Determinants of Intraparenchymal Infusion Distributions: Modeling and Analyses of Human Glioblastoma Trials

Abstract
1. Introduction
2. Materials and Methods
2.1. Simulation Models and Methods
2.1.1. Mathematical Modeling of CED
2.1.2. Simulation Methods
2.2. Experimental Methods
2.2.1. Infusion Protocols
2.2.2. Imaging Protocols
2.2.3. Concentration Measurement
2.2.4. Distribution Volume Measurement
3. Results
3.1. Descriptive Results
3.2. Statistics of Infusions and Simulations
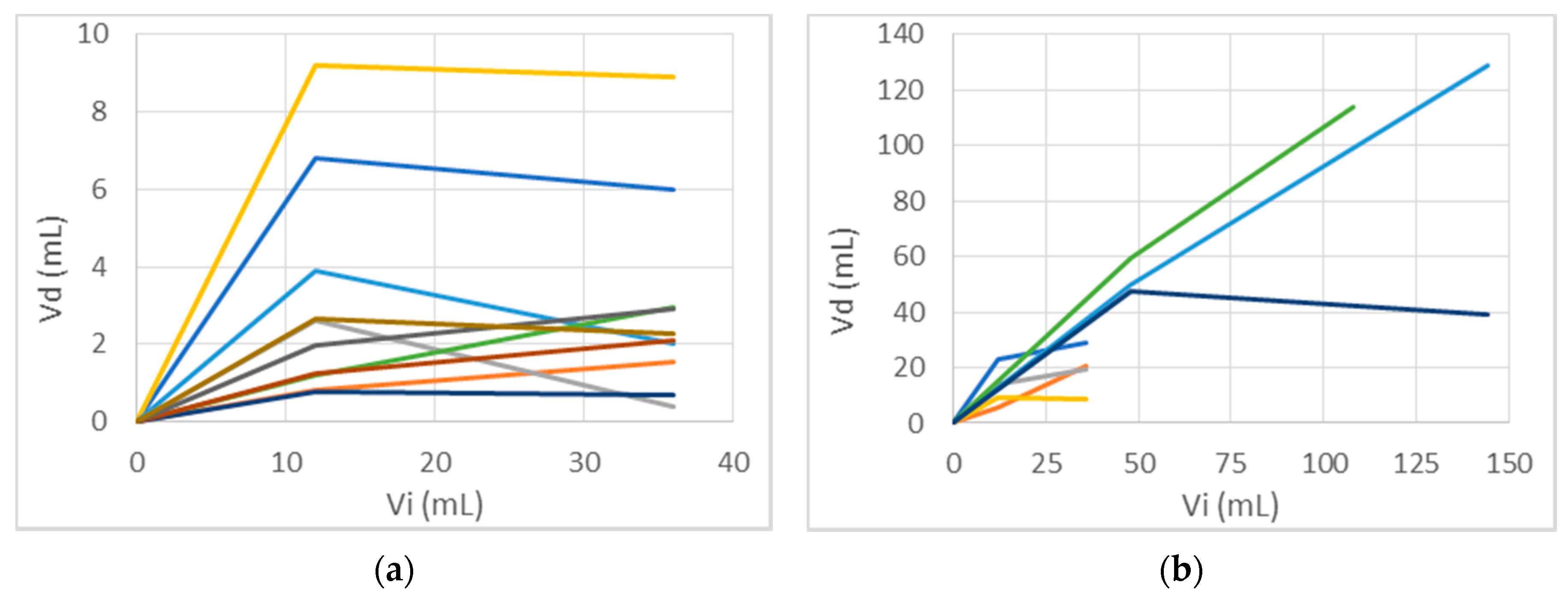
3.2.1. Total Distribution Volume
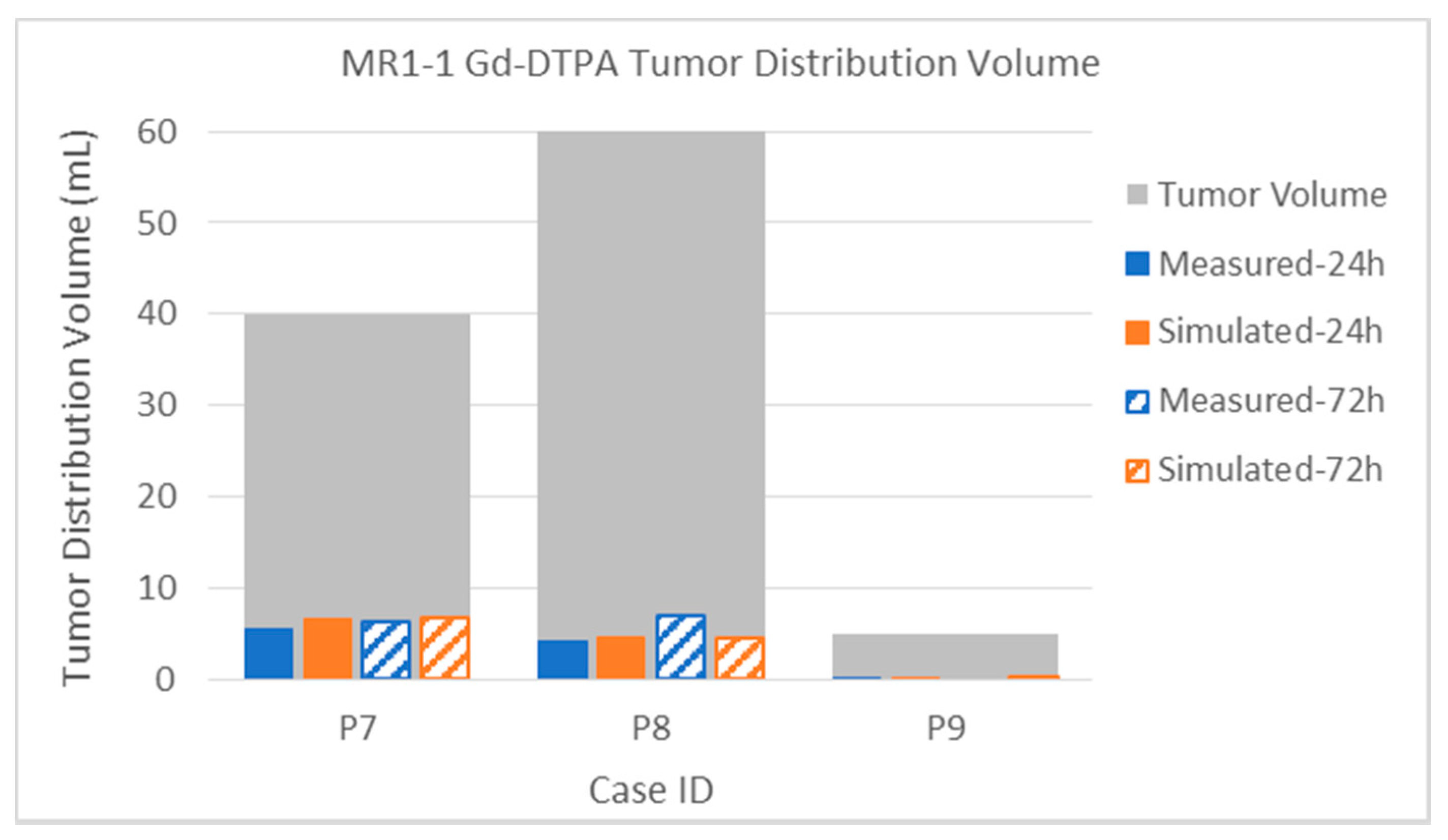
3.2.2. Tumor Distribution Volume
3.2.3. Error Measure
4. Discussion
4.1. Quantitative Results
4.2. Qualitative Conclusions
- Anisotropic distribution arises from expansion of the intercellular space and corresponding increase in hydraulic conductivity rather than from cell geometry and directionality of the brain pathways.
- Pre-infusion DTI does not directly predict distribution anisotropy.
- Advection is size-independent, over a range of sizes from small molecules to large proteins and beyond.
- Capillary losses result in widely varying residence times in tissue.
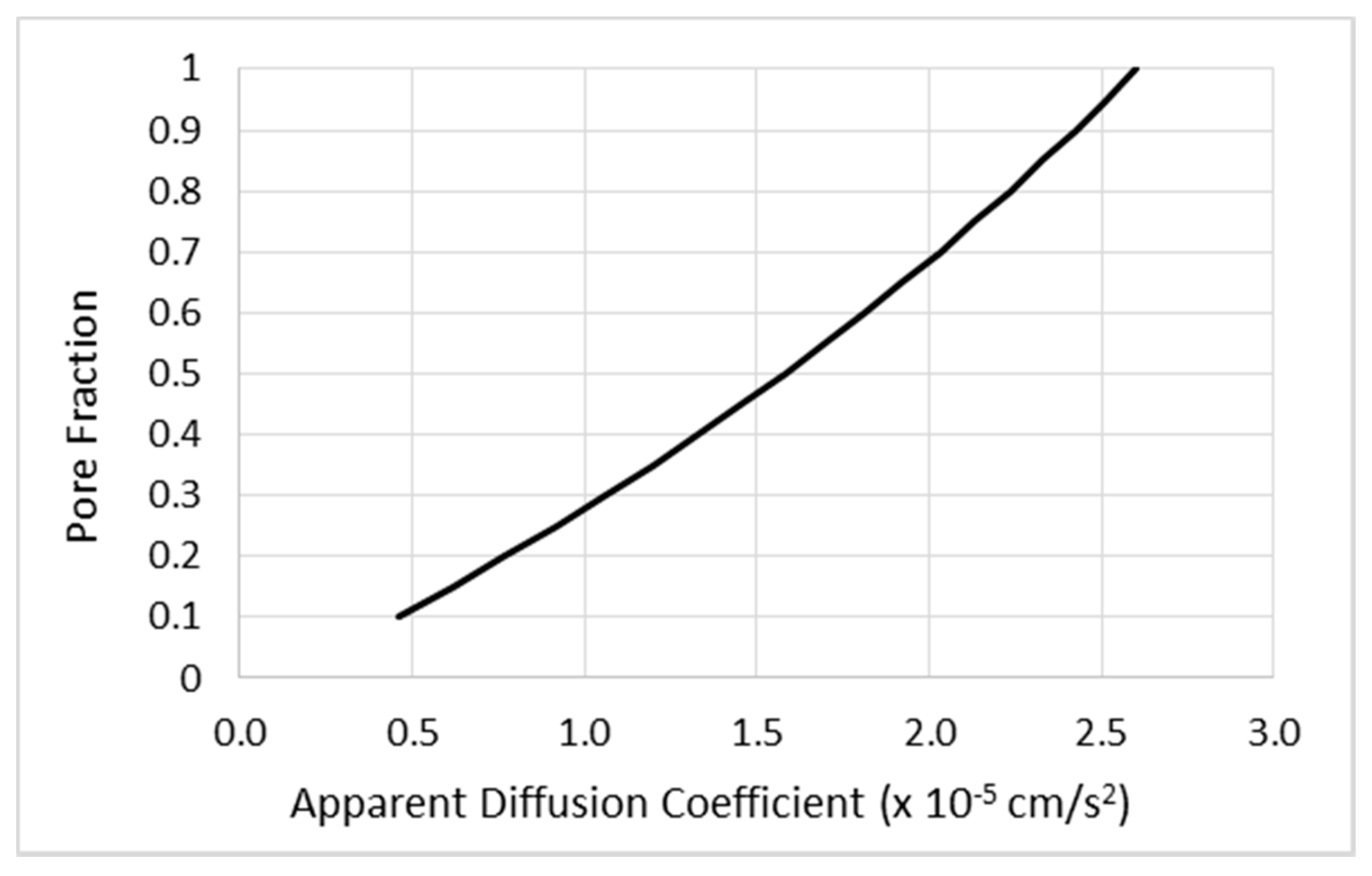
- Diffusion effects are not a significant part of CED infusion distribution for “biologics” or protein-sized molecules.
- Saturation of distribution volume.
4.3. Phenomena Omitted
- Perivascular and glymphatic spaces
- The tumor interstitium
- Binding of drug molecules.
5. Conclusions
- Interstitium responses to infusion of fluid: Simulations and planning can reduce variability. Figure 18 illustrates an approach we hope to pursue and test in the future. An a priori plan (top) is used, in conjunction with neurosurgical expertise, to position the catheters to obtain best coverage of the target region. The infusion proceeds and a surrogate tracer is imaged (if conjugating the actual therapeutic particle with an imaging reagent is not possible).
- Losses and binding: Compromised BBB has significant effects on small molecules, so that their distribution is quite different from that of larger particles in areas of high BBB permeability. The principal uncertainty in quantification of the DCE is the arterial input function, as has been noted many times in the literature (see also Section 5.1.2). To quantify the losses of a range of small chemotherapeutic reagents, such as being used in other CED studies currently (see, e.g., the reviews [30,31]), more pharmacokinetic studies will be needed, as proposed in [44,45]. However, for larger biological agents, it appears these losses are not so significant that we are unable to obtain reasonably accurate simulations. We discuss binding effects above.
- Unpredictability of device-tissue interaction: We show several examples of unpredictability where we discuss our results (Section 3). The device–tissue interaction, as well as the effects of catheter geometry, are hard to model. Backflow and bubbles are both unpredictable and have large impacts on the subsequent distributions. As noted, our backflow model was an approximation to only one geometry of catheter—the cylindrical endport—and did not account for pre-stress. Both of these have very significant effects on backflow (see, for example, [32,46]). In addition, porous catheters, deemed useful for delivery over a large area with advantages of bridging over sinks [47], require a quite different model for outflow from the catheter [48]. However, in these large infusions in human brain, the effects matter less as long as the infusion does not start close to CSF spaces. It would be more robust to measure the flux at the start of the infusions for distribution estimates. We have developed and tested these ideas in animal models: the experiments have been reported in [36,49,50]; and the theory behind it as well as some of the results have appeared in [24]. The protocol for the studies reported here did not allow for application of these ideas, as discussed in Section 2.2.2, nor were the methods sufficiently developed at the time of these trials for implementation. When implemented, this would likely avoid the unpredictability arising from having to model either the specific catheter or its backflow characteristics. We mention above that priming and other methods can reduce or nearly eliminate large bubbles, but a more practicable method needs to be found. Thus, we conclude that backflow is device and device–tissue interaction-dependent and unpredictable, and it can have a large impact on the subsequent distribution. If the infusion is not close to the CSF, this tends to matter less as infusions become larger. However, backflow and device–tissue interaction will matter in small infusions and can be incorporated into the planning system by the real-time MR methods referenced above.
5.1. Future Studies
5.1.1. Modeling and Simulations
5.1.2. Imaging
5.1.3. Delivery, Devices, and Device-Tissue Interaction
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
Appendix A. Table of Data for All Infusions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CaseID | Distribution Volume (mL) | Coverage (%) | Tumor Volume (mL) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| [n catheters] | Enhancing Tumor | Margin | Total | Enhancing Tumor | Margin | Enhancing | Margin | |||||||
| 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | |||||
| MR1-1 P07 | Gad | Measured | 5.6 | 6.4 | 38.2 | 45.8 | 48.3 | 70.4 | 14.0% | 16.0% | 22.7% | 27.2% | 40.0 | 168.1 |
| [4] | Simulated | 6.7 | 6.7 | 30.8 | 37.5 | 37.7 | 45.2 | 16.8% | 16.8% | 18.3% | 22.3% | 40.0 | 168.1 | |
| 124I | Measured | 15.3 | 21.7 | 31.3 | 73.6 | 50.1 | 128.6 | 38.3% | 54.3% | 18.6% | 43.8% | 40.0 | 168.1 | |
| Simulated | 22.9 | 26.4 | 45.4 | 70.9 | 69.5 | 104.4 | 57.3% | 66.0% | 27.0% | 42.2% | 40.0 | 168.1 | ||
| 24 h | 54 hr | 24 h | 54 hr | 24 h | 54 hr | 24 h | 54 hr | 24 h | 54 hr | |||||
| MR1-1 P08 | Gad | Measured | 4.3 | 7.0 | 9.8 | 16.7 | 15.4 | 28.2 | 7.2% | 11.7% | 5.3% | 9.0% | 60.0 | 186.2 |
| [4] | Simulated | 4.6 | 4.6 | 11.0 | 12.0 | 15.8 | 17.0 | 7.7% | 7.7% | 5.9% | 6.4% | 60.0 | 186.2 | |
| 124I | Measured | 29.0 | 34.5 | 25.9 | 64.8 | 59.5 | 113.9 | 48.3% | 57.5% | 13.9% | 34.8% | 60.0 | 186.2 | |
| Simulated | 25.7 | 25.8 | 26.8 | 40.2 | 53.6 | 69.2 | 42.8% | 43.0% | 14.4% | 21.6% | 60.0 | 186.2 | ||
| 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | |||||
| MR1-1 P09 | Gad | Measured | 0.1 | 0.0 | 24.2 | 0.0 | 29.4 | 0.0 | 2.0% | 0.0% | 45.1% | 0.0% | 5.0 | 53.6 |
| [4] | Simulated | 0.3 | 0.3 | 24.5 | 25.6 | 31.8 | 34.7 | 6.0% | 6.0% | 45.7% | 47.8% | 5.0 | 53.6 | |
| 124I | Measured | 2.1 | 2.2 | 35.2 | 32.5 | 47.3 | 39.4 | 42.0% | 44.0% | 65.7% | 60.6% | 5.0 | 53.6 | |
| Simulated | 1.4 | 1.4 | 28.3 | 30.3 | 37.8 | 42.9 | 28.0% | 28.0% | 52.8% | 56.5% | 5.0 | 53.6 | ||
| 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | |||||
| D2C7 P1001 | Gad | Measured | 1.0 | 0.6 | 1.9 | 0.0 | 3.3% | 0.0% | 0.6% | 0.0% | 30.1 | 95.0 | ||
| [2] | Simulated | 4.2 | 4.2 | 1.7 | 1.7 | 5.9 | 5.9 | 14.0% | 14.0% | 1.8% | 1.8% | 30.1 | 95.0 | |
| 124I | Simulated | 20.4 | 26.2 | 12.9 | 31.1 | 33.3 | 57.3 | 67.8% | 87.0% | 13.6% | 32.7% | 30.1 | 95.0 | |
| 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | |||||
| D2C7 P1002 | Gad | Measured | 1.5 | 2.0 | 5.3 | 4.0 | 6.8 | 6.0 | 15.0% | 20.0% | 6.2% | 4.7% | 10.0 | 86.0 |
| [1] | Simulated | 2.9 | 2.9 | 4.2 | 4.2 | 7.1 | 7.1 | 29.0% | 29.0% | 4.9% | 4.9% | 10.0 | 86.0 | |
| 124I | Measured | 4.7 | 3.3 | 18.1 | 25.8 | 22.8 | 29.1 | 47.0% | 33.0% | 21.0% | 30.0% | 10.0 | 86.0 | |
| Simulated | 5.5 | 7.5 | 11.2 | 19.6 | 16.7 | 27.1 | 55.0% | 75.0% | 13.0% | 22.8% | 10.0 | 86.0 | ||
| 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | |||||
| D2C7 P1004 | Gad | Measured | 0.8 | 1.5 | 0.1 | 0.1 | 0.8 | 1.6 | 9.9% | 19.0% | 0.1% | 0.1% | 7.9 | 81.0 |
| [1] | Simulated | 1.2 | 1.2 | 0.2 | 0.2 | 1.4 | 1.4 | 15.2% | 15.2% | 0.2% | 0.2% | 7.9 | 81.0 | |
| 124I | Measured | 4.6 | 7.7 | 1.2 | 11.3 | 5.8 | 20.8 | 58.2% | 97.0% | 1.5% | 14.0% | 7.9 | 81.0 | |
| Simulated | 7.4 | 7.4 | 11.3 | 21.9 | 20.0 | 32.0 | 93.0% | 94.1% | 14.0% | 27.0% | 7.9 | 81.0 | ||
| 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | |||||
| D2C7 P1005 | Gad | Measured | 0.7 | 0.1 | 1.5 | 0.2 | 2.6 | 0.4 | 10.0% | 1.5% | 1.6% | 0.2% | 6.6 | 96.0 |
| [1] | Simulated | 0.6 | 0.6 | 0.5 | 0.5 | 1.1 | 1.1 | 9.1% | 9.1% | 0.5% | 0.5% | 6.6 | 96.0 | |
| 124I | Measured | 4.9 | 5.0 | 9.1 | 14.4 | 14.0 | 19.4 | 74.2% | 75.8% | 9.5% | 15.0% | 6.6 | 96.0 | |
| Simulated | 2.4 | 2.9 | 3.9 | 6.1 | 6.3 | 9.0 | 36.4% | 43.9% | 4.1% | 6.4% | 6.6 | 96.0 | ||
| 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | |||||
| D2C7 P1006 | Gad | Measured | 0.6 | 1.2 | 0.2 | 0.4 | 0.8 | 1.6 | 7.2% | 14.5% | 0.2% | 0.4% | 8.3 | 94.3 |
| [1] | Simulated | 0.5 | 0.5 | 0.1 | 0.1 | 0.6 | 0.6 | 5.4% | 5.4% | 0.1% | 0.1% | 8.3 | 94.3 | |
| 124I | Measured | 4.8 | 4.2 | 3.8 | 4.0 | 9.2 | 8.9 | 57.8% | 50.6% | 4.0% | 4.2% | 8.3 | 94.3 | |
| Simulated | 3.3 | 3.9 | 2.3 | 3.8 | 5.6 | 7.7 | 39.8% | 47.0% | 2.4% | 4.0% | 8.3 | 94.3 | ||
| 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | |||||
| D2C7 P1007 | Gad | Measured | 1.0 | 0.2 | 2.9 | 1.8 | 3.9 | 2.0 | 26.3% | 5.3% | 4.9% | 3.1% | 3.8 | 59.0 |
| [1] | Simulated | 0.7 | 0.7 | 2.0 | 2.0 | 2.7 | 2.7 | 18.4% | 18.4% | 3.4% | 3.4% | 3.8 | 59.0 | |
| 124I | Simulated | 2.4 | 3.2 | 7.7 | 10.6 | 10.1 | 13.8 | 63.2% | 84.2% | 13.1% | 18.0% | 3.8 | 59.0 | |
| 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | |||||
| D2C7 P1008 | Gad | Measured | 1.6 | 1.5 | 0.8 | 1.2 | 2.4 | 2.7 | 26.7% | 25.0% | 1.6% | 2.4% | 6.0 | 50.0 |
| [1] | ||||||||||||||
| 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | |||||
| D2C7 P1025 | Gad | Measured | 0.10 | 1.44 | 0.00 | 1.90 | 0.0% | 7.2% | 0.0% | 3.0% | 1.4 | 47.4 | ||
| [1] | Simulated | 0.16 | 0.16 | 2.36 | 2.37 | 2.52 | 2.53 | 11.6% | 11.6% | 5.0% | 5.0% | 1.4 | 47.4 | |
| 124I | Simulated | 0.97 | 0.98 | 5.77 | 6.79 | 6.74 | 7.82 | 70.3% | 71.0% | 12.2% | 14.3% | 1.4 | 47.4 | |
| 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | |||||
| D2C7 P1026 | Gad | Measured | 0.08 | 0.11 | 1.13 | 2.84 | 1.21 | 2.95 | 1.0% | 1.3% | 1.9% | 4.8% | 8.2 | 59.5 |
| [1] | Simulated | 0.45 | 0.45 | 0.80 | 0.84 | 1.32 | 1.36 | 5.5% | 5.5% | 1.3% | 1.4% | 8.2 | 59.5 | |
| 124I | Simulated | 6.66 | 6.80 | 9.48 | 11.14 | 17.06 | 18.86 | 80.9% | 82.6% | 15.9% | 18.7% | 8.2 | 59.5 | |
| 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | |||||
| D2C7 P1031 | Gad | Measured | 0.45 | 0.22 | 0.05 | 0.36 | 0.80 | 0.70 | 22.2% | 10.8% | 0.1% | 0.7% | 2.0 | 50.2 |
| [1] | Simulated | 0.50 | 0.50 | 0.40 | 0.40 | 1.24 | 1.24 | 24.6% | 24.6% | 0.8% | 0.8% | 2.0 | 50.2 | |
| 124I | Simulated | 1.96 | 1.97 | 10.44 | 14.08 | 12.82 | 16.52 | 96.6% | 97.0% | 20.8% | 28.0% | 2.0 | 50.2 | |
| 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | |||||
| D2C7 P1032 | Gad | Measured | 0.64 | 1.08 | 0.55 | 1.03 | 1.25 | 2.11 | 25.3% | 42.7% | 1.3% | 2.3% | 2.5 | 43.9 |
| [1] | Simulated | 1.45 | 1.45 | 1.92 | 1.96 | 3.50 | 3.50 | 57.3% | 57.3% | 4.4% | 4.5% | 2.5 | 43.9 | |
| 124I | Simulated | 2.53 | 2.53 | 10.10 | 15.05 | 12.63 | 17.58 | 100.0% | 100.0% | 23.0% | 34.3% | 2.5 | 43.9 | |
| 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | |||||
| D2C7 P1034 | Gad | Measured | 0.99 | 1.75 | 0.19 | 0.95 | 1.96 | 2.90 | 4.2% | 7.4% | 0.2% | 0.9% | 23.7 | 102.0 |
| [1] | Simulated | 1.48 | 1.48 | 0.00 | 0.00 | 1.86 | 1.86 | 6.2% | 6.2% | 0.0% | 0.0% | 23.7 | 102.0 | |
| 124I | Simulated | 15.73 | 22.94 | 2.06 | 9.54 | 18.79 | 33.71 | 66.4% | 96.8% | 2.0% | 9.4% | 23.7 | 102.0 | |
| 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | |||||
| D2C7 P1037 | Gad | Measured | 1.90 | 1.73 | 0.61 | 0.44 | 2.65 | 2.26 | 34.5% | 31.5% | 0.9% | 0.6% | 5.5 | 70.2 |
| [1] | Simulated | 1.53 | 1.53 | 0.27 | 0.27 | 1.80 | 1.80 | 27.8% | 27.8% | 0.4% | 0.4% | 5.5 | 70.2 | |
| 124I | Simulated | 3.46 | 4.16 | 2.88 | 5.18 | 6.41 | 9.39 | 62.9% | 75.6% | 4.1% | 7.4% | 5.5 | 70.2 | |
| 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | 24 h | 72 h | |||||
| D2C7 P1041 | Gad | Measured | 1.61 | 4.78 | 3.02 | 9.52 | 4.74 | 15.41 | 6.2% | 18.4% | 1.6% | 5.0% | 26.0 | 191.0 |
| [2] | Simulated | 1.46 | 1.46 | 3.21 | 3.49 | 5.44 | 5.71 | 5.6% | 5.6% | 1.7% | 1.8% | 26.0 | 191.0 | |
| 124I | Simulated | 15.76 | 20.89 | 36.04 | 76.69 | 54.50 | 102.65 | 60.6% | 80.3% | 18.9% | 40.2% | 26.0 | 191.0 | |
Appendix B. Potential Applications of Models of Flow and Transport
| Model/Simulate/Predict: | Application: |
|---|---|
| Resting hydraulic conductivity of tissue | Plan catheter placement surgeries |
| Extracellular volume increase under forced infusion | Plan safe convection-enhanced delivery |
| Convective transport of macromolecules | Direct drug delivery planning |
| Convective transport of viral delivery agents | Viral vector delivery planning |
| Intra-cerebral pressure distributions, normal brain | Baseline patient-specific atlas |
| Endogenous bulk flow pathways in normal brain | Predict deposition routes for plaque etc. |
| Intra-cerebral pressure distributions, edematous brain | Plan chemotherapies, direct drug delivery |
| Endogenous bulk flow pathways in edematous brain | Pathways map for cell and other transport |
| Intra-cerebral pressure across trauma and other brain insults | Treat and stage brain insults and injuries |
| Convective (passive) transport of cells | Likely sites of glioblastoma recurrence |
| Distance interactions and signals for cell migration | Planning stem cell delivery and migration |
| Glioblastoma cells: proliferation, bulk flow, diffusion | Stage/treat disease, plan radiotherapy, etc. |
| Backflow from cylindrical catheters | Catheter design and placement |
| Backflow from novel catheters (stepped, spherical, etc.) | Other device design, e.g., balloons in tissue |
| Tissue mechanics and response to pressure | Plan neurosurgery allowing brain shift, etc. |
| Tissue response to mechanics and ultrasound | Enhance penetration of CSF–brain barriers |
| Streaming under ultrasound | Enhance drug delivery |
| Tissue mechanics with cell growth | Cancer cell proliferation; |
| Small and macro molecules across the vascular barrier | Plan dosage for CNS applications |
| Transport + metabolism + kinetics using receptor densities | Plan dosage for drugs with toxicity |
References
- Raghavan, R.; Brady, M.L. Predictive models for pressure-driven fluid infusions into brain parenchyma. Phys. Med. Biol. 2011, 56, 6179–6204. [Google Scholar] [CrossRef]
- Raghavan, R.; Brady, M.; Sampson, J. Delivering therapy to target: Improving the odds for successful drug development. Ther. Deliv. 2016, 7, 457–481. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.W.; Cheng, Y.-C.N.; Haacke, E.M.; Thompson, M.R.; Venkatesan, R. Magnetic Resonance Imaging: Physical Principles and Sequence Design, 2nd ed.; John Wiley and Sons: Hoboken, NJ, USA, 2014. [Google Scholar]
- Mehta, A.I.; Choi, B.D.; Ajay, D.; Raghavan, R.; Brady, M.; Friedman, A.H.; Pastan, I.; Bigner, D.D.; Sampson, J.H. Convection Enhanced Delivery of Macromolecules for Brain Tumors. Curr. Drug Discov. Technol. 2012, 9, 305–310. [Google Scholar] [CrossRef]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef]
- Iliff, J.J.; Lee, H.; Yu, M.; Feng, T.; Logan, J.; Nedergaard, M.; Benveniste, H. Brain-wide pathway for waste clearance captured by contrast-enhanced MRI. J. Clin. Investig. 2013, 123, 1299–1309. [Google Scholar] [CrossRef]
- Iliff, J.J.; Wang, M.; Zeppenfeld, D.M.; Venkataraman, A.; Plog, B.A.; Liao, Y.; Deane, R.; Nedergaard, M. Cerebral arterial pulsation drives paravascular CSF-interstitial fluid exchange in the murine brain. J. Neurosci. 2013, 33, 18190–18199. [Google Scholar] [CrossRef]
- Smith, A.J.; Verkman Alan, S. The “glymphatic” mechanism for solute clearance in Alzheimer’s disease: Game changer or unproven speculation? FASEB J. 2018, 32, 543–551. [Google Scholar] [CrossRef]
- Albargothy, N.J.; Johnston David, A.; MacGregor-Sharp, M.; Weller, R.O.; Verma, A.; Hawkes, C.A.; Carare, R.O. Convective influx/glymphatic system: Tracers injected into the CSF enter and leave the brain along separate periarterial basement membrane pathways. Acta Neuropathol. 2018, 136, 139–152. [Google Scholar] [CrossRef]
- Abbott, J.N.; Pizzo Michelle, E.; Preston Jane, E.; Janigro, D.; Thorne, R.G. The role of brain barriers in fluid movement in the CNS: Is there a ‘glymphatic’ system? Acta Neuropathol. 2018, 135, 387–407. [Google Scholar] [CrossRef]
- Zhang, X.-Y.; Luck, J.; Dewhirst, M.W.; Yuan, F. Interstitial hydraulic conductivity in a fibrosarcoma. Am. J. Physiol. 2000, 279, H2726–H2734. [Google Scholar] [CrossRef]
- Raghavan, R.; Zalutsky, M.R.; Howell, R.W. A model for optimizing delivery of targeted radionuclide therapies into resection cavity margins for the treatment of primary brain cancers. Biomed. Phys. Eng. Express 2017, 3, 035005. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, H.F. Theory of Linear Poroelasticity with Applications to Geomechanics and Hydrogeology; Princeton University Press: Princeton, NJ, USA, 2000. [Google Scholar]
- Vargova, L.; Homola, A.; Zamecnik, J.; Tichy, M.; Benes, V.; Sykova, E. Diffusion of parameters of the extracellular space in human gliomas. GLIA 2003, 42, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Kang, H.; Xu, Q.; Chen, M.J.; Liao, Y.; Thiyagarajan, M.; O’Donnell, J.; Christensen, D.J.; Nicholson, C.; Iliff, J.J.; et al. Sleep drives metabolite clearance from the adult brain. Science 2013, 342, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; O’Donnell, J.; Xu, Q.; Kang, N.; Goldman, N.; Nedergaard, M. Changes in the composition of brain interstitial ions control the sleep-wake cycle. Science 2016, 80, 550–555. [Google Scholar] [CrossRef]
- Bear, J. Dynamics of Fluids in Porous Media; Dover Publications: New York, NY, USA, 1988. [Google Scholar]
- Neeves, K.B. Convection-Enhanced Drug Delivery: Porous Media Models and Microfluidic Devices. Ph.D. Thesis, Cornell University, Ithaca, NY, USA, June 2006. [Google Scholar]
- Raghavan, R.; Grossman, S. A Treatment Simulator for Brain Diseases and Method of Use Therof. US Patent 9,265,577, 23 February 2016. [Google Scholar]
- Morrison, P.F.; Laske, D.W.; Bobo, H.; Oldfield, E.H.; Dedrick, R.L. High flow microinfusion: Tissue penetration and pharmacodynamics. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1994, 266, R292–R305. [Google Scholar] [CrossRef]
- Morrison, P.F.; Chen, M.Y.; Chadwick, R.S.; Lonser, R.R.; Oldfield, E.H. Focal delivery during direct infusion to brain: Role of flow rate, catheter diameter, and tissue mechanics. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1999, 277, R1218–R1229. [Google Scholar] [CrossRef]
- Raghavan, R.; Mikaelian, S.; Brady, M.; Chen, Z.J. Fluid infusions from catheters into elastic tissue I: Azimuthally symmetric backflow in homogeneous media. Phys. Med. Biol. 2010, 55, 281–304. [Google Scholar] [CrossRef]
- Sillay, K.; Hinchman, A.; Kumbier, L.; Schomberg, D.; Ross, C.; Kubota, K.; Brady, M.; Brodsky, E.; Miranpuri, G.; Raghavan, R. Strategies for the delivery of multiple collinear infusion clouds in convection-enhanced delivery in the treatment of Parkinson’s disease. Stereotact Funct Neurosurg. 2013, 91, 153–161. [Google Scholar] [CrossRef]
- Raghavan, R.; Brady Martin, L. Efficient numerical solutions of Neumann problems in inhomogeneous media from their probabilistic representations and applications. Eng. Rep. 2020, 2, 12108. [Google Scholar] [CrossRef]
- Bao, X.; Pastan, I.; Bigner, D.D. Chandramohan, Vidyalakshmi EGFR/EGFRvIII-targeted immunotoxin therapy for the treatment of glioblastomas via convection-enhanced delivery. Recept. Clin. Investig. 2016, 3. [Google Scholar] [CrossRef]
- Chandramohan, V.; Bao, X.; Yu, X.; Parker, S.; McDowall, C.; Yu, Y.-R.; Healy, P.; Desjardins, A.; Gunn, M.D.; Gromeier, M.; et al. Improved efficacy against malignant brain tumors with EGFRwt/EGFRvIII targeting immunotoxin and checkpoint inhibitor combinations. J. Immunother Cancer 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Gromeier, M.; Nair, S.K. Recombinant Poliovirus for Cancer Immunotherapy. Annu. Rev. Med. 2018, 69, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, A.; Gromeier, M.; Herndon, J.E.; Beaubier, N.; Bolognesi, D.P.; Friedman, A.H.; Friedman, H.S.; McSherry, F.; Muscat, A.M.; Nair, S.; et al. Recurrent Glioblastoma Treated with Recombinant Poliovirus. N. Engl. J. Med. 2018, 379, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Deoni, S.C.L.; Peters, T.M.; Rutt, B.K. Determination of optimal angles for variable nutation proton magnetic spin-lattice, T1, and spin-spin, T2, relaxation times measurement. Magn. Reson. Med. 2004, 51, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Rothrock Robert, J.; Canoll, P.; Bruce, J.N. Convection-enhanced delivery for targeted delivery of antiglioma agents: The translational experience. J. Drug Deliv. 2013, 2013, 7. Available online: https://www.hindawi.com/journals/jdd/2013/107573/ (accessed on 18 September 2020). [CrossRef][Green Version]
- Saito, R.; Tominaga, T. Convection-enhanced delivery of therapeutics for malignant gliomas. Neuorologia Medico-Chir. 2017, 57, 8–16. [Google Scholar] [CrossRef]
- Brady, M.L.; Raghavan, R.; Singh, D.; Anand, P.J.; Fleisher, A.S.; Mata, J.; Broaddus, W.C.; Olbricht, W.L. In vivo performance of a microfabricated catheter for intraparenchymal delivery. J. Neurosci. Methods 2014, 229, 76–83. [Google Scholar] [CrossRef]
- Hersh, D.S.; Nguyen, B.A.; Dancy, J.G.; Adapa, A.R.; Winkles, J.A.; Woodworth, G.F.; Kim, A.J.; Frenkel, V. Pulsed ultrasound expands the extracellular and perivascular spaces of the brain. Brain Res. 2016, 1646, 543–550. [Google Scholar] [CrossRef]
- Raghavan, R. Theory for acoustic streaming in soft porous matter and its applications to ultrasound-enhanced convective delivery. J. Ther. Ultrasound 2018, 6, 26. [Google Scholar] [CrossRef]
- Ray, L.; Iliff, J.J.; Heys, J.J. Analysis of convective and diffusive transport in the brain interstitium. Fluids Barriers CNS 2019, 16, 6. [Google Scholar] [CrossRef]
- Brady, M.; Raghavan, R.; Wang, P.; Olsen, M.; Brodsky, E.K.; Oakes, T.; Alexander, A.L.; Block, W.F. Real-time MR Brain Infusion Monitoring Enables Accurate Prediction of End Drug Distribution. In Proceedings of the ISMRM 26th Annual Meeting and Exhibition, Paris, France, 16–21 June 2018. [Google Scholar]
- Brady, M.L.; Raghavan, R.; Alexander, A.; Kubota, K.; Sillay, K.; Emborg, M.E. Pathways of infusate loss during convection-enhanced delivery into the putamen nucleus. Stereotact Funct Neurosurg. 2013, 91, 69–78. [Google Scholar] [CrossRef]
- Papadopoulos, M.C.; Saadoun, S.; Binder, D.K.; Manley, G.K.; Krishna, S.; Verkman, A.S. Molecular mechanisms of brain tumor edema. Neuroscience 2004, 129, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Baxter, L.T.; Jain, R.K. Transport of fluid and macromolecules in tumors I: Role of interstitial pressure and convection. Microvasc. Res. 1989, 37, 77–104. [Google Scholar] [CrossRef]
- Baxter, L.T.; Jain, R.K. Transport of fluid and macromolecules in tumors II: Role of heterogeneous perfusion and lymphatics. Microvasc. Res. 1990, 40, 246–263. [Google Scholar] [CrossRef]
- Baxter, L.T.; Jain, R.K. Transport of fluid and macromolecules in tumors III: Role of binding and metabolism. Microvasc. Res. 1991, 41, 5–23. [Google Scholar]
- Baxter, L.T.; Jain, R.K. Transport of fluid and macromolecules in tumors IV: A microscopic model of the perivascular distribution. Microvasc. Res. 1991, 41, 252–272. [Google Scholar] [CrossRef]
- Raghavan, R. Interstitial flow, pathological states, and stem cell delivery in the brain. ResearchGate 2019. [Google Scholar] [CrossRef]
- Grossman, S.; Desai, A.; Pitz, M.; Blakeley, J.; Portnow, J.; Raghavan, R. Modeling the Entry of Systemically Administered Antineoplastic Agents Into Brain Tumors, Brain Adjacent to Tumor, and Normal Brain. Abstracts for the Thirteenth Annual Meeting of the Society for Neuro-Oncology. Neuro. Oncol. 2008, 10, 838. [Google Scholar] [CrossRef]
- Grossman, S.; Desai, A.; Pitz, M.; Blakeley, J.; Portnow, J.; Brady, M.; Raghavan, R. Creation of a Chemotherapy Simulator to Predict the Concentrations of Systemically Administered Chemotherapy Within Brain Tumor Tissue. Abstracts from the Third Quadrennial Meeting of the World Federation of Neuro-Oncology (WFNO) and the Sixth Meeting of the Asian Society for Neuro-Oncology (ASNO). Neuro. Oncol. 2009, 11, 906–907. [Google Scholar] [CrossRef][Green Version]
- Brady, M.L.; Raghavan, R.; Grabow, B.; Block, W. Backflow Mitigation in Stepped Catheters, with Appendix on theory for flow redirection and pre-stress by R. Raghavan. ResearchGate 2019. [Google Scholar] [CrossRef]
- Brady, M.L.; Raghavan, R.; Mata, J.; Wilson, M.; Wilson, S.; Odland, R.M.; Broaddus, W.C. Large-volume infusions into the brain: A comparative study of catheter designs. Stereotact Funct Neurosurg. 2017, 96, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, R.; Odland, R. Theory of porous catheters and their applications in intraparenchymal infusions. Biomed. Phys. Eng. Express 2017, 3, 025008. [Google Scholar] [CrossRef] [PubMed]
- Brady, M.; Raghavan, R.; Alexander, A.L.; Block, W.F. Controlling brain infusion distributions: Moving from surgical planning to real-time MR Guidance. In Proceedings of the ISMRM 25th Annual Meeting, Honolulu, HI, USA, 22–27 April 2017; p. 5539. [Google Scholar]
- Brady, M.; Raghavan, R.; Wang, P.; Olsen, M.; Brodsky, E.K.; Oakes, T.; Alexander, A.L.; Block, W. Informing Efficiacy in Brain Drug Trials: The Potential of Real-time MR Brain Infusion Monitoring. In Proceedings of the ISMRM Workshop on High-Value MRI, Washington, DC, USA, 18–20 February 2018. [Google Scholar]
- Olson, J.J.; Zhang, Z.; Dillehay, D.; Stubbs, J. Assessment of a balloon-tipped catheter modified for intracerebral convection-enhanced delivery. J. Neuro. Oncol. 2008, 89, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, R.; Brady, M.L. Delivery of Materials into Margins of Tissue Cavities. US Patent 9,757,543 B2, 12 September 2017. [Google Scholar]


















| Symbol | Stands For |
|---|---|
| p | Fluid pressure in the interstitium; function of position (tissue place) but not time |
| Darcy velocity in tissue (a vector, or more precisely, a covector); function of position | |
| c | The interstitial concentration of a free or unbound particle (therapeutic or marker molecule); function of position and time |
| b | The interstitial concentration of the bound particle (therapeutic); as for c |
| K | The hydraulic conductivity in tissue (a second rank symmetric positive definite tensor, which is a matrix in a specific coordinate system); can depend on position |
| D | The diffusion tensor of the particle in tissue (a second rank symmetric positive definite tensor, which is a matrix in a specific coordinate system); as for D |
| The interstitial volume fraction, also known as the pore fraction particularly in the geophysical literature of porous media. Depends on position (tissue type) | |
| α | In Section 2.1.1 (porous medium physics related) poroelastic coefficient known as the Biot–Willis parameter; a constant |
| (The square root of) capillary conductance; depends on position according to BBB compromise | |
| B | Generally, a bulk modulus of elasticity; specifically a drained bulk modulus (Section 2.1.1); here a constant |
| G | Shear modulus of tissue; constant |
| A loss rate of the free particle or molecule, being a sum of the rates due to capillary loss and to irreversible degradation (which add in the linear approximation). This and rates below are all taken constant | |
| Irreversible endocytosis rate constant of the bound therapeutic particle | |
| Linearized binding rate constant of the free particle | |
| Linearized unbinding rate constant of the bound particle | |
| B | The concentration of binding sites |
| div, grad | The divergence and gradient operators in vector calculus |
| Type of Data | # of Infusions | Bias (%) | L1 Error (%) |
|---|---|---|---|
| MR tracer total volume in MR1-1 | 5 | −22.1 | 26 |
| MR tracer volume in tumor in MR1-1 | 5 | −2.1 | 18.3 |
| PET tracer total volume in MR1-1 | 6 | −14 | 18.96 |
| PET tracer volume in tumor in MR1-1 | 6 | −1.5 | 17.8 |
| MR tracer total volume in D2C7 | 23 | −9.5 | 37.6 |
| MR tracer volume in tumor in D2C7 | 23 | 3.0 | 49.6 |
| PET tracer total volume in D2C7 | 8 | −3.8 | 42.9 |
| PET tracer volume in tumor in D2C7 | 8 | 2.8 | 37 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brady, M.; Raghavan, R.; Sampson, J. Determinants of Intraparenchymal Infusion Distributions: Modeling and Analyses of Human Glioblastoma Trials. Pharmaceutics 2020, 12, 895. https://doi.org/10.3390/pharmaceutics12090895
Brady M, Raghavan R, Sampson J. Determinants of Intraparenchymal Infusion Distributions: Modeling and Analyses of Human Glioblastoma Trials. Pharmaceutics. 2020; 12(9):895. https://doi.org/10.3390/pharmaceutics12090895
Chicago/Turabian StyleBrady, Martin, Raghu Raghavan, and John Sampson. 2020. "Determinants of Intraparenchymal Infusion Distributions: Modeling and Analyses of Human Glioblastoma Trials" Pharmaceutics 12, no. 9: 895. https://doi.org/10.3390/pharmaceutics12090895
APA StyleBrady, M., Raghavan, R., & Sampson, J. (2020). Determinants of Intraparenchymal Infusion Distributions: Modeling and Analyses of Human Glioblastoma Trials. Pharmaceutics, 12(9), 895. https://doi.org/10.3390/pharmaceutics12090895