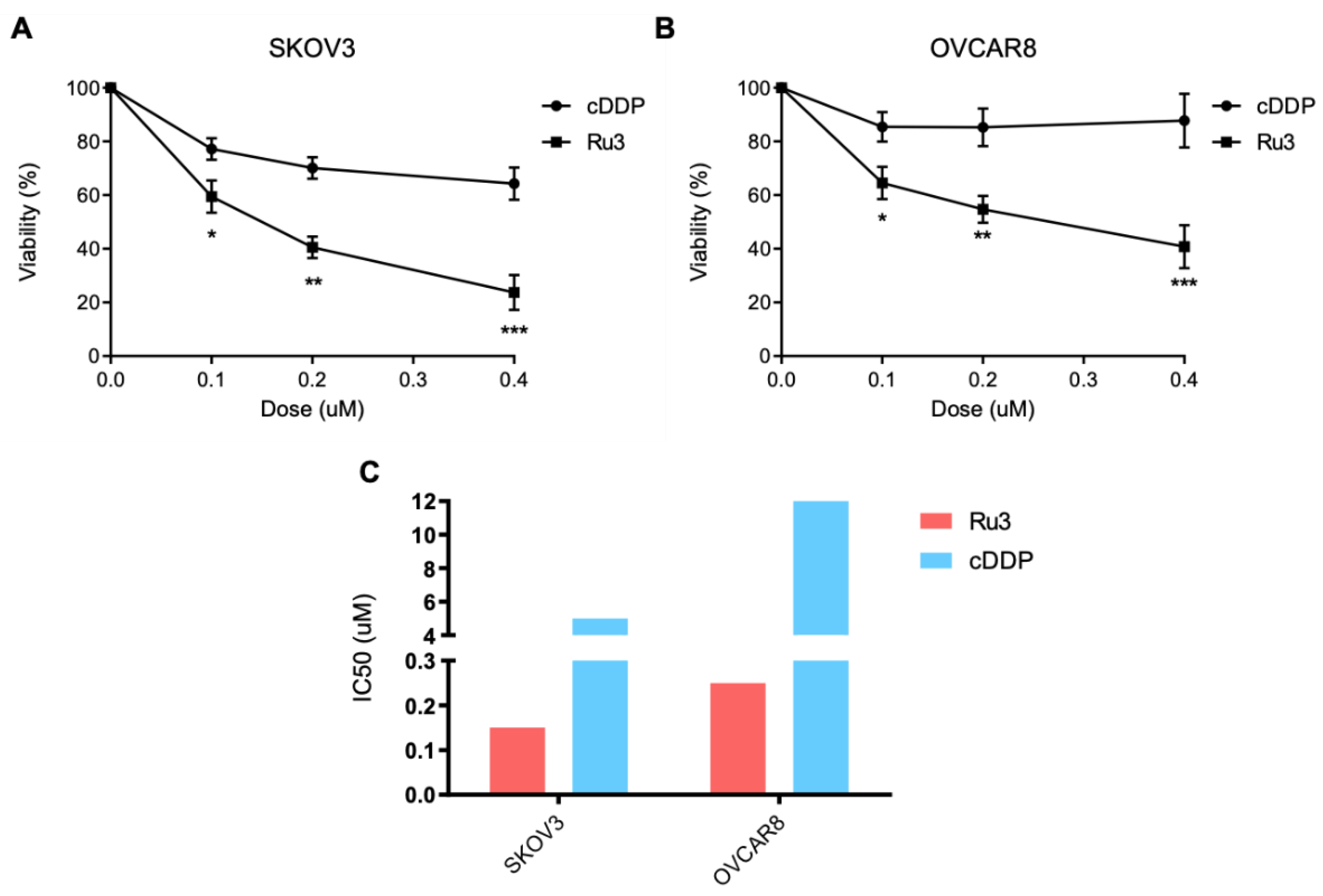
3.7. Ru3 Is Also a Therapeutic Alternative in Ovarian Cancer
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Baliu-Piqué, M.; Pandiella, A.; Ocana, A. Breast Cancer Heterogeneity and Response to Novel Therapeutics. Cancers 2020, 12, 3271. [Google Scholar] [CrossRef]
- Aldea, M.; Andre, F.; Marabelle, A.; Dogan, S.; Barlesi, F.; Soria, J.-C. Overcoming Resistance to Tumor-Targeted and Immune-Targeted Therapies. Cancer Discov. 2021, 11, 874–899. [Google Scholar] [CrossRef]
- García-Alonso, S.; Ocaña, A.; Pandiella, A. Trastuzumab Emtansine: Mechanisms of Action and Resistance, Clinical Progress, and Beyond; Cell Press: Cambridge, MA, USA, 2020; Volume 6. [Google Scholar]
- Perou, C.M.; Børresen-Dale, A.-L. Systems Biology and Genomics of Breast Cancer. Cold Spring Harb. Perspect. Biol. 2011, 3, a003293. [Google Scholar] [CrossRef]
- Ocaña, A.; Pandiella, A. Identifying Breast Cancer Druggable Oncogenic Alterations: Lessons Learned and Future Targeted Options. Clin. Cancer Res. 2008, 14, 961–970. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ocana, A.; Pandiella, A. Targeting Oncogenic Vulnerabilities in Triple Negative Breast Cancer: Biological Bases and Ongoing Clinical Studies. Oncotarget 2017, 8, 22218–22234. [Google Scholar] [CrossRef] [PubMed]
- Schneider, B.P.; Winer, E.P.; Foulkes, W.D.; Garber, J.; Perou, C.M.; Richardson, A.; Sledge, G.W.; Carey, L.A. Triple-Negative Breast Cancer: Risk Factors to Potential Targets. Clin. Cancer Res. 2008, 14, 8010–8018. [Google Scholar] [CrossRef] [PubMed]
- Tutt, A.; Tovey, H.; Cheang, M.C.U.; Kernaghan, S.; Kilburn, L.; Gazinska, P.; Owen, J.; Abraham, J.; Barrett, S.; Barrett-Lee, P.; et al. Carboplatin in BRCA1/2-Mutated and Triple-Negative Breast Cancer BRCAness Subgroups: The TNT Trial. Nat. Med. 2018, 24, 628–637. [Google Scholar] [CrossRef]
- Tchounwou, P.B.; Dasari, S.; Noubissi, F.K.; Ray, P.; Kumar, S. Advances in Our Understanding of the Molecular Mechanisms of Action of Cisplatin in Cancer Therapy. J. Exp. Pharmacol. 2021, 13, 303–328. [Google Scholar] [CrossRef]
- Murray, B.S.; Dyson, P.J. Recent Progress in the Development of Organometallics for the Treatment of Cancer. Curr. Opin. Chem. Biol. 2020, 56, 28–34. [Google Scholar] [CrossRef]
- Yousuf, I.; Bashir, M.; Arjmand, F.; Tabassum, S. Advancement of Metal Compounds as Therapeutic and Diagnostic Metallodrugs: Current Frontiers and Future Perspectives. Coord. Chem. Rev. 2021, 445, 214104. [Google Scholar] [CrossRef]
- Chang, L.; Ruiz, P.; Ito, T.; Sellers, W.R. Targeting Pan-Essential Genes in Cancer: Challenges and Opportunities. Cancer Cell 2021, 39, 466–479. [Google Scholar] [CrossRef] [PubMed]
- Anthony, E.J.; Bolitho, E.M.; Bridgewater, H.E.; Carter, O.W.L.; Donnelly, J.M.; Imberti, C.; Lant, E.C.; Lermyte, F.; Needham, R.J.; Palau, M.; et al. Metallodrugs Are Unique: Opportunities and Challenges of Discovery and Development. Chem. Sci. 2020, 11, 12888–12917. [Google Scholar] [CrossRef] [PubMed]
- Santi, M.; Mapanao, A.K.; Biancalana, L.; Marchetti, F.; Voliani, V. Ruthenium Arene Complexes in the Treatment of 3D Models of Head and Neck Squamous Cell Carcinomas. Eur. J. Med. Chem. 2021, 212, 113143. [Google Scholar] [CrossRef] [PubMed]
- Thota, S.; Rodrigues, D.A.; Crans, D.C.; Barreiro, E.J. Ru(II) Compounds: Next-Generation Anticancer Metallotherapeutics? J. Med. Chem. 2018, 61, 5805–5821. [Google Scholar] [CrossRef] [PubMed]
- Rausch, M.; Dyson, P.J.; Nowak-Sliwinska, P. Recent Considerations in the Application of RAPTA-C for Cancer Treatment and Perspectives for Its Combination with Immunotherapies. Adv. Ther. 2019, 2, 1900042. [Google Scholar] [CrossRef]
- Elsayed, S.A.; Harrypersad, S.; Sahyon, H.A.; El-Magd, M.A.; Walsby, C.J. Ruthenium(II)/(III) DMSO-Based Complexes of 2-Aminophenyl Benzimidazole with In Vitro and In Vivo Anticancer Activity. Molecules 2020, 25, 4284. [Google Scholar] [CrossRef]
- Alessio, E.; Messori, L. NAMI-A and KP1019/1339, Two Iconic Ruthenium Anticancer Drug Candidates Face-to-Face: A Case Story in Medicinal Inorganic Chemistry. Molecules 2019, 24, 1995. [Google Scholar] [CrossRef]
- Trondl, R.; Heffeter, P.; Kowol, C.R.; Jakupec, M.A.; Berger, W.; Keppler, B.K. NKP-1339, the First Ruthenium-Based Anticancer Drug on the Edge to Clinical Application. Chem. Sci. 2014, 5, 2925–2932. [Google Scholar] [CrossRef]
- Leijen, S.; Burgers, S.A.; Baas, P.; Pluim, D.; Tibben, M.; van Werkhoven, E.; Alessio, E.; Sava, G.; Beijnen, J.H.; Schellens, J.H.M. Phase I/II Study with Ruthenium Compound NAMI-A and Gemcitabine in Patients with Non-Small Cell Lung Cancer after First Line Therapy. Investig. New Drugs 2015, 33, 201–214. [Google Scholar] [CrossRef]
- Dittrich, C.; Scheulen, M.E.; Jaehde, U.; Kynast, B.; Gneist, M.; Richly, H.; Schaad, S.; Arion, V.; Keppler, B.K. Phase I and Pharmacokinetic Study of Sodium Trans-[Tetrachlorobis(1H-Indazole)Ruthenate(III)]/Indazolehydrochloride (1:1.1) (FFC14A, KP1019) in Patients with Solid Tumors—A Study of the CESAR Central European Society for Anticancer Drug Research—EW. Cancer Res. 2005, 65, 110. [Google Scholar]
- Chamberlain, S.; Cole, H.D.; Roque, J.; Bellnier, D.; McFarland, S.A.; Shafirstein, G. TLD1433-Mediated Photodynamic Therapy with an Optical Surface Applicator in the Treatment of Lung Cancer Cells In Vitro. Pharmaceuticals 2020, 13, 137. [Google Scholar] [CrossRef] [PubMed]
- Theralase Inc. A Phase II Clinical Study of Intravesical Photodynamic Therapy in Patients With BCG-Unresponsive Non-Muscle Invasive Bladder Cancer (“NMIBC”) Or Patients Who Are Intolerant to BCG Therapy (“Study”). 2021. Available online: https://Clinicaltrials.gov (accessed on 17 June 2021).
- Gasser, G.; Ott, I.; Metzler-Nolte, N. Organometallic Anticancer Compounds. J. Med. Chem. 2011, 54, 3–25. [Google Scholar] [CrossRef] [PubMed]
- Bergamo, A.; Masi, A.; Peacock, A.F.A.; Habtemariam, A.; Sadler, P.J.; Sava, G. In Vivo Tumour and Metastasis Reduction and in Vitro Effects on Invasion Assays of the Ruthenium RM175 and Osmium AFAP51 Organometallics in the Mammary Cancer Model. J. Inorg. Biochem. 2010, 104, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.E.; Aird, R.E.; Murdoch, P.D.S.; Chen, H.; Cummings, J.; Hughes, N.D.; Parsons, S.; Parkin, A.; Boyd, G.; Jodrell, D.I.; et al. Inhibition of Cancer Cell Growth by Ruthenium(II) Arene Complexes. J. Med. Chem. 2001, 44, 3616–3621. [Google Scholar] [CrossRef] [PubMed]
- Murray, B.S.; Babak, M.V.; Hartinger, C.G.; Dyson, P.J. The Development of RAPTA Compounds for the Treatment of Tumors. Coord. Chem. Rev. 2016, 306, 86–114. [Google Scholar] [CrossRef]
- Steel, T.R.; Walsh, F.; Wieczorek-Błauż, A.; Hanif, M.; Hartinger, C.G. Monodentately-Coordinated Bioactive Moieties in Multimodal Half-Sandwich Organoruthenium Anticancer Agents. Coord. Chem. Rev. 2021, 439, 213890. [Google Scholar] [CrossRef]
- Małecka, M.; Skoczyńska, A.; Goodman, D.M.; Hartinger, C.G.; Budzisz, E. Biological Properties of Ruthenium(II)/(III) Complexes with Flavonoids as Ligands. Coord. Chem. Rev. 2021, 436, 213849. [Google Scholar] [CrossRef]
- Swaminathan, S.; Haribabu, J.; Kalagatur, N.K.; Nikhil, M.; Balakrishnan, N.; Bhuvanesh, N.S.P.; Kadirvelu, K.; Kolandaivel, P.; Karvembu, R. Tunable Anticancer Activity of Furoylthiourea-Based RuII–Arene Complexes and Their Mechanism of Action. Chem. A Eur. J. 2021, 27, 7418–7433. [Google Scholar] [CrossRef]
- Maji, M.; Acharya, S.; Maji, S.; Purkait, K.; Gupta, A.; Mukherjee, A. Differences in Stability, Cytotoxicity, and Mechanism of Action of Ru(II) and Pt(II) Complexes of a Bidentate N,O Donor Ligand. Inorg. Chem. 2020, 59, 10262–10274. [Google Scholar] [CrossRef]
- Mukherjee, A.; Acharya, S.; Purkait, K.; Chakraborty, K.; Bhattacharjee, A.; Mukherjee, A. Effect of N,N Coordination and RuII Halide Bond in Enhancing Selective Toxicity of a Tyramine-Based RuII (p-Cymene) Complex. Inorg. Chem. 2020, 59, 6581–6594. [Google Scholar] [CrossRef]
- Song, L.; Bai, H.; Liu, C.; Gong, W.; Wang, A.; Wang, L.; Zhao, Y.; Zhao, X.; Wang, H. Synthesis, Biomacromolecular Interactions, Photodynamic NO Releasing and Cellular Imaging of Two [RuCl(Qn)(Lbpy)(NO)]X Complexes. Molecules 2021, 26, 2545. [Google Scholar] [CrossRef]
- Munteanu, A.-C.; Notaro, A.; Jakubaszek, M.; Cowell, J.; Tharaud, M.; Goud, B.; Uivarosi, V.; Gasser, G. Synthesis, Characterization, Cytotoxic Activity, and Metabolic Studies of Ruthenium(II) Polypyridyl Complexes Containing Flavonoid Ligands. Inorg. Chem. 2020, 59, 4424–4434. [Google Scholar] [CrossRef]
- Golbaghi, G.; Castonguay, A. Rationally Designed Ruthenium Complexes for Breast Cancer Therapy. Molecules 2020, 25, 265. [Google Scholar] [CrossRef]
- Broomfield, L.M.; Alonso-Moreno, C.; Martin, E.; Shafir, A.; Posadas, I.; Ceña, V.; Castro-Osma, J.A. Aminophosphine Ligands as a Privileged Platform for Development of Antitumoral Ruthenium(Ii) Arene Complexes. Dalton Trans. 2017. [Google Scholar] [CrossRef]
- Corrales Sanchez, V.; Nieto-Jiménez, C.; Castro-Osma, J.A.; De Andrés, F.; Pacheco-Liñán, P.J.; Bravo, I.; Rodriguez Farinas, N.; Niza, E.; Domínguez-Jurado, E.; Lara-Sánchez, A. Screening and Preliminary Biochemical and Biological Studies of [RuCl (p-Cymene)(N, N-Bis (Diphenylphosphino)-Isopropylamine)][BF4] in Breast Cancer Models. ACS Omega 2019, 4, 13005–13014. [Google Scholar] [CrossRef]
- Sheldrick, G. Sadabs, Program for Empirical Absorption Correction of Area Detector Data; University of Göttingen: Göttingen, Germany, 1996; undefined. [Google Scholar]
- Altomare, A.; Burla, M.C.; Camalli, M.; Cascarano, G.L.; Giacovazzo, C.; Guagliardi, A.; Moliterni, A.G.G.; Polidori, G.; Spagna, R. SIR97: A New Tool for Crystal Structure Determination and Refinement. J. Appl. Cryst. 1999, 32, 115–119. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX Suite for Small-Molecule Single-Crystal Crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: New York, NY, USA; ISBN ISBN 978-0-387-31278-1.
- Zhang, M.; Lv, Q.; Yue, N.; Wang, H. Study of Fluorescence Quenching Mechanism between Quercetin and Tyrosine-H2O2-Enzyme Catalyzed Product. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2009, 72, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.Z.; Lin, Z.; Yang, L.J.; Wang, C.; Bai, C.L. Investigation of the Interaction between Acridine Orange and Bovine Serum Albumin. Talanta 1998, 47, 1223–1229. [Google Scholar] [CrossRef]
- Rimoldi, I.; Facchetti, G.; Lucchini, G.; Castiglioni, E.; Marchianò, S.; Ferri, N. In Vitro Anticancer Activity Evaluation of New Cationic Platinum(II) Complexes Based on Imidazole Moiety. Bioorg. Med. Chem. 2017, 25, 1907–1913. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, I.D.L.; Marszaukowski, F.; Ribeiro, R.; de Lazaro, S.R.; de Oliveira, K.M.; Batista, A.A.; Castellen, P.; Wrobel, E.; Garcia, J.R.; Boeré, R.T.; et al. Synthesis and Characterization of H6-p-Cymene Ruthenium(II) Complexes Containing Alkyl- and Methoxy-Substituted Triarylphosphines. J. Organomet. Chem. 2021, 931, 121599. [Google Scholar] [CrossRef]
- Higuera-Padilla, A.R.; Batista, A.A.; Colina-Vegas, L.; Villarreal, W.; Colnago, L.A. Synthesis of the [(H6-p-Cymene)Ru(Dppb)Cl]PF6 Complex and Catalytic Activity in the Transfer Hydrogenation of Ketones. J. Coord. Chem. 2017, 70, 3541–3551. [Google Scholar] [CrossRef]
- Sari, O.; Schüttler, A.; Lönnecke, P.; Bednarski, P.J.; Hey-Hawkins, E.; Karakus, M. Synthesis, Structure and in Vitro Anticancer Activity of Ruthenium(II) and Platinum(II) Complexes with Chiral Aminophosphine Ligands. Transit. Met. Chem. 2021, 46, 299–305. [Google Scholar] [CrossRef]
- Diaz, A.A.; Young, J.D.; Khan, M.A.; Wehmschulte, R.J. Facile Synthesis of Unsymmetrical 9-Phospha- and 9-Arsafluorenes. Inorg. Chem. 2006, 45, 5568–5575. [Google Scholar] [CrossRef]
- Mazzeo, P.P.; Bacchi, A.; Pelagatti, P. Crystal Engineering Guidelines for Ruthenium Based Wheel-and-Axle Compounds. Coord. Chem. Rev. 2020, 414, 213302. [Google Scholar] [CrossRef]
- Biancalana, L.; Zacchini, S.; Ferri, N.; Lupo, M.G.; Pampaloni, G.; Marchetti, F. Tuning the Cytotoxicity of Ruthenium(II) Para-Cymene Complexes by Mono-Substitution at a Triphenylphosphine/Phenoxydiphenylphosphine Ligand. Dalton Trans. 2017, 46, 16589–16604. [Google Scholar] [CrossRef]
- Adhireksan, Z.; Davey, G.E.; Campomanes, P.; Groessl, M.; Clavel, C.M.; Yu, H.; Nazarov, A.A.; Yeo, C.H.F.; Ang, W.H.; Dröge, P.; et al. Ligand Substitutions between Ruthenium-Cymene Compounds Can Control Protein versus DNA Targeting and Anticancer Activity. Nat. Commun. 2014, 5, 3462. [Google Scholar] [CrossRef]
- Martínez-Alonso, M.; Busto, N.; Jalón, F.A.; Manzano, B.R.; Leal, J.M.; Rodríguez, A.M.; García, B.; Espino, G. Derivation of Structure-Activity Relationships from the Anticancer Properties of Ruthenium(II) Arene Complexes with 2-Aryldiazole Ligands. Inorg. Chem. 2014, 53, 11274–11288. [Google Scholar] [CrossRef]
- Pettinari, R.; Marchetti, F.; Condello, F.; Pettinari, C.; Lupidi, G.; Scopelliti, R.; Mukhopadhyay, S.; Riedel, T.; Dyson, P.J. Ruthenium(II)–Arene RAPTA Type Complexes Containing Curcumin and Bisdemethoxycurcumin Display Potent and Selective Anticancer Activity. Organometallics 2014, 33, 3709–3715. [Google Scholar] [CrossRef]
- Cimas, F.J.; Niza, E.; Juan, A.; Noblejas-López, M.D.M.; Bravo, I.; Lara-Sanchez, A.; Alonso-Moreno, C.; Ocaña, A. Controlled Delivery of BET-PROTACs: In Vitro Evaluation of MZ1-Loaded Polymeric Antibody Conjugated Nanoparticles in Breast Cancer. Pharmaceutics 2020, 12, 986. [Google Scholar] [CrossRef] [PubMed]
- Niza, E.; Noblejas-lópez, M.D.M.; Bravo, I.; Nieto-jiménez, C.; Castro-osma, J.A.; Canales-vázquez, J.; Lara-sanchez, A.; Moya, E.M.G.; Burgos, M.; Ocaña, A.; et al. Trastuzumab-Targeted Biodegradable Nanoparticles for Enhanced Delivery of Dasatinib in HER2+ Metastasic Breast Cancer. Nanomaterials 2019, 9, 1793. [Google Scholar] [CrossRef] [PubMed]
- Niza, E.; Nieto-Jiménez, C.; Noblejas-López, M.D.M.; Bravo, I.; Castro-Osma, J.A.; De La Cruz-Martínez, F.; Martínez de Sarasa Buchaca, M.; Posadas, I.; Canales-Vázquez, J.; Lara-Sanchez, A. Poly (Cyclohexene Phthalate) Nanoparticles for Controlled Dasatinib Delivery in Breast Cancer Therapy. Nanomaterials 2019, 9, 1208. [Google Scholar] [CrossRef] [PubMed]
- Manzano, A.; Ocaña, A. Antibody-Drug Conjugates: A Promising Novel Therapy for the Treatment of Ovarian Cancer. Cancers 2020, 12, 2223. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | IC50 (nM) | ||
|---|---|---|---|
| Ru0 | Ru2 | Ru3 | |
| HACAT | - | - | 350 |
| MCF7 | 300 | 45 | 15 |
| T47D | 450 | 60 | 10 |
| HS578T | - | - | 250 |
| BT549 | - | - | 45 |
| SKBR3 | 700 | 80 | 60 |
| BT474 | 800 | 180 | 120 |
| OVCAR8 | - | - | 300 |
| SKOV3 | - | - | 170 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Domínguez-Jurado, E.; Cimas, F.J.; Castro-Osma, J.A.; Juan, A.; Lara-Sánchez, A.; Rodríguez-Diéguez, A.; Shafir, A.; Ocaña, A.; Alonso-Moreno, C. Tuning the Cytotoxicity of Bis-Phosphino-Amines Ruthenium(II) Para-Cymene Complexes for Clinical Development in Breast Cancer. Pharmaceutics 2021, 13, 1559. https://doi.org/10.3390/pharmaceutics13101559
Domínguez-Jurado E, Cimas FJ, Castro-Osma JA, Juan A, Lara-Sánchez A, Rodríguez-Diéguez A, Shafir A, Ocaña A, Alonso-Moreno C. Tuning the Cytotoxicity of Bis-Phosphino-Amines Ruthenium(II) Para-Cymene Complexes for Clinical Development in Breast Cancer. Pharmaceutics. 2021; 13(10):1559. https://doi.org/10.3390/pharmaceutics13101559
Chicago/Turabian StyleDomínguez-Jurado, Elena, Francisco J. Cimas, José Antonio Castro-Osma, Alberto Juan, Agustín Lara-Sánchez, Antonio Rodríguez-Diéguez, Alexandr Shafir, Alberto Ocaña, and Carlos Alonso-Moreno. 2021. "Tuning the Cytotoxicity of Bis-Phosphino-Amines Ruthenium(II) Para-Cymene Complexes for Clinical Development in Breast Cancer" Pharmaceutics 13, no. 10: 1559. https://doi.org/10.3390/pharmaceutics13101559
APA StyleDomínguez-Jurado, E., Cimas, F. J., Castro-Osma, J. A., Juan, A., Lara-Sánchez, A., Rodríguez-Diéguez, A., Shafir, A., Ocaña, A., & Alonso-Moreno, C. (2021). Tuning the Cytotoxicity of Bis-Phosphino-Amines Ruthenium(II) Para-Cymene Complexes for Clinical Development in Breast Cancer. Pharmaceutics, 13(10), 1559. https://doi.org/10.3390/pharmaceutics13101559