Solid Lipid Nanoparticles and Nanostructured Lipid Carriers as Smart Drug Delivery Systems in the Treatment of Glioblastoma Multiforme



Abstract
1. Introduction
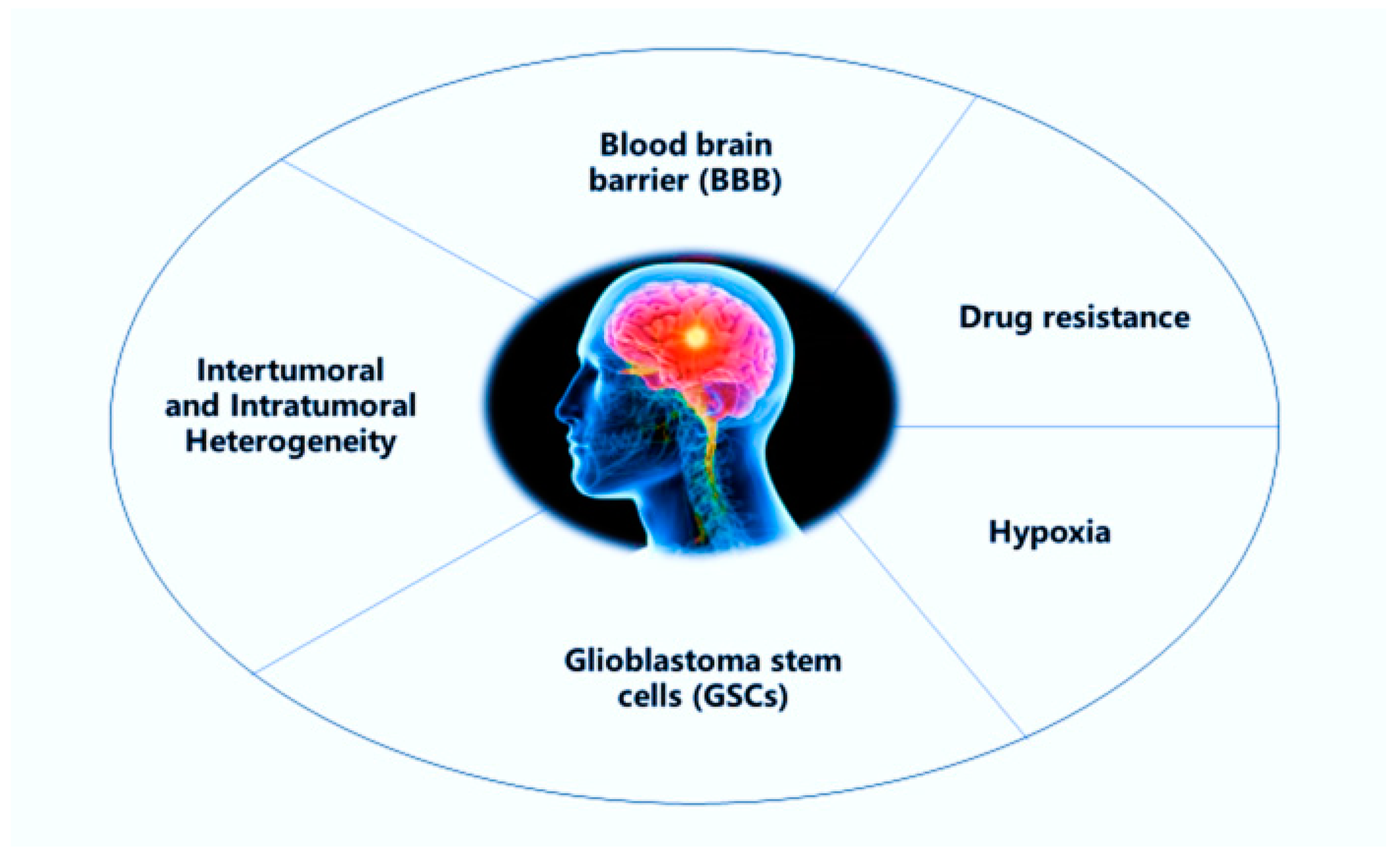
2. Challenges in the Treatment of Glioblastoma Multiforme (GBM)
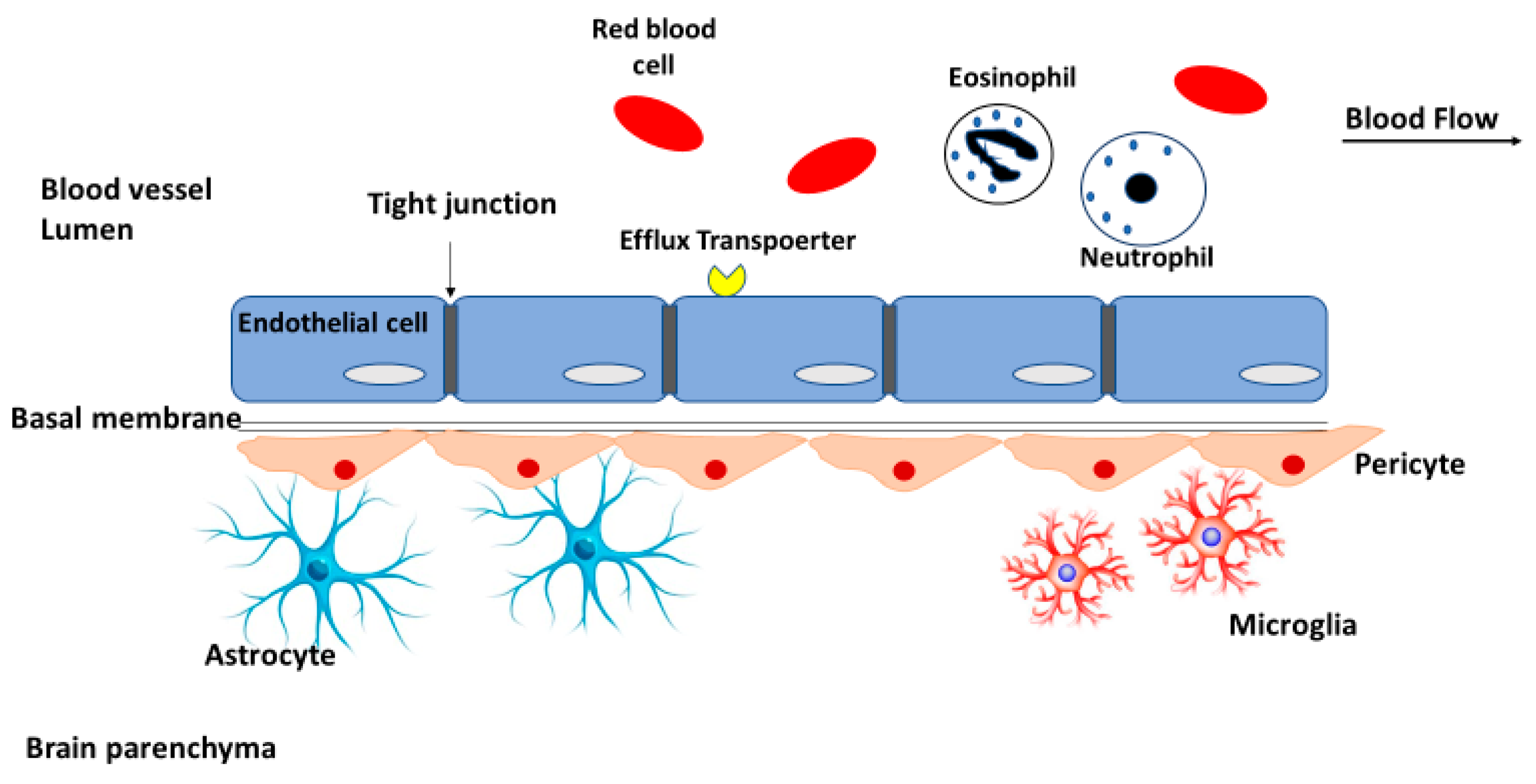
2.1. Blood-Brain Barrier
2.2. Intertumoral and Intratumoral Heterogeneity
2.2.1. Intertumoral Heterogeneity
2.2.2. Intratumoral Heterogeneity
2.3. Glioblastoma Stem Cells
2.4. Drug Efflux
2.5. Hypoxia
3. Solid Lipid Nanoparticles (SLNs) and Nanostructured Lipid Carriers (NLCs) for Targeting Glioblastoma
3.1. SLN and NLC as Smart Drug Delivery Systems in the Treatment of GBM
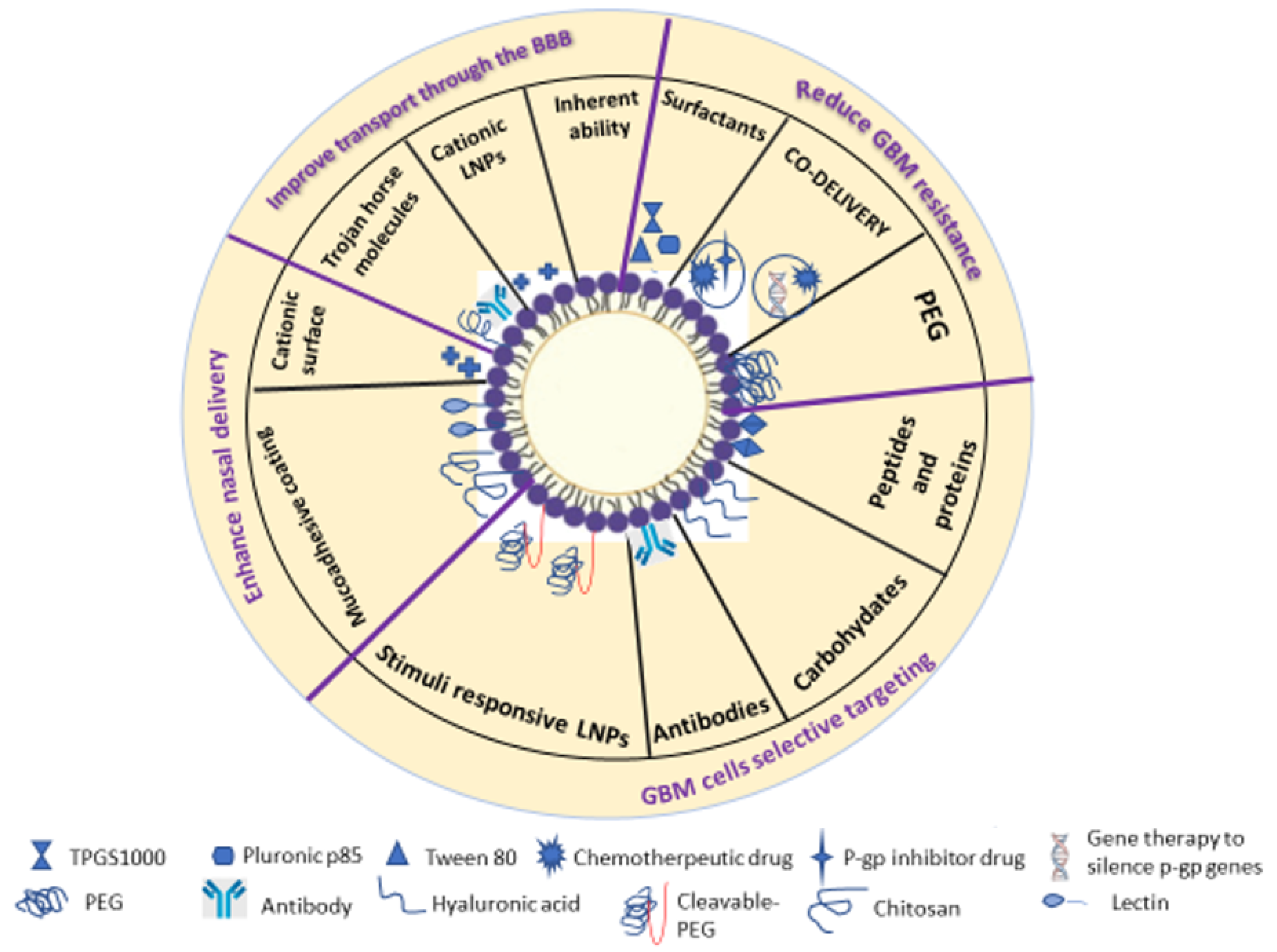
3.2. Modification Strategies to Enhance Crossing the Blood-Brain Barrier (BBB) in GBM Treatment
3.3. Modification Strategies Against GBM Cell Resistance
3.4. Strategies for Selective Targeting of GBM Cells
3.5. Modified Lipid Nanoparticles for Nose-to-Brain Delivery
4. Future Perspectives of SLNs and NLCs for Glioblastoma Treatment
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; Farah, P.; Ondracek, A.; Chen, Y.; Wolinsky, Y.; Stroup, N.E.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2006–2010. Neuro Oncol. 2013, 15, ii1–ii56. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Koshy, M.; Villano, J.L.; Dolecek, T.A.; Howard, A.; Mahmood, U.; Chmura, S.J.; Weichselbaum, R.R.; McCarthy, B.J. Improved survival time trends for glioblastoma using the SEER 17 population-based registries. J. Neurooncol. 2012, 107, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, R.K. Drug delivery systems, CNS protection, and the blood brain barrier. Biomed Res. Int. 2014. [Google Scholar] [CrossRef]
- Pathak, Y.V. Surface Modification of Nanoparticles for Targeted Drug Delivery; Springer: Berlin/Heidelberg, Germany, 2019; ISBN 978-3-030-06115-9. [Google Scholar]
- Dolatabadi, J.E.N.; Omidi, Y. Solid lipid-based nanocarriers as efficient targeted drug and gene delivery systems. TrAC Trends Anal. Chem. 2016, 77, 100–108. [Google Scholar] [CrossRef]
- Müller, R.H.; Radtke, M.; Wissing, S.A. Nanostructured lipid matrices for improved microencapsulation of drugs. Int. J. Pharm. 2002, 242, 121–128. [Google Scholar] [CrossRef]
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee, S.U. Glioblastoma Multiforme: A review of its epidemiology and pathogenesis through clinical presentation and treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3–9. [Google Scholar] [CrossRef]
- Simpson, L.; Galanis, E. Recurrent glioblastoma multiforme: Advances in treatment and promising drug candidates. Expert Rev. Anticancer Ther. 2006, 6, 1593–1607. [Google Scholar] [CrossRef]
- Noch, E.K.; Ramakrishna, R.; Magge, R. Challenges in the treatment of glioblastoma: Multisystem mechanisms of therapeutic resistance. World Neurosurg. 2018, 116, 505–517. [Google Scholar] [CrossRef]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Persidsky, Y.; Ramirez, S.H.; Haorah, J.; Kanmogne, G.D. Blood-brain barrier: Structural components and function under physiologic and pathologic conditions. J. Neuroimmune Pharmacol. 2006, 1, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, L. Modern methods for delivery of drugs across the blood–brain barrier. Adv. Drug Deliv. Rev. 2012, 64, 640–665. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Drug transport across the blood–brain barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef] [PubMed]
- Erickson, M.A.; Banks, W.A. Neuroimmune axes of the blood–brain barriers and blood–brain interfaces: Bases for physiological regulation, disease states, and pharmacological interventions. Pharmacol. Rev. 2018, 70, 278–314. [Google Scholar] [CrossRef]
- Hervé, F.; Ghinea, N.; Scherrmann, J.-M. CNS delivery via adsorptive transcytosis. AAPS J. 2008, 10, 455–472. [Google Scholar] [CrossRef]
- Bickel, U. Antibody delivery through the blood-brain barrier. Adv. Drug Deliv. Rev. 1995, 15, 53–72. [Google Scholar] [CrossRef]
- Silva, S.; Almeida, A.J.; Vale, N. Combination of cell-penetrating peptides with nanoparticles for therapeutic application: A review. Biomolecules 2019, 9, 22. [Google Scholar] [CrossRef]
- Vivès, E.; Brodin, P.; Lebleu, B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem. 1997, 272, 16010–16017. [Google Scholar] [CrossRef]
- Binda, E.; Visioli, A.; Reynolds, B.; Vescovi, A.L. Heterogeneity of cancer-initiating cells within glioblastoma. Front. Biosci. 2012, 4, 1235–1248. [Google Scholar] [CrossRef]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef]
- Johnson, B.E.; Mazor, T.; Hong, C.; Barnes, M.; Aihara, K.; McLean, C.Y.; Fouse, S.D.; Yamamoto, S.; Ueda, H.; Tatsuno, K.; et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 2014, 343, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Laug, D.; Glasgow, S.M.; Deneen, B. A glial blueprint for gliomagenesis. Nat. Rev. Neurosci. 2018, 19, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Daniel, P.; Sabri, S.; Chaddad, A.; Meehan, B.; Jean-Claude, B.; Rak, J.; Abdulkarim, B.S. Temozolomide induced hypermutation in glioma: Evolutionary mechanisms and therapeutic opportunities. Front. Oncol. 2019, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-C.; Bai, H.; Sun, Q.; Zhao, Y.; Lv, Y.; Zhou, J.; Liang, C.; Chen, Y.; Liang, D.; Zheng, H. Multiregional radiomics profiling from multiparametric MRI: Identifying an imaging predictor of IDH1 mutation status in glioblastoma. Cancer Med. 2018, 7, 5999–6009. [Google Scholar] [CrossRef] [PubMed]
- Eskilsson, E.; Rosland, G.V.; Talasila, K.M.; Knappskog, S.; Keunen, O.; Sottoriva, A.; Foerster, S.; Solecki, G.; Taxt, T.; Jirik, R.; et al. EGFRvIII mutations can emerge as late and heterogenous events in glioblastoma development and promote angiogenesis through Src activation. Neuro-oncology 2016, 18, 1644–1655. [Google Scholar] [CrossRef]
- Szerlip, N.J.; Pedraza, A.; Chakravarty, D.; Azim, M.; McGuire, J.; Fang, Y.; Ozawa, T.; Holland, E.C.; Huse, J.T.; Jhanwar, S.; et al. Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc. Natl. Acad. Sci. USA 2012, 109, 3041–3046. [Google Scholar] [CrossRef]
- Mosrati, M.A.; Malmström, A.; Lysiak, M.; Krysztofiak, A.; Hallbeck, M.; Milos, P.; Hallbeck, A.-L.; Bratthäll, C.; Strandéus, M.; Stenmark-Askmalm, M.; et al. TERT promoter mutations and polymorphisms as prognostic factors in primary glioblastoma. Oncotarget 2015, 6, 16663–16673. [Google Scholar] [CrossRef]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar]
- Zhu, T.S.; Costello, M.A.; Talsma, C.E.; Flack, C.G.; Crowley, J.G.; Hamm, L.L.; He, X.; Hervey-Jumper, S.L.; Heth, J.A.; Muraszko, K.M.; et al. Endothelial cells create a stem cell niche in glioblastoma by providing notch ligands that nurture self-renewal of cancer stem-like cells. Cancer Res. 2011, 71, 6061–6072. [Google Scholar] [CrossRef]
- Filatova, A.; Acker, T.; Garvalov, B.K. The cancer stem cell niche(s): The crosstalk between glioma stem cells and their microenvironment. Biochim. Biophys. Acta 2013, 1830, 2496–2508. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Barr, J.; Kong, L.-Y.; Wang, Y.; Wu, A.; Sharma, A.K.; Gumin, J.; Henry, V.; Colman, H.; Sawaya, R.; et al. Glioma-associated cancer-initiating cells induce immunosuppression. Clin. Cancer Res. 2010, 16, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Dundar, T.T.; Hatiboglu, M.A.; Ergul, Z.; Seyithanoglu, M.H.; Sozen, E.; Tuzgen, S.; Kaynar, M.Y.; Karaoz, E. Glioblastoma stem cells and comparison of isolation methods. J. Clin. Med. Res. 2019, 11, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wakeman, T.P.; Latha, J.D.; Hjelmeland, A.B.; Wang, X.-F.; White, R.R.; Rich, J.N.; Sullenger, B.A. Notch promotes radioresistance of glioma stem cells. Stem Cells 2010, 28, 17–28. [Google Scholar] [CrossRef]
- Li, W.-Q.; Li, Y.-M.; Tao, B.-B.; Lu, Y.-C.; Hu, G.-H.; Liu, H.-M.; He, J.; Xu, Y.; Yu, H.-Y. Downregulation of ABCG2 expression in glioblastoma cancer stem cells with miRNA-328 may decrease their chemoresistance. Med. Sci. Monit. 2010, 16, HY27-30. [Google Scholar]
- Haar, C.P.; Hebbar, P.; Wallace, G.C.; Das, A.; Vandergrift, W.A.; Smith, J.A.; Giglio, P.; Patel, S.J.; Ray, S.K.; Banik, N.L. Drug resistance in glioblastoma: A mini review. Neurochem. Res. 2012, 37, 1192–1200. [Google Scholar] [CrossRef]
- De Vries, N.A.; Buckle, T.; Zhao, J.; Beijnen, J.H.; Schellens, J.H.M.; van Tellingen, O. Restricted brain penetration of the tyrosine kinase inhibitor erlotinib due to the drug transporters P-gp and BCRP. Investig. New Drugs 2012, 30, 443–449. [Google Scholar] [CrossRef]
- Munoz, J.L.; Walker, N.D.; Scotto, K.W.; Rameshwar, P. Temozolomide competes for P-glycoprotein and contributes to chemoresistance in glioblastoma cells. Cancer Lett. 2015, 367, 69–75. [Google Scholar] [CrossRef]
- Vaupel, P.; Kelleher, D.K.; Höckel, M. Oxygen status of malignant tumors: Pathogenesis of hypoxia and significance for tumor therapy. Semin. Oncol. 2001, 28, 29–35. [Google Scholar] [CrossRef]
- Monteiro, A.R.; Hill, R.; Pilkington, G.J.; Madureira, P.A. The role of hypoxia in glioblastoma invasion. Cells 2017, 6, 45. [Google Scholar] [CrossRef]
- Semenza, G.L. Expression of hypoxia-inducible factor 1: Mechanisms and consequences. Biochem. Pharmacol. 2000, 59, 47–53. [Google Scholar] [CrossRef]
- Vlaminck, B.; Toffoli, S.; Ghislain, B.; Demazy, C.; Raes, M.; Michiels, C. Dual effect of echinomycin on hypoxia-inducible factor-1 activity under normoxic and hypoxic conditions. FEBS J. 2007, 274, 5533–5542. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.-H.; Ma, Z.-X.; Huang, G.-H.; Xu, Q.-F.; Xiang, Y.; Li, N.; Sidlauskas, K.; Zhang, E.E.; Lv, S.-Q. Downregulation of HIF-1a sensitizes U251 glioma cells to the temozolomide (TMZ) treatment. Exp. Cell Res. 2016, 343, 148–158. [Google Scholar] [CrossRef]
- Chou, C.-W.; Wang, C.-C.; Wu, C.-P.; Lin, Y.-J.; Lee, Y.-C.; Cheng, Y.-W.; Hsieh, C.-H. Tumor cycling hypoxia induces chemoresistance in glioblastoma multiforme by upregulating the expression and function of ABCB1. Neuro-oncology 2012, 14, 1227–1238. [Google Scholar] [CrossRef]
- Del Rowe, J.; Scott, C.; Werner-Wasik, M.; Bahary, J.P.; Curran, W.J.; Urtasun, R.C.; Fisher, B. Single-arm, open-label phase II study of intravenously administered tirapazamine and radiation therapy for glioblastoma multiforme. J. Clin. Oncol. 2000, 18, 1254–1259. [Google Scholar] [CrossRef] [PubMed]
- Tapeinos, C.; Battaglini, M.; Ciofani, G. Advances in the design of solid lipid nanoparticles and nanostructured lipid carriers for targeting brain diseases. J. Control. Release 2017, 264, 306–332. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Badde, S.; Kamble, R.; Pokharkar, V. Development and characterization of liposomal drug delivery system for Nimesulide. Int. J. Pharm. Pharm. Sci. 2010, 2, 87–89. [Google Scholar]
- Patidar, A.; Thakur, D.S.; Kumar, P.; Verma, J. A review on novel lipid based nanocarriers. Int. J. Pharm. Pharm. Sci. 2010, 2, 30–35. [Google Scholar]
- Beloqui, A.; Solinís, M.Á.; Rodríguez-Gascón, A.; Almeida, A.J.; Préat, V. Nanostructured lipid carriers: Promising drug delivery systems for future clinics. Nanomedicine 2016, 12, 143–161. [Google Scholar] [CrossRef]
- Gaspar, D.P.; Faria, V.; Quintas, J.P.; Almeida, A.J. Targeted delivery of lipid nanoparticles by means of surface chemical modification. Curr. Org. Chem. 2017, 21. [Google Scholar] [CrossRef]
- Bertrand, N.; Wu, J.; Xu, X.; Kamaly, N.; Farokhzad, O.C. Cancer nanotechnology: The impact of passive and active targeting in the era of modern cancer biology. Adv. Drug Deliv. Rev. 2014, 66, 2–25. [Google Scholar] [CrossRef] [PubMed]
- Hossen, S.; Hossain, M.K.; Basher, M.K.; Mia, M.N.H.; Rahman, M.T.; Uddin, M.J. Smart nanocarrier-based drug delivery systems for cancer therapy and toxicity studies: A review. J. Adv. Res. 2019, 15, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Xu, C.-L.; Liu, C.-M. Drug delivery strategies to enhance the permeability of the blood-brain barrier for treatment of glioma. Drug Des. Dev. Ther. 2015, 9, 2089–2100. [Google Scholar] [CrossRef] [PubMed]
- Mendes, M.; Miranda, A.; Cova, T.; Gonçalves, L.; Almeida, A.J.; Sousa, J.J.; do Vale, M.L.C.; Marques, E.F.; Pais, A.; Vitorino, C. Modeling of ultra-small lipid nanoparticle surface charge for targeting glioblastoma. Eur. J. Pharm. Sci. 2018, 117, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Kadari, A.; Pooja, D.; Gora, R.H.; Gudem, S.; Kolapalli, V.R.M.; Kulhari, H.; Sistla, R. Design of multifunctional peptide collaborated and docetaxel loaded lipid nanoparticles for antiglioma therapy. Eur. J. Pharm. Biopharm. 2018, 132, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.-C.; Chao, I.-W. Conjugation of melanotransferrin antibody on solid lipid nanoparticles for mediating brain cancer malignancy. Biotechnol. Prog. 2016, 32, 480–490. [Google Scholar] [CrossRef]
- Lu, W. Adsorptive-mediated brain delivery systems. Curr. Pharm. Biotechnol. 2012, 13, 2340–2348. [Google Scholar] [CrossRef]
- Liu, Z.; Zhao, H.; Shu, L.; Zhang, Y.; Okeke, C.; Zhang, L.; Li, J.; Li, N. Preparation and evaluation of Baicalin-loaded cationic solid lipid nanoparticles conjugated with OX26 for improved delivery across the BBB. Drug Dev. Ind. Pharm. 2015, 41, 353–361. [Google Scholar] [CrossRef]
- Kuo, Y.-C.; Liang, C.-T. Inhibition of human brain malignant glioblastoma cells using carmustine-loaded catanionic solid lipid nanoparticles with surface anti-epithelial growth factor receptor. Biomaterials 2011, 32, 3340–3350. [Google Scholar] [CrossRef]
- Kuo, Y.-C.; Lee, I.-H. Delivery of doxorubicin to glioblastoma multiforme in vitro using solid lipid nanoparticles with surface aprotinin and melanotransferrin antibody for enhanced chemotherapy. J. Taiwan Inst. Chem. Eng. 2016, 61, 32–45. [Google Scholar] [CrossRef]
- Agarwal, A.; Majumder, S.; Agrawal, H.; Majumdar, S.; Agrawal, G.P. Cationized albumin conjugated solid lipid nanoparticles as vectors for brain delivery of an anti-cancer drug. Curr. Nanosci. 2011, 7, 71–80. [Google Scholar] [CrossRef]
- Kabanov, A.V.; Batrakova, E.V.; Alakhov, V.Y. An essential relationship between ATP depletion and chemosensitizing activity of Pluronic block copolymers. J. Control. Release 2003, 91, 75–83. [Google Scholar] [CrossRef]
- Tang, J.; Ji, H.; Ren, J.; Li, M.; Zheng, N.; Wu, L. Solid lipid nanoparticles with TPGS and Brij 78: A co-delivery vehicle of curcumin and piperine for reversing P-glycoprotein-mediated multidrug resistance in vitro. Oncol. Lett. 2017, 13, 389–395. [Google Scholar] [CrossRef]
- Estella-Hermoso de Mendoza, A.; Préat, V.; Mollinedo, F.; Blanco-Prieto, M.J. In vitro and in vivo efficacy of edelfosine-loaded lipid nanoparticles against glioma. J. Control. Release 2011, 156, 421–426. [Google Scholar] [CrossRef]
- Vijayakumar, M.R.; Kumari, L.; Patel, K.K.; Vuddanda, P.R.; Vajanthri, K.Y.; Mahto, S.K.; Singh, S. Intravenous administration of trans-resveratrol-loaded TPGS-coated solid lipid nanoparticles for prolonged systemic circulation, passive brain targeting and improved in vitro cytotoxicity against C6 glioma cell lines. RSC Adv. 2016, 6, 50336–50348. [Google Scholar] [CrossRef]
- Wang, S.-W.; Monagle, J.; McNulty, C.; Putnam, D.; Chen, H. Determination of P-glycoprotein inhibition by excipients and their combinations using an integrated high-throughput process. J. Pharm. Sci. 2004, 93, 2755–2767. [Google Scholar] [CrossRef]
- Madan, J.; Pandey, R.S.; Jain, V.; Katare, O.P.; Chandra, R.; Katyal, A. Poly (ethylene)-glycol conjugated solid lipid nanoparticles of noscapine improve biological half-life, brain delivery and efficacy in glioblastoma cells. Nanomedicine 2013, 9, 492–503. [Google Scholar] [CrossRef]
- Venishetty, V.K.; Komuravelli, R.; Kuncha, M.; Sistla, R.; Diwan, P.V. Increased brain uptake of docetaxel and ketoconazole loaded folate-grafted solid lipid nanoparticles. Nanomedicine 2013, 9, 111–121. [Google Scholar] [CrossRef]
- Gandhi, N.S.; Tekade, R.K.; Chougule, M.B. Nanocarrier mediated delivery of siRNA/miRNA in combination with chemotherapeutic agents for cancer therapy: Current progress and advances. J. Control. Release 2014, 194, 238–256. [Google Scholar] [CrossRef]
- Saad, M.; Garbuzenko, O.B.; Minko, T. Co-delivery of siRNA and an anticancer drug for treatment of multidrug-resistant cancer. Nanomedicine 2008, 3, 761–776. [Google Scholar] [CrossRef]
- Fernandes, C.; Suares, D.; Yergeri, M.C. Tumor microenvironment targeted nanotherapy. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Lu, C.; Zhang, X.; Li, J.; Jiang, H. Targeted delivery of etoposide to cancer cells by folate-modified nanostructured lipid drug delivery system. Drug Deliv. 2016, 23, 1838–1845. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kuo, Y.-C.; Lee, C.-H. Inhibition against growth of glioblastoma multiforme in vitro using etoposide-loaded solid lipid nanoparticles with p-aminophenyl-α-d-manno-pyranoside and folic acid. J. Pharm. Sci. 2015, 104, 1804–1814. [Google Scholar] [CrossRef] [PubMed]
- Sanna, V.; Pala, N.; Sechi, M. Targeted therapy using nanotechnology: Focus on cancer. Int. J. Nanomed. 2014, 9, 467–483. [Google Scholar] [CrossRef]
- Song, S.; Mao, G.; Du, J.; Zhu, X. Novel RGD containing, temozolomide-loading nanostructured lipid carriers for glioblastoma multiforme chemotherapy. Drug Deliv. 2016, 23, 1404–1408. [Google Scholar] [CrossRef] [PubMed]
- Chou, L.Y.T.; Ming, K.; Chan, W.C.W. Strategies for the intracellular delivery of nanoparticles. Chem. Soc. Rev. 2011, 40, 233–245. [Google Scholar] [CrossRef]
- Singh, I.; Swami, R.; Pooja, D.; Jeengar, M.K.; Khan, W.; Sistla, R. Lactoferrin bioconjugated solid lipid nanoparticles: A new drug delivery system for potential brain targeting. J. Drug Target. 2016, 24, 212–223. [Google Scholar] [CrossRef]
- Hayward, S.L.; Wilson, C.L.; Kidambi, S. Hyaluronic acid-conjugated liposome nanoparticles for targeted delivery to CD44 overexpressing glioblastoma cells. Oncotarget 2016, 7, 34158–34171. [Google Scholar] [CrossRef]
- Shen, H.; Shi, S.; Zhang, Z.; Gong, T.; Sun, X. Coating solid lipid nanoparticles with hyaluronic acid enhances antitumor activity against melanoma stem-like cells. Theranostics 2015, 5, 755–771. [Google Scholar] [CrossRef]
- Tran, T.H.; Choi, J.Y.; Ramasamy, T.; Truong, D.H.; Nguyen, C.N.; Choi, H.-G.; Yong, C.S.; Kim, J.O. Hyaluronic acid-coated solid lipid nanoparticles for targeted delivery of vorinostat to CD44 overexpressing cancer cells. Carbohydr. Polym. 2014, 114, 407–415. [Google Scholar] [CrossRef]
- Singh, D.; Srivastava, S.K.; Chaudhuri, T.K.; Upadhyay, G. Multifaceted role of matrix metalloproteinases (MMPs). Front. Mol. Biosci. 2015, 2. [Google Scholar] [CrossRef] [PubMed]
- Bruun, J.; Larsen, T.B.; Jølck, R.I.; Eliasen, R.; Holm, R.; Gjetting, T.; Andresen, T.L. Investigation of enzyme-sensitive lipid nanoparticles for delivery of siRNA to blood-brain barrier and glioma cells. Int. J. Nanomed. 2015, 10, 5995–6008. [Google Scholar] [CrossRef]
- Mo, R.; Gu, Z. Tumor microenvironment and intracellular signal-activated nanomaterials for anticancer drug delivery. Mater. Today 2016, 19, 274–283. [Google Scholar] [CrossRef]
- Chuang, C.-H.; Wu, P.-C.; Tsai, T.-H.; Fang, Y.-P.; Tsai, Y.-H.; Cheng, T.-C.; Huang, C.-C.; Huang, M.-Y.; Chen, F.-M.; Hsieh, Y.-C.; et al. Development of pH-sensitive cationic pegylated solid lipid nanoparticles for selective cancer-targeted therapy. J. Biomed. Nanotechnol. 2017, 13, 192–203. [Google Scholar] [CrossRef]
- Kim, C.H.; Sa, C.-K.; Goh, M.S.; Lee, E.S.; Kang, T.H.; Yoon, H.Y.; Battogtokh, G.; Ko, Y.T.; Choi, Y.W. pH-sensitive PEGylation of RIPL peptide-conjugated nanostructured lipid carriers: Design and in vitro evaluation. Int. J. Nanomed. 2018, 13, 6661–6675. [Google Scholar] [CrossRef] [PubMed]
- Saito, G.; Swanson, J.A.; Lee, K.-D. Drug delivery strategy utilizing conjugation via reversible disulfide linkages: Role and site of cellular reducing activities. Adv. Drug Deliv. Rev. 2003, 55, 199–215. [Google Scholar] [CrossRef]
- McNeeley, K.; Karathanasis, E.; Annapragada, A.; Bellamkonda, R. Masking and triggered unmasking of targeting ligands on nanocarriers to improve drug delivery to brain tumors. Biomaterials 2009, 30, 3986–3995. [Google Scholar] [CrossRef]
- Pires, A.; Fortuna, A.; Alves, G.; Falcão, A. Intranasal drug delivery: How, why and what for? J. Pharm. Pharm. Sci. 2009, 12, 288–311. [Google Scholar] [CrossRef]
- Marttin, E.; Schipper, N.G.; Verhoef, J.C.; Merkus, F.W.H. Nasal mucociliary clearance as a factor in nasal drug delivery. Adv. Drug Deliv. Rev. 1998, 29, 13–38. [Google Scholar] [CrossRef]
- Charlton, S.; Jones, N.S.; Davis, S.S.; Illum, L. Distribution and clearance of bioadhesive formulations from the olfactory region in man: Effect of polymer type and nasal delivery device. Eur. J. Pharm. Sci. 2007, 30, 295–302. [Google Scholar] [CrossRef]
- Haffejee, N.; Du Plessis, J.; Müller, D.G.; Schultz, C.; Kotzé, A.F.; Goosen, C. Intranasal toxicity of selected absorption enhancers. Pharmazie 2001, 56, 882–888. [Google Scholar] [PubMed]
- Madane, R.G.; Mahajan, H.S. Curcumin-loaded nanostructured lipid carriers (NLCs) for nasal administration: Design, characterization, and in vivo study. Drug Deliv. 2016, 23, 1326–1334. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Imam, S.S.; Aqil, M.; Ahad, A.; Sultana, Y.; Ali, A.; Khan, K. Brain targeting of temozolomide via the intranasal route using lipid-based nanoparticles: Brain pharmacokinetic and scintigraphic analyses. Mol. Pharm. 2016, 13, 3773–3782. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhao, X.; Du, J.; Liu, M.; Feng, J.; Hu, K. Improved brain delivery of pueraria flavones via intranasal administration of borneol-modified solid lipid nanoparticles. Nanomedicine 2019, 14, 2105–2119. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, X.; Du, S.; Wu, Q.; Yao, Z.; Zhai, Y. The in situ and in vivo study on enhancing effect of borneol in nasal absorption of Geniposide in rats. Arch. Pharm. Res. 2010, 33, 691–696. [Google Scholar] [CrossRef]
- Gartziandia, O.; Herran, E.; Pedraz, J.L.; Carro, E.; Igartua, M.; Hernandez, R.M. Chitosan coated nanostructured lipid carriers for brain delivery of proteins by intranasal administration. Colloids Surf. B 2015, 134, 304–313. [Google Scholar] [CrossRef]
- Bies, C.; Lehr, C.-M.; Woodley, J.F. Lectin-mediated drug targeting: History and applications. Adv. Drug Deliv. Rev. 2004, 56, 425–435. [Google Scholar] [CrossRef]
- Broadwell, R.D.; Balin, B.J. Endocytic and exocytic pathways of the neuronal secretory process and trans-synaptic transfer of wheat germ agglutinin-horseradish peroxidase in vivo. J. Comp. Neurol. 1985, 242, 632–650. [Google Scholar] [CrossRef]
- Thorne, R.G.; Emory, C.R.; Ala, T.A.; Frey, W.H. Quantitative analysis of the olfactory pathway for drug delivery to the brain. Brain Res. 1995, 692, 278–282. [Google Scholar] [CrossRef]
- Fu, T.; Burbage, C.; Tagge, E.P.; Brothers, T.; Willingham, M.C.; Frankel, A.E. Ricin toxin contains three lectin sites which contribute to its in vivo toxicity. Int. J. Immunopharmacol. 1996, 18, 685–692. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Strategies to Enhance Crossing the BBB | ||||
| Formulation | Cargo/drug | Ligand | Target | Ref |
| SLN | Docetaxel | Angiopep-2 | lipoprotein receptor related protein 1 (LRP1) | [56] |
| SLN | Etoposide | melanotransferrin antibody (MA) | Melanotransferrin | [57] |
| Cationic SLN | Biacalin | OX26 monoclonal antibody | Transferrin receptor (TfR) | [59] |
| Cationic SLN | Carmustine | Anti-EGFR | EGFR | [60] |
| SLN | Doxorubicin | Aprotinin, melanotransferrin antibody | low-density lipoprotein receptor (LDLR) related protein (LRP), melanotransferrin | [61] |
| SLN | Methotrexate | Bovine serum albumin (BSA) | Negative charge of BBB endothelial cells membrane | [62] |
| Modification Strategies Against GBM Cells Resistance | ||||
| Formulation | Cargo/drug | Strategy | Target | Ref |
| SLN | Edelfosine | Tween® 80 | P-gp efflux | [65] |
| SLN | Trans-Resveratrol | TPGS | P-gp efflux | [66] |
| SLN | Noscapine | PEG | P-gp efflux | [68] |
| SLN | Curcumin, Piperine | TPGS and Brij 78 | MDR effect | [64] |
| Folate SLN | Docetaxel | Ketoconazol | P-gp efflux | [69] |
| Strategies for Selective Targeting of GBM Cells | ||||
| NLC | Etoposide | Folic acid | Folate receptor | [73] |
| NLC | Etoposide | Folic acid, ρ-aminophenyl-α-d-manno-pyranoside (APMP) | Folate receptor, glucose transporter 1 | [74] |
| SLN | Carmustine | Cetuximab | EGFR | [60] |
| NLC | Temozolomide | RGD peptide | Integrin receptors | [76] |
| SLN | Docetaxel | Lactoferrin | Lactoferrin receptors | [78] |
| SLN | Vorinostat | Hyaluronic acid | CD44 | [81] |
| LNP | siRNA | PEGylated (poly (ethylene glycol)) cleavable lipopeptide | MMPs | [83] |
| Cationic SLN | camptothecin | Cleavable PEG | Tumor low pH | [85] |
| Modified Lipid Nanoparticles for Nose-to-Brain Delivery | ||||
| SLN | Pueraria flavone | Borneol | Improve crossing the BBB and permeability through nasal mucosa | [95] |
| NLC | Proteins | Chitosan | Prolonged interact with nasal mucosa | [97] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jnaidi, R.; Almeida, A.J.; Gonçalves, L.M. Solid Lipid Nanoparticles and Nanostructured Lipid Carriers as Smart Drug Delivery Systems in the Treatment of Glioblastoma Multiforme. Pharmaceutics 2020, 12, 860. https://doi.org/10.3390/pharmaceutics12090860
Jnaidi R, Almeida AJ, Gonçalves LM. Solid Lipid Nanoparticles and Nanostructured Lipid Carriers as Smart Drug Delivery Systems in the Treatment of Glioblastoma Multiforme. Pharmaceutics. 2020; 12(9):860. https://doi.org/10.3390/pharmaceutics12090860
Chicago/Turabian StyleJnaidi, Raneem, António José Almeida, and Lídia M. Gonçalves. 2020. "Solid Lipid Nanoparticles and Nanostructured Lipid Carriers as Smart Drug Delivery Systems in the Treatment of Glioblastoma Multiforme" Pharmaceutics 12, no. 9: 860. https://doi.org/10.3390/pharmaceutics12090860
APA StyleJnaidi, R., Almeida, A. J., & Gonçalves, L. M. (2020). Solid Lipid Nanoparticles and Nanostructured Lipid Carriers as Smart Drug Delivery Systems in the Treatment of Glioblastoma Multiforme. Pharmaceutics, 12(9), 860. https://doi.org/10.3390/pharmaceutics12090860