Encapsulation of Nedaplatin in Novel PEGylated Liposomes Increases Its Cytotoxicity and Genotoxicity against A549 and U2OS Human Cancer Cells


 , , and
, , and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods
2.1. Reagents
2.2. Liposome Preparation
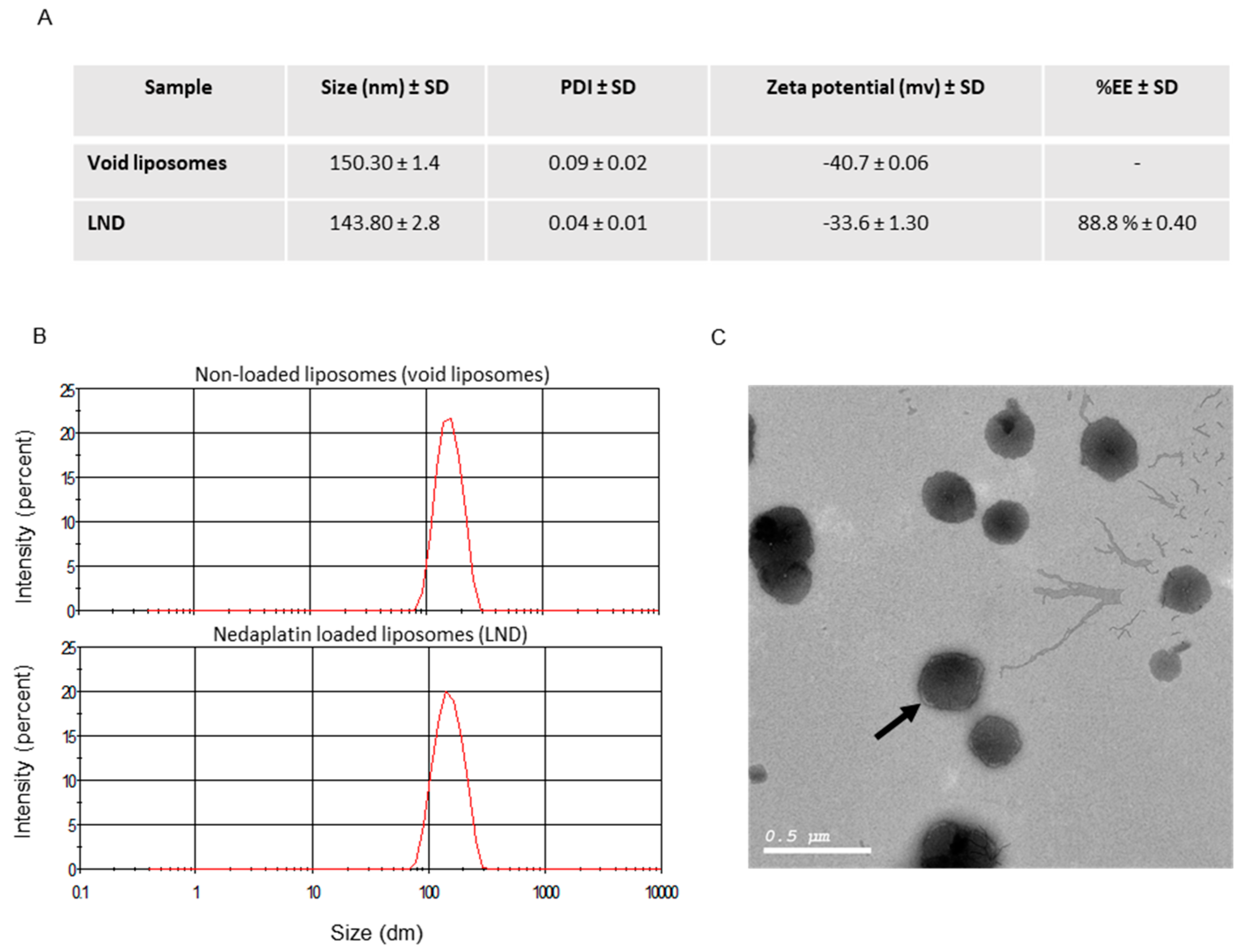
2.3. Liposome Characterization
2.3.1. Entrapment Efficiency
2.3.2. Particle Size and Zeta Potential
2.3.3. Morphology
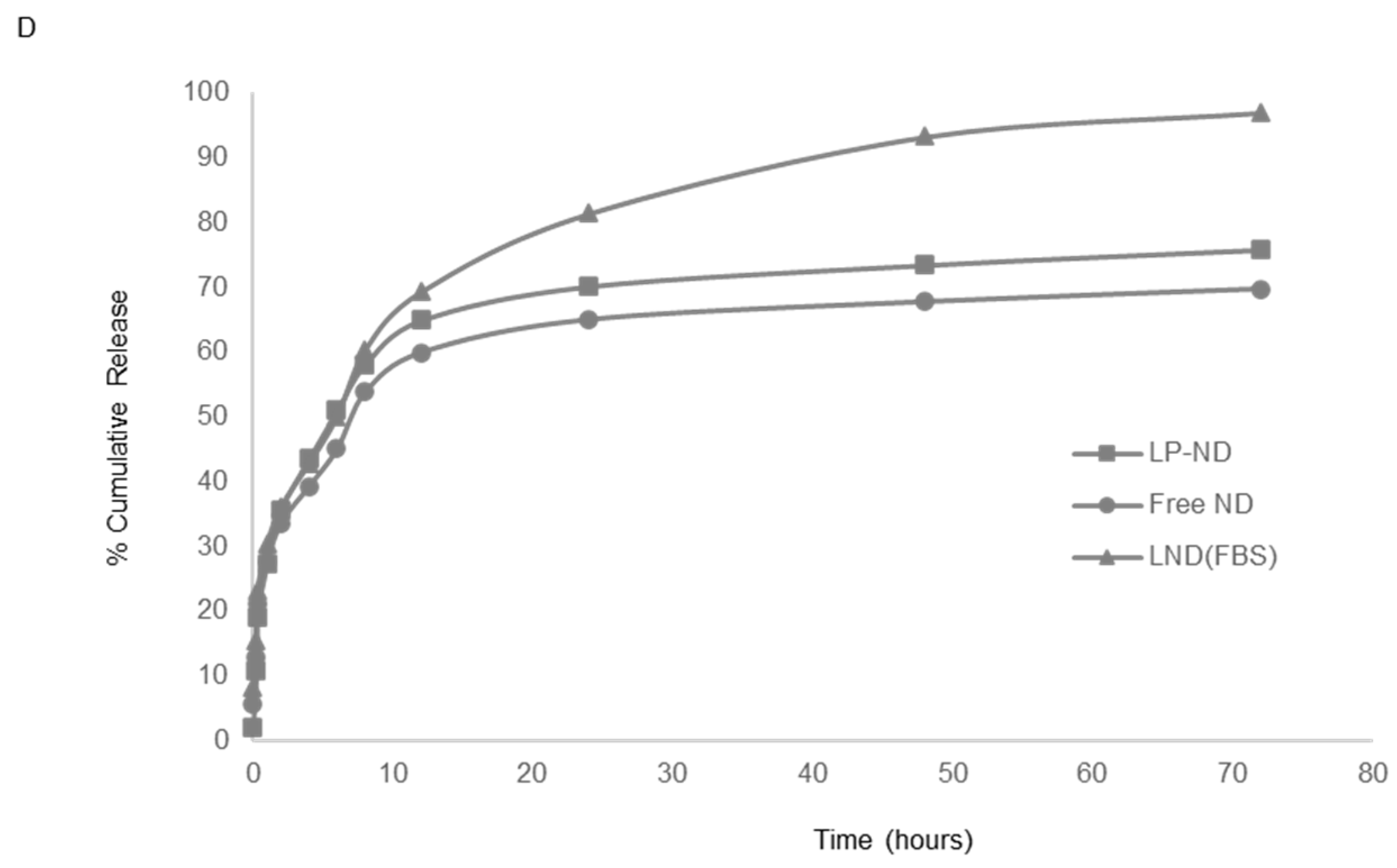
2.3.4. In Vitro Drug Release
2.3.5. Cell Culture
2.4. Cell Proliferation/Antiproliferation/Cytotoxicity Assay
MTT Assay
2.5. Genotoxicity
2.5.1. Micronucleus (MNi) Formation Assay
2.5.2. γH2AX/53BP1 Immunofluorescence Foci Formation Assay
2.6. Statistical Analysis
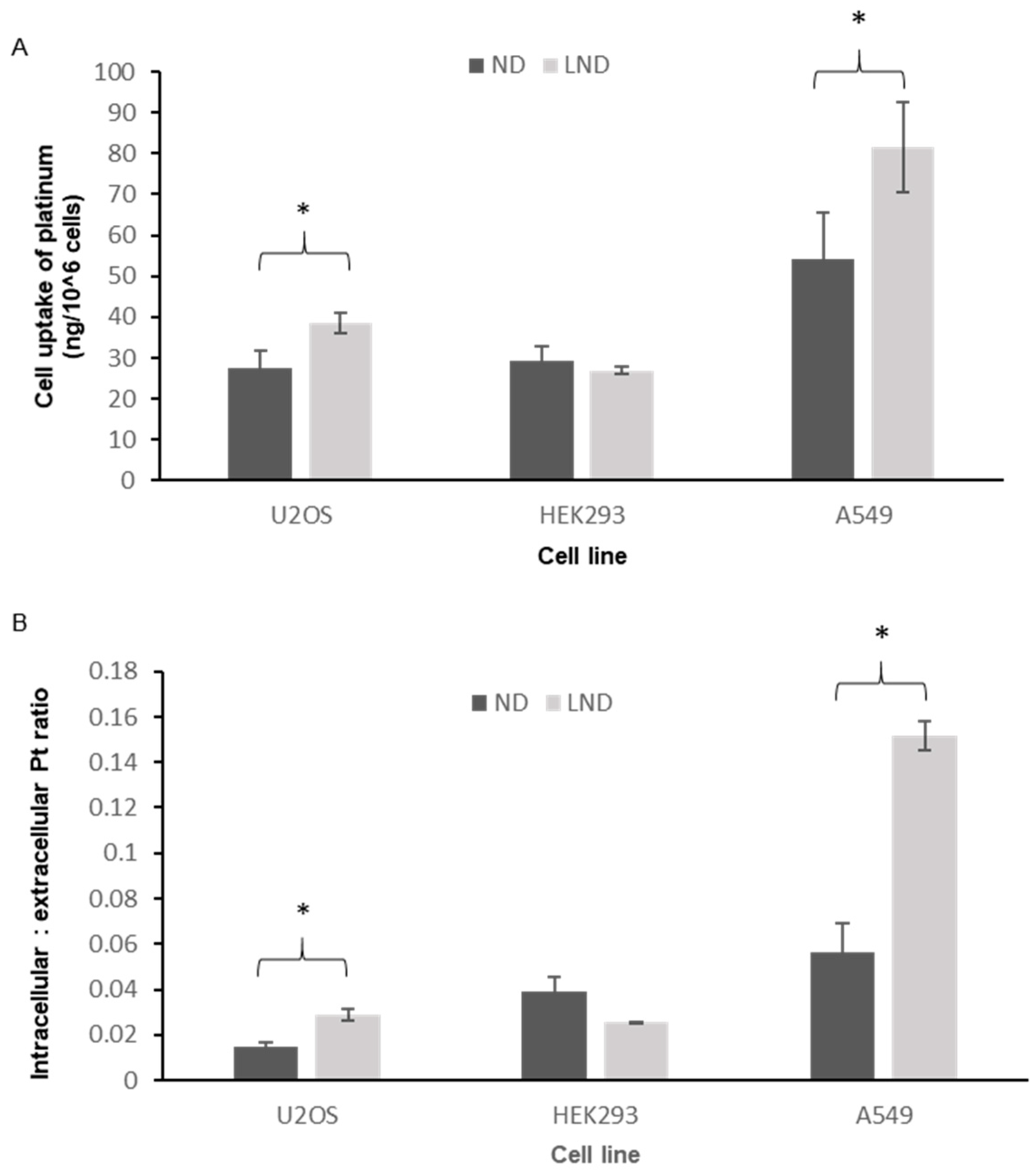
2.7. Uptake of Platinum by the Cell Lines
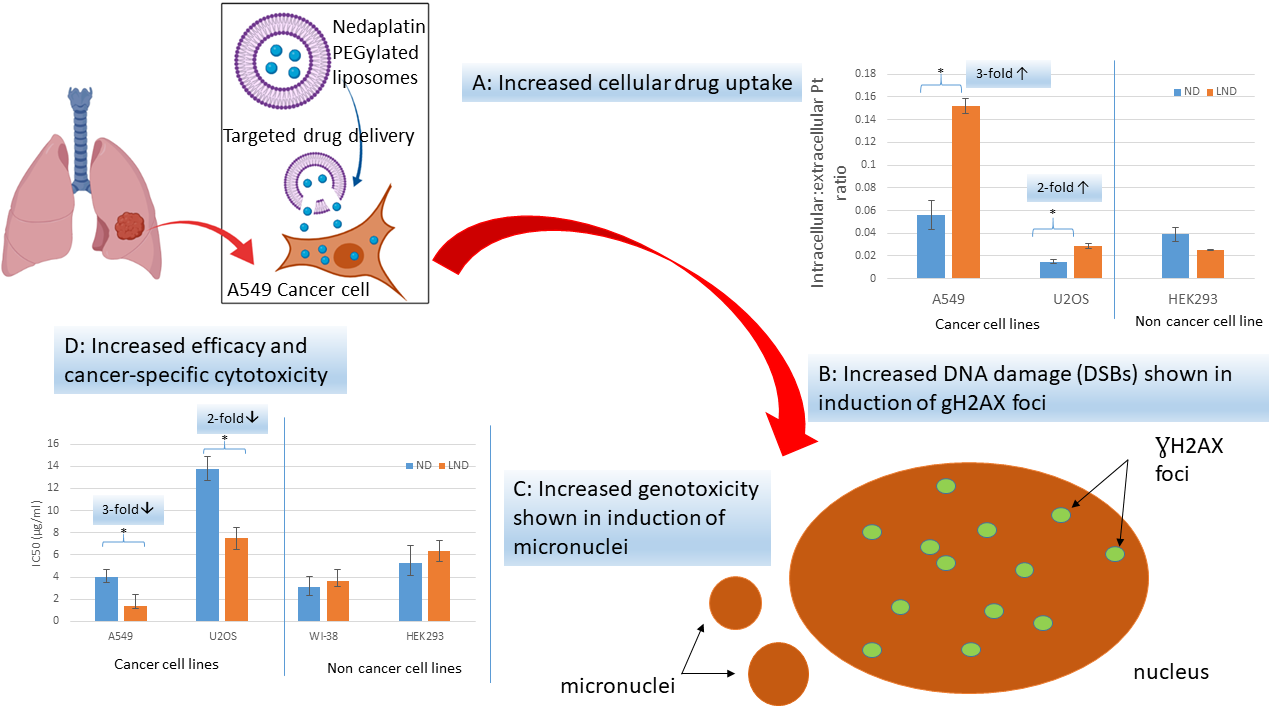
3. Results and Discussion
3.1. Preparation of Stealth Liposomes Containing ND
3.2. In Vitro Drug Release
3.3. Intracellular Accumulation of Platinum
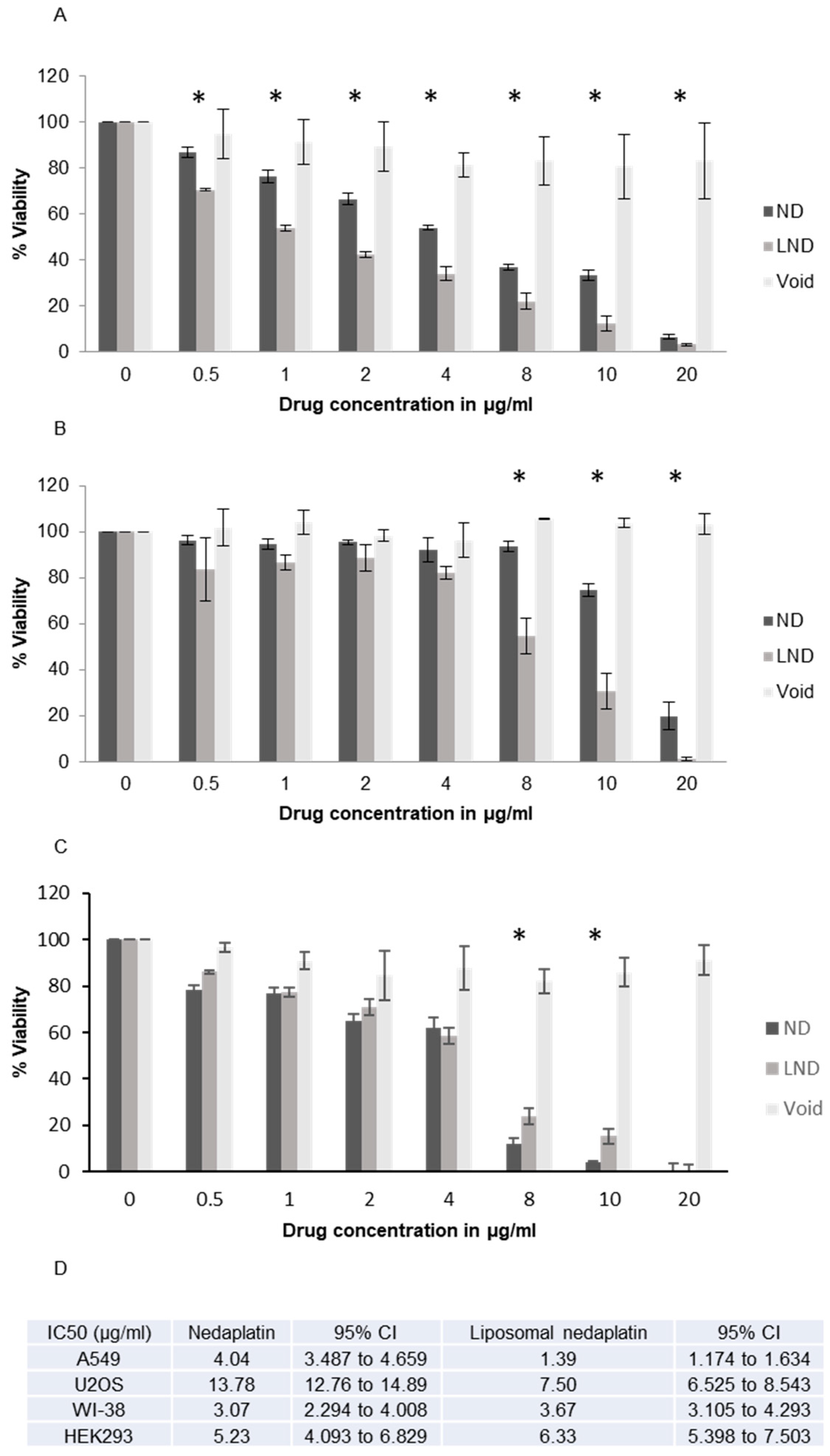
3.4. Cytotoxicity
3.5. Genotoxicity
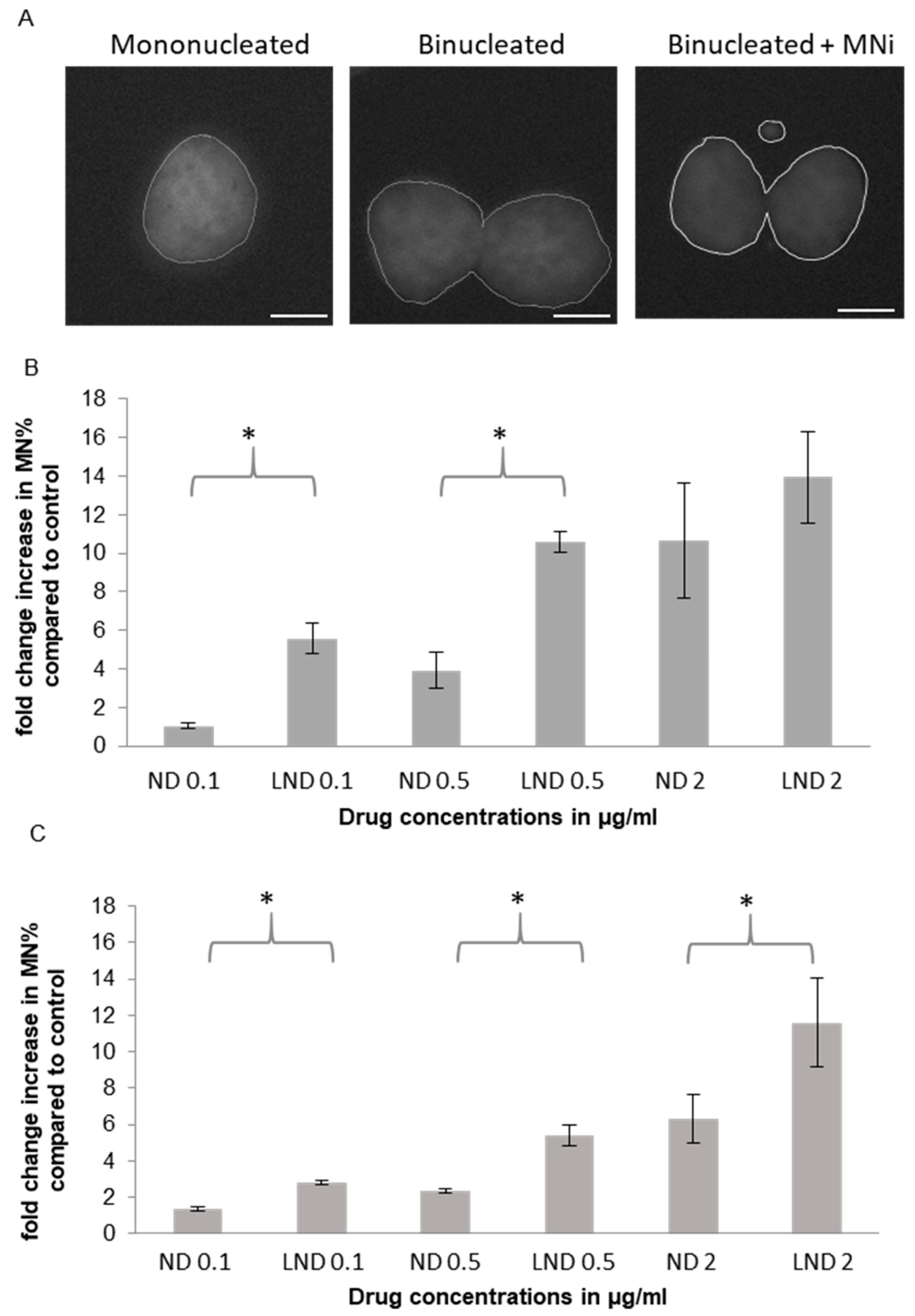
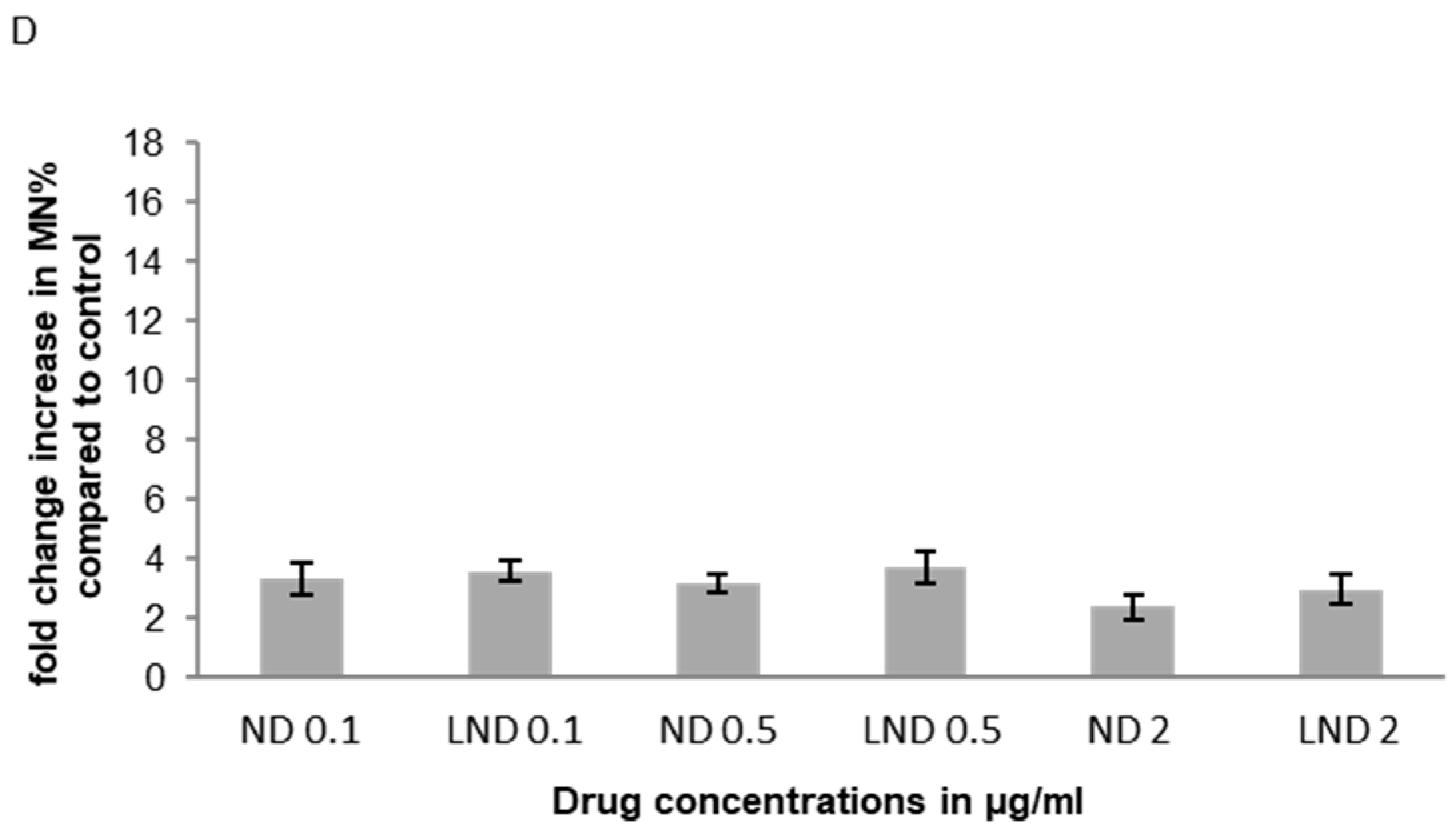
3.5.1. Micronucleus (MNi) Formation Assay
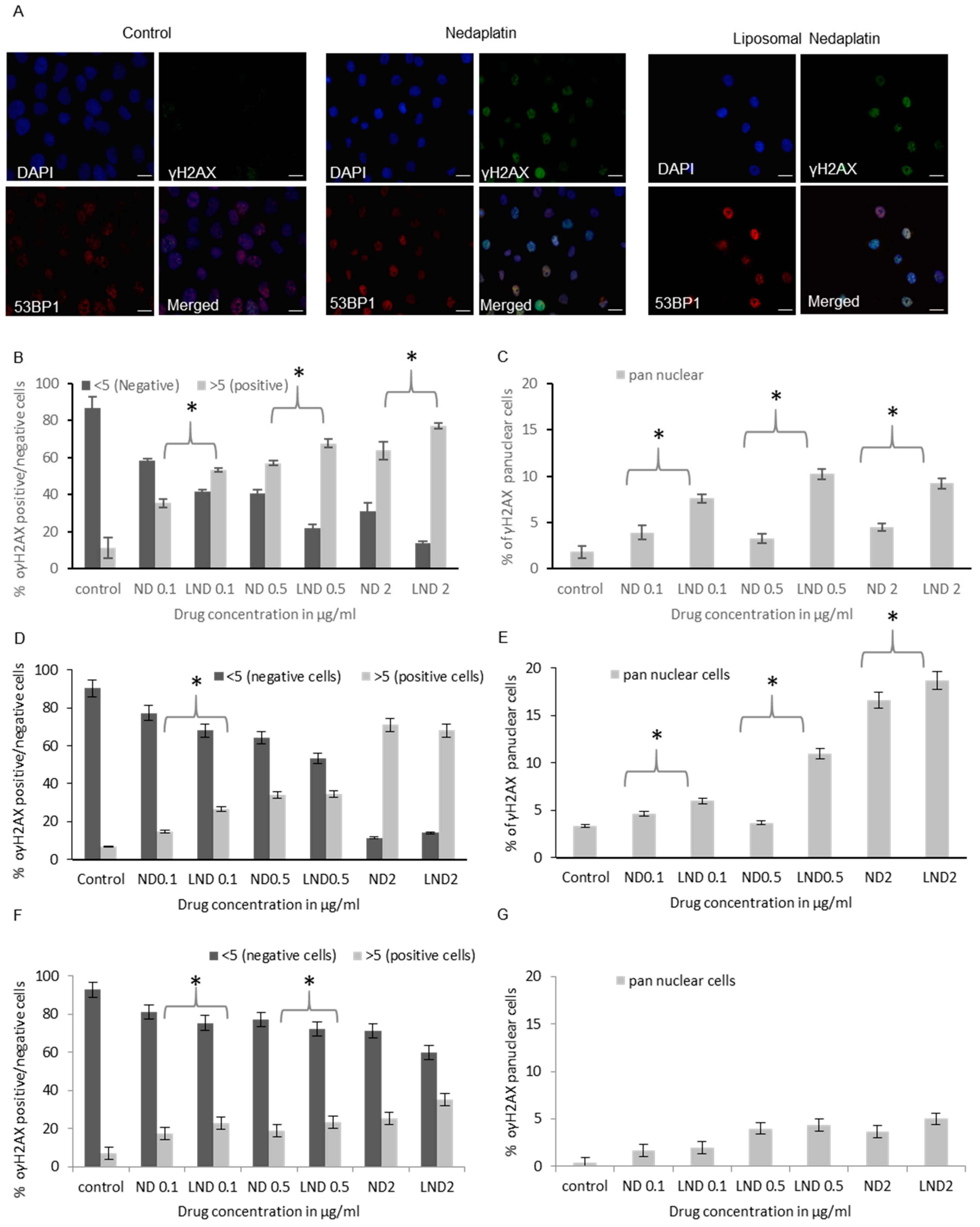
3.5.2. γH2AX Foci Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hang, Z.; Cooper, M.A.; Ziora, Z.M. Platinum-based anticancer drugs encapsulated liposome and polymeric micelle formulation in clinical trials. Biochem. Compd. 2016, 4, 1. [Google Scholar] [CrossRef]
- Latosińska, J.N.; Latosińska, M. Anticancer Drug Discovery—From Serendipity to Rational Design; IntechOpen Limited: London, UK, 2013. [Google Scholar] [CrossRef]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef]
- Chaney, S.G.; Campbell, S.L.; Temple, B.; Bassett, E.; Wu, Y.; Faldu, M. Protein interactions with platinum–DNA adducts: From structure to function. J. Inorg. Biochem. 2004, 98, 1551–1559. [Google Scholar] [CrossRef]
- Barry, M.A.; Behnke, C.A.; Eastman, A. Activation of programmed cell death (apoptosis) by cisplatin, other anticancer drugs, toxins and hyperthermia. Biochem. Pharmacol. 1990, 40, 2353–2362. [Google Scholar] [CrossRef]
- Andrews, P.A. Cancer Treatment and research. Cancer Treat. 1994, 73, 217–248. [Google Scholar] [CrossRef]
- Cressey, D. The resistance against resistance. Nature 2013. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Dilruba, S.; Kalayda, G.V. Platinum-based drugs: Past, present and future. Cancer Chemoth. Pharm. 2016, 77, 1103–1124. [Google Scholar] [CrossRef]
- Raymond, E.; Faivre, S.; Chaney, S.; Woynarowski, J.; Cvitkovic, E. Cellular and molecular pharmacology of oxaliplatin. Mol. Cancer Ther. 2002, 1, 227–235. [Google Scholar]
- Ali, I.; Wani, W.A.; Saleem, K.; Haque, A. Platinum compounds: A hope for future cancer chemotherapy. Anti-Cancer Agent 2013, 13, 296–306. [Google Scholar] [CrossRef]
- Liu, D.; He, C.; Wang, A.Z.; Lin, W. Application of liposomal technologies for delivery of platinum analogs in oncology. Int. J. Nanomed. 2013, 8, 3309–3319. [Google Scholar] [CrossRef][Green Version]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal formulations in clinical use: An updated review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef]
- Blanchard, P.; Tao, Y. Nedaplatin in nasopharyngeal cancer: The rebirth of platinum salts? Lancet Oncol. 2018, 19, 429–431. [Google Scholar] [CrossRef]
- Chitapanarux, I.; Charoentum, C.; Traisathit, P. Nedaplatin: A new paradigm in nasopharyngeal cancer. Ann. Nasopharynx. Cancer 2018, 2, 10. [Google Scholar] [CrossRef]
- Liu, Y.; Yu, S.; Liu, S.; Cao, H.; Ma, R.; Wu, J.; Feng, J. Comparison of nedaplatin-based versus cisplatin-based chemotherapy for advanced non-small cell lung cancer among East Asian populations: A meta-analysis. Sci. Rep. UK 2015, 5, 10516. [Google Scholar] [CrossRef]
- Tang, L.-Q.; Chen, D.-P.; Guo, L.; Mo, H.-Y.; Huang, Y.; Guo, S.-S.; Qi, B.; Tang, Q.-N.; Wang, P.; Li, X.-Y.; et al. Concurrent chemoradiotherapy with nedaplatin versus cisplatin in stage II–IVB nasopharyngeal carcinoma: An open-label, non-inferiority, randomised phase 3 trial. Lancet Oncol. 2018, 19, 461–473. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, Y.; Wang, Z.-Q.; Sun, P.; Wang, D.-S.; Jiang, Y.-X.; Zhang, D.-S.; Wang, F.-H.; Xu, R.-H.; Li, Y.-H. Efficacy and safety of cisplatin-based versus nedaplatin-based regimens for the treatment of metastatic/recurrent and advanced esophageal squamous cell carcinoma: A systematic review and meta-analysis: Cisplatin versus nedaplatin in ESCC. Dis. Esophagus 2016. [Google Scholar] [CrossRef]
- Zhong, L.-Z.; Xu, H.-Y.; Zhao, Z.-M.; Zhang, G.-M.; Lin, F.-W. Comparison of efficacy and toxicity between nedaplatin and cisplatin in treating malignant pleural effusion. Oncotargets Ther. 2018, 11, 5509–5512. [Google Scholar] [CrossRef]
- Su, X.-Y.; Yin, H.-T.; Li, S.-Y.; Huang, X.-E.; Tan, H.-Y.; Dai, H.-Y.; Shi, F.-F. Intervention effects of nedaplatin and cisplatin on proliferation and apoptosis of human tumour cells in vitro. Asian Pac. J. Cancer Prev. 2012, 13, 4531–4536. [Google Scholar] [CrossRef]
- Nallamothu, R.; Wood, G.C.; Kiani, M.F.; Moore, B.M.; Horton, F.P.; Thoma, L.A. A targeted liposome delivery system for combretastatin A4: Formulation optimization through drug loading and in vitro release studies. PDA J. Pharm. Sci. Technol. 2006, 60, 144–155. [Google Scholar]
- McKay, K. New techniques in the pharmacokinetic analysis of cancer drugs. II. The ultratrace determination of platinum in biological samples by inductively coupled plasma-mass spectrometry. Cancer Surv. 1993, 17, 407–414. [Google Scholar] [PubMed]
- Hossann, M.; Wang, T.; Wiggenhorn, M.; Schmidt, R.; Zengerle, A.; Winter, G.; Eibl, H.; Peller, M.; Reiser, M.; Issels, R.D.; et al. Size of thermosensitive liposomes influences content release. J. Control. Release 2010, 147, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Devarajan, P.; Tarabishi, R.; Mishra, J.; Ma, Q.; Kourvetaris, A.; Vougiouka, M.; Boulikas, T. Low renal toxicity of lipoplatin compared to cisplatin in animals. Anticancer Res. 2004, 24, 2193–2200. [Google Scholar] [PubMed]
- Newman, M.S.; Colbern, G.T.; Working, P.K.; Engbers, C.; Amantea, M.A. Comparative pharmacokinetics, tissue distribution, and therapeutic effectiveness of cisplatin encapsulated in long-circulating, pegylated liposomes (SPI-077) in tumor-bearing mice. Cancer Chemoth. Pharm. 1999, 43, 1–7. [Google Scholar] [CrossRef]
- Tian, Y.; Wu, K.; Liu, Q.; Han, N.; Zhang, L.; Chu, Q.; Chen, Y. Modification of platinum sensitivity by KEAP1/NRF2 signals in non-small cell lung cancer. J. Hematol. Oncol. 2016, 9, 83. [Google Scholar] [CrossRef]
- WANG, H.; ZHU, X.; HUANG, J.; CHEN, P.; HAN, S.; YAN, X. Nedaplatin sensitization of cisplatin-resistant human non-small cell lung cancer cells. Oncol. Lett. 2016, 11, 2566–2572. [Google Scholar] [CrossRef]
- Zhang, C.; Li, Y.-L.; Weng, X.; Li, L.-Y.; Zhou, M.-X.; Zhang, D.-Y.; Lin, N.-M. Nedaplatin enhanced apoptotic effects of ABT-737 in human cancer cells via Mcl-1 inhibition. Oncol. Lett. 2016, 12, 4195–4202. [Google Scholar] [CrossRef][Green Version]
- Tsai, H.-C.; Huang, C.-Y.; Su, H.-L.; Tang, C.-H. CCN2 enhances resistance to cisplatin-mediating cell apoptosis in human osteosarcoma. PLoS ONE 2014, 9, e90159. [Google Scholar] [CrossRef]
- Santos, G.C.D.; Mendonça, L.M.; Antonucci, G.A.; Santos, A.C.D.; Antunes, L.M.G.; Bianchi, M.d.L.P. Protective effect of bixin on cisplatin-induced genotoxicity in PC12 cells. Food Chem. Toxicol. 2011, 50, 335–340. [Google Scholar] [CrossRef]
- Chondrou, V.; Trochoutsou, K.; Panayides, A.; Efthimiou, M.; Stephanou, G.; Demopoulos, N.A. Combined study on clastogenic, aneugenic and apoptotic properties of doxorubicin in human cells in vitro. J. Biol. Res. Thessalon. Greece 2018, 25, 17. [Google Scholar] [CrossRef]
- Bankoglu, E.E.; Tschopp, O.; Schmitt, J.; Burkard, P.; Jahn, D.; Geier, A.; Stopper, H. Role of PTEN in Oxidative Stress and DNA Damage in the Liver of Whole-Body Pten Haplodeficient Mice. PLoS ONE 2016, 11, e0166956. [Google Scholar] [CrossRef] [PubMed]
- Lorge, E.; Hayashi, M.; Albertini, S.; Kirkland, D. Comparison of different methods for an accurate assessment of cytotoxicity in the in vitro micronucleus test. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2008, 655, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Norppa, H.; Falck, G.C.-M. What do human micronuclei contain? Mutagenesis 2003, 18, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Utani, K.; Kohno, Y.; Okamoto, A.; Shimizu, N. Emergence of micronuclei and their effects on the fate of cells under replication stress. PLoS ONE 2010, 5, e10089. [Google Scholar] [CrossRef]
- Löbrich, M.; Shibata, A.; Beucher, A.; Fisher, A.; Ensminger, M.; Goodarzi, A.A.; Barton, O.; Jeggo, P.A. gammaH2AX foci analysis for monitoring DNA double-strand break repair: Strengths, limitations and optimization. Cell Cycle Georget. Tex. 2010, 9, 662–669. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA Double-stranded Breaks Induce Histone H2AX Phosphorylation on Serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef]
- Rothkamm, K.; Horn, S.; Scherthan, H.; Rössler, U.; Amicis, A.D.; Barnard, S.; Kulka, U.; Lista, F.; Meineke, V.; Braselmann, H.; et al. Laboratory intercomparison on the γ-H2AX foci assay. Radiat. Res. 2013, 180, 149–155. [Google Scholar] [CrossRef]
- Celeste, A.; Petersen, S.; Romanienko, P.J.; Fernandez-Capetillo, O.; Chen, H.T.; Sedelnikova, O.A.; Reina-San-Martin, B.; Coppola, V.; Meffre, E.; Difilippantonio, M.J.; et al. Genomic instability in mice lacking histone H2AX. Science 2002, 296, 922–927. [Google Scholar] [CrossRef]
- Celeste, A.; Difilippantonio, S.; Difilippantonio, M.J.; Fernandez-Capetillo, O.; Pilch, D.R.; Sedelnikova, O.A.; Eckhaus, M.; Ried, T.; Bonner, W.M.; Nussenzweig, A. H2AX haploinsufficiency modifies genomic stability and tumor susceptibility. Cell 2003, 114, 371–383. [Google Scholar] [CrossRef]
- Bassing, C.H.; Suh, H.; Ferguson, D.O.; Chua, K.F.; Manis, J.; Eckersdorff, M.; Gleason, M.; Bronson, R.; Lee, C.; Alt, F.W. Histone H2AX. Cell 2003, 114, 359–370. [Google Scholar] [CrossRef]
- Mogi, S.; Oh, D.H. γ-H2AX formation in response to interstrand crosslinks requires XPF in human cells. DNA Repair 2006, 5, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Marti, T.M.; Hefner, E.; Feeney, L.; Natale, V.; Cleaver, J.E. H2AX phosphorylation within the G1 phase after UV irradiation depends on nucleotide excision repair and not DNA double-strand breaks. Proc. Natl. Acad. Sci. USA 2006, 103, 9891–9896. [Google Scholar] [CrossRef] [PubMed]
- de Feraudy, S.; Revet, I.; Bezrookove, V.; Feeney, L.; Cleaver, J.E. A minority of foci or pan-nuclear apoptotic staining of gammaH2AX in the S phase after UV damage contain DNA double-strand breaks. Proc. Natl. Acad. Sci. USA 2010, 107, 6870–6875. [Google Scholar] [CrossRef] [PubMed]
- FitzGerald, J.E.; Grenon, M.; Lowndes, N.F. 53BP1: Function and mechanisms of focal recruitment. Biochem. Soc. Trans. 2009, 37, 897–904. [Google Scholar] [CrossRef]
- Noon, A.T.; Goodarzi, A.A. 53BP1-mediated DNA double strand break repair: Insert bad pun here. DNA Repair 2011, 10, 1071–1076. [Google Scholar] [CrossRef]
- Bothmer, A.; Robbiani, D.F.; Feldhahn, N.; Gazumyan, A.; Nussenzweig, A.; Nussenzweig, M.C. 53BP1 regulates DNA resection and the choice between classical and alternative end joining during class switch recombination. J. Exp. Med. 2010, 207, 855–865. [Google Scholar] [CrossRef]
- Clingen, P.H.; Wu, J.Y.-H.; Miller, J.; Mistry, N.; Chin, F.; Wynne, P.; Prise, K.M.; Hartley, J.A. Histone H2AX phosphorylation as a molecular pharmacological marker for DNA interstrand crosslink cancer chemotherapy. Biochem. Pharmacol. 2008, 76, 19–27. [Google Scholar] [CrossRef]
- Olive, P.L.; Banáth, J.P. Kinetics of H2AX phosphorylation after exposure to cisplatin. Cytom. Part B Clin. Cytom. 2009, 76, 79–90. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Shafie, S.; Fahmy, S.A.; Ziko, L.; Elzahed, N.; Shoeib, T.; Kakarougkas, A. Encapsulation of Nedaplatin in Novel PEGylated Liposomes Increases Its Cytotoxicity and Genotoxicity against A549 and U2OS Human Cancer Cells. Pharmaceutics 2020, 12, 863. https://doi.org/10.3390/pharmaceutics12090863
El-Shafie S, Fahmy SA, Ziko L, Elzahed N, Shoeib T, Kakarougkas A. Encapsulation of Nedaplatin in Novel PEGylated Liposomes Increases Its Cytotoxicity and Genotoxicity against A549 and U2OS Human Cancer Cells. Pharmaceutics. 2020; 12(9):863. https://doi.org/10.3390/pharmaceutics12090863
Chicago/Turabian StyleEl-Shafie, Salma, Sherif Ashraf Fahmy, Laila Ziko, Nada Elzahed, Tamer Shoeib, and Andreas Kakarougkas. 2020. "Encapsulation of Nedaplatin in Novel PEGylated Liposomes Increases Its Cytotoxicity and Genotoxicity against A549 and U2OS Human Cancer Cells" Pharmaceutics 12, no. 9: 863. https://doi.org/10.3390/pharmaceutics12090863
APA StyleEl-Shafie, S., Fahmy, S. A., Ziko, L., Elzahed, N., Shoeib, T., & Kakarougkas, A. (2020). Encapsulation of Nedaplatin in Novel PEGylated Liposomes Increases Its Cytotoxicity and Genotoxicity against A549 and U2OS Human Cancer Cells. Pharmaceutics, 12(9), 863. https://doi.org/10.3390/pharmaceutics12090863