Independent Tailoring of Dose and Drug Release via a Modularized Product Design Concept for Mass Customization

 , ,
, ,  ,
,
Abstract
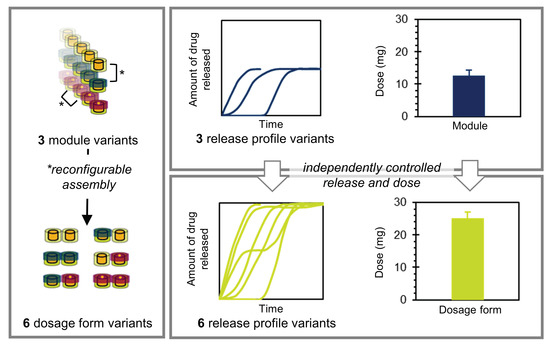
1. Introduction
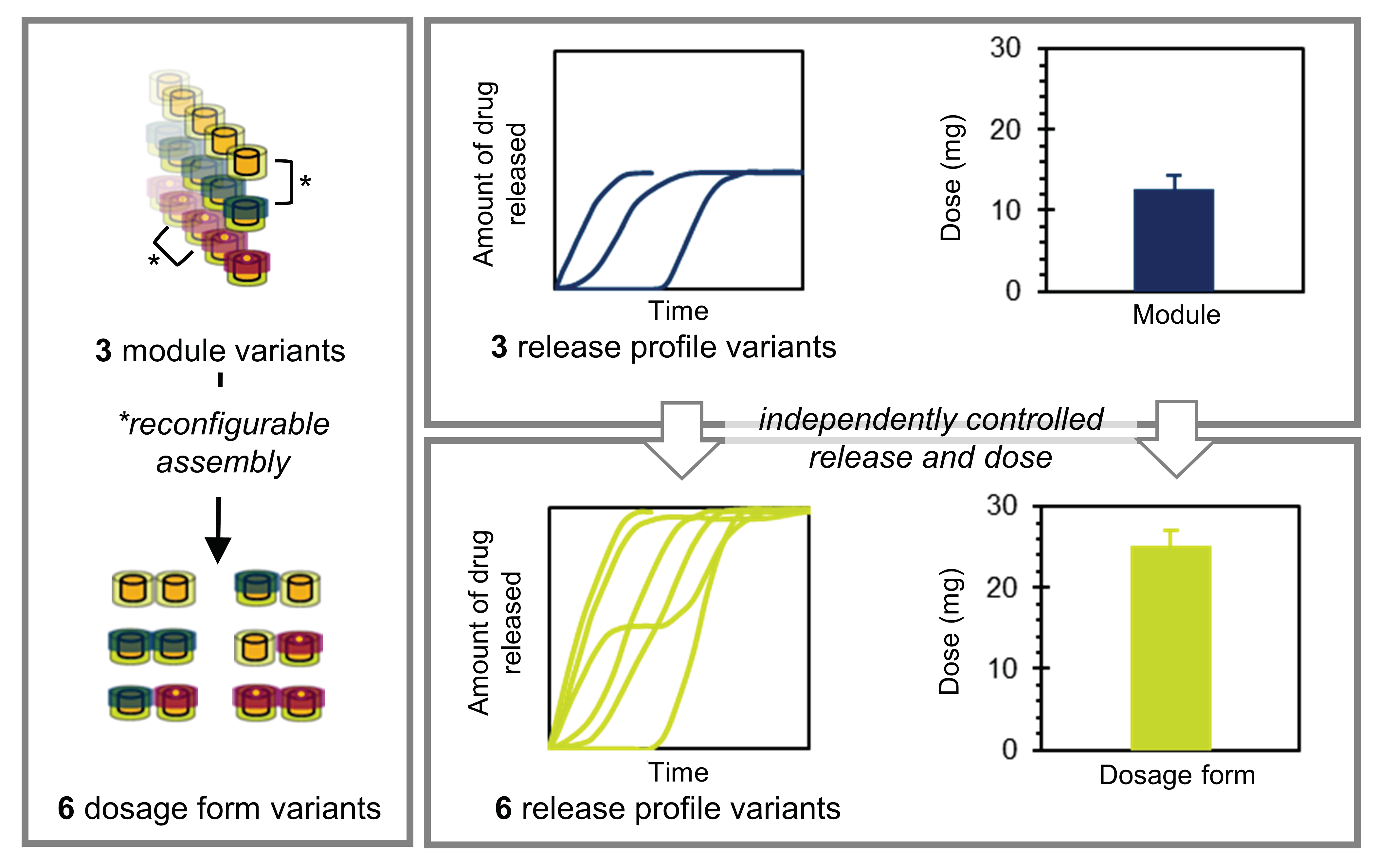
1.1. Theoretical Considerations: Pharmaceutical Product Modularization
1.2. Design of the Proposed Product Concept
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Technical Realization of the Product Concept
2.2.2. Fused Deposition Modeling (FDM)
2.2.3. Melt Extrusion of the Drug-Containing Filament
2.2.4. Melt Molding of Drug-Containing Cores
2.2.5. Thermal Characterization
2.2.6. X-Ray Computed Microtomography (X-Ray µCT)
2.2.7. In Vitro Drug Release and Drug Content Homogeneity
3. Results
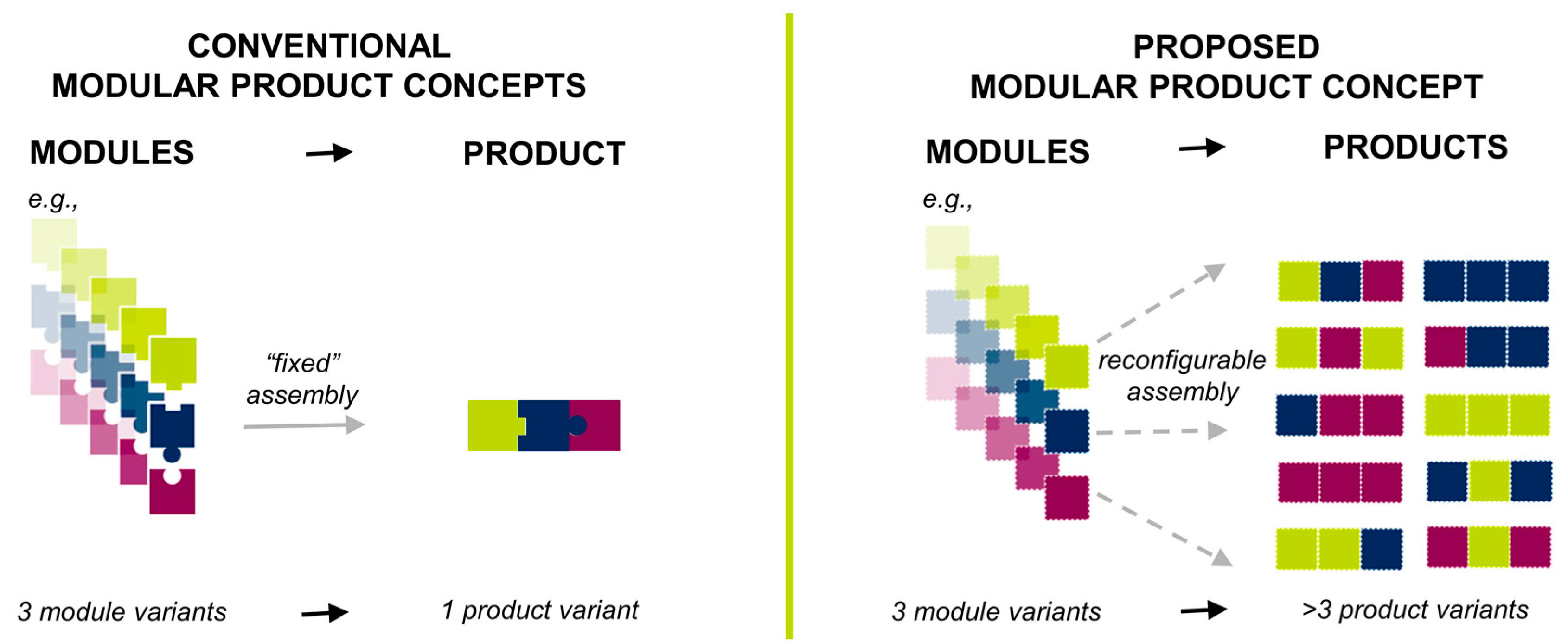
3.1. Realization of the Product Concept
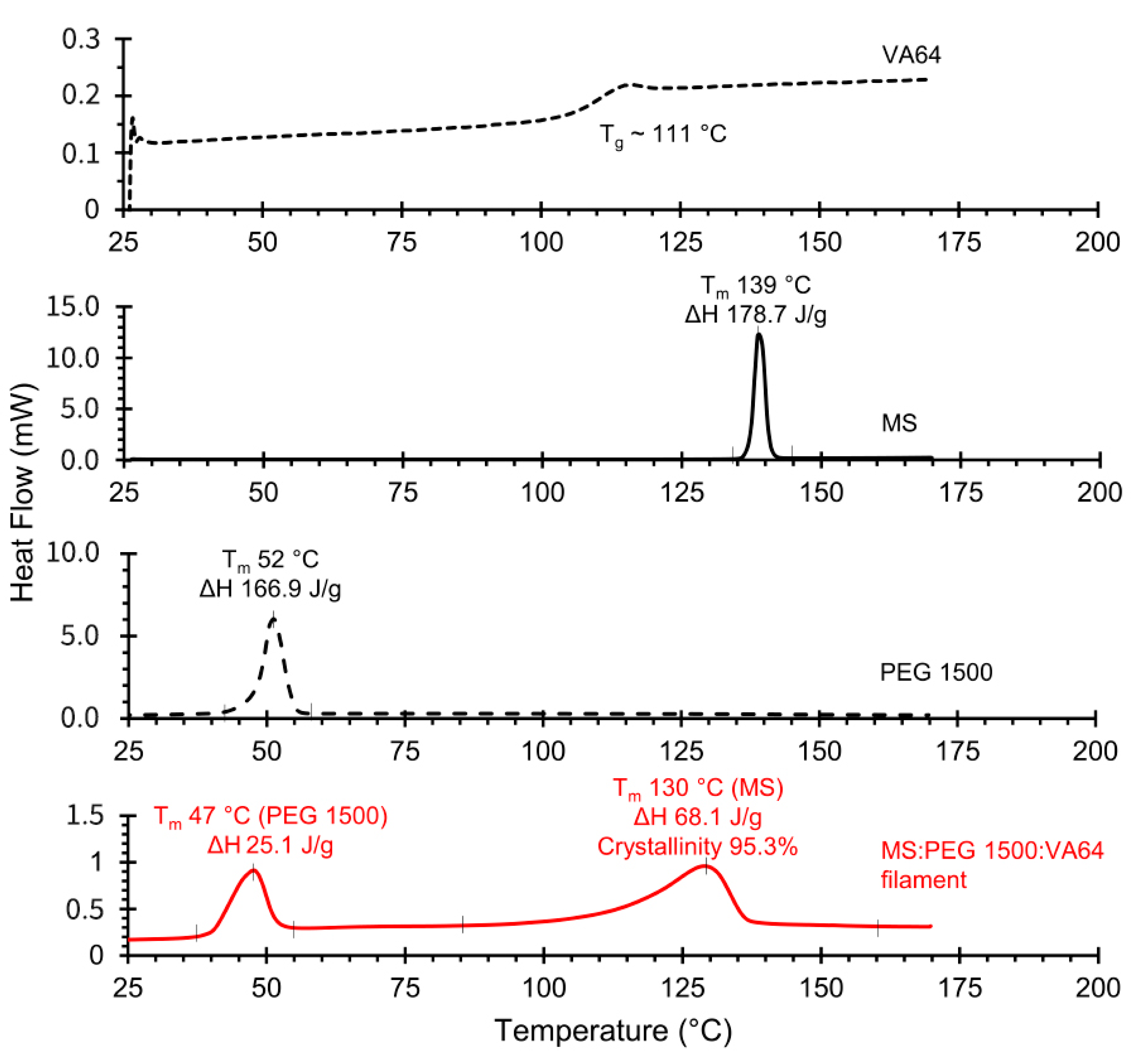
3.2. Thermal Characterization
3.3. X-Ray Computed Microtomography
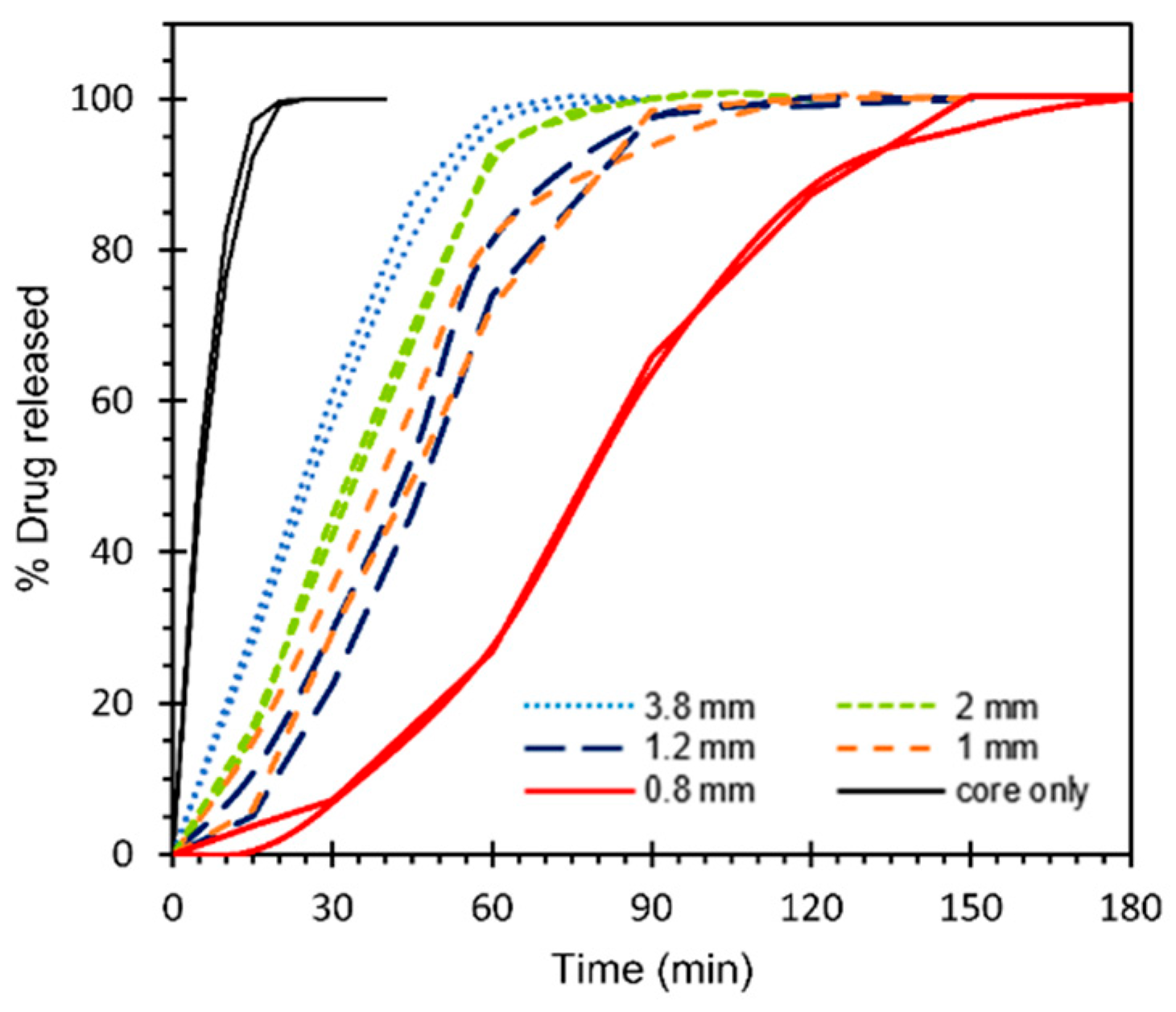
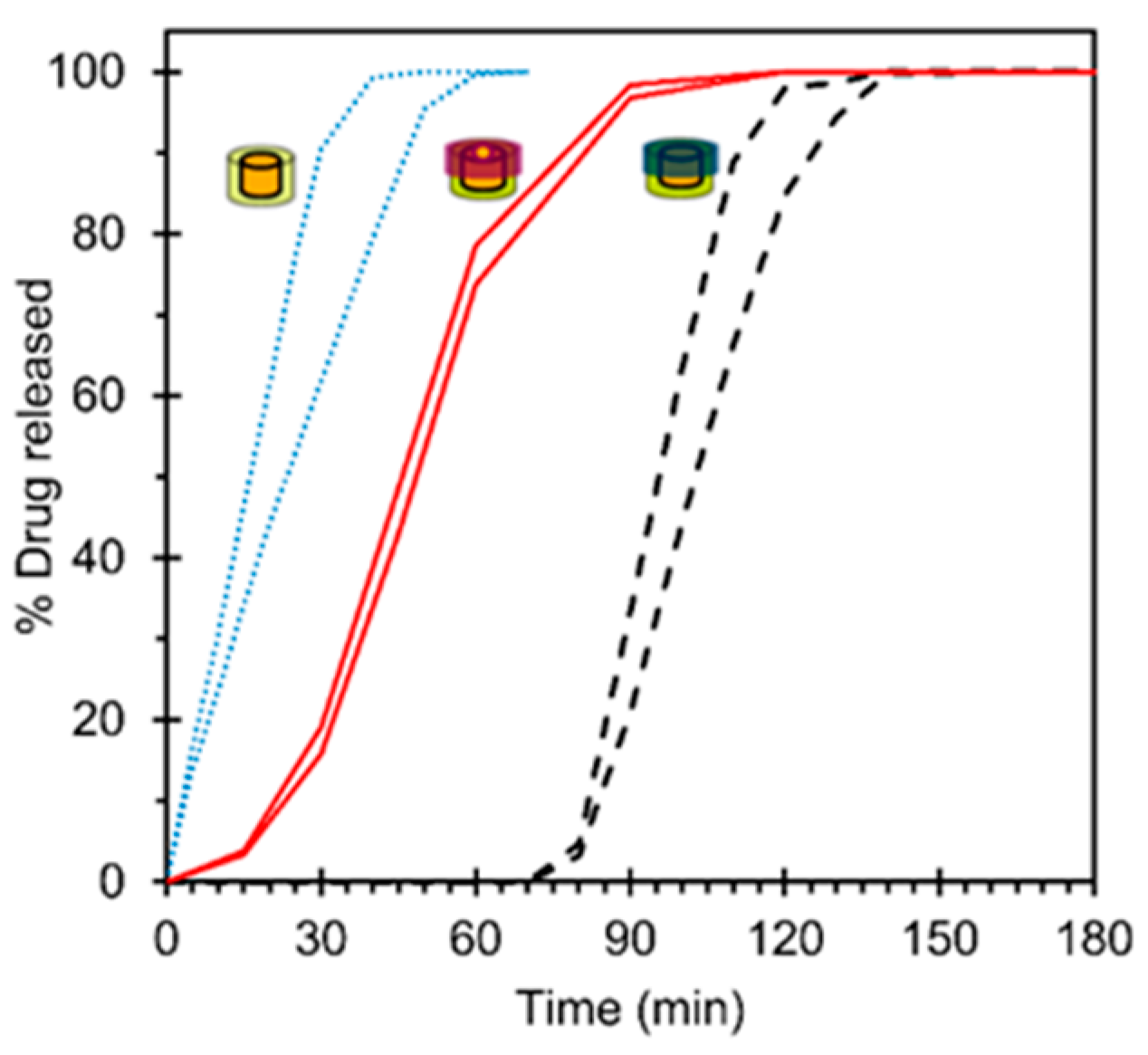
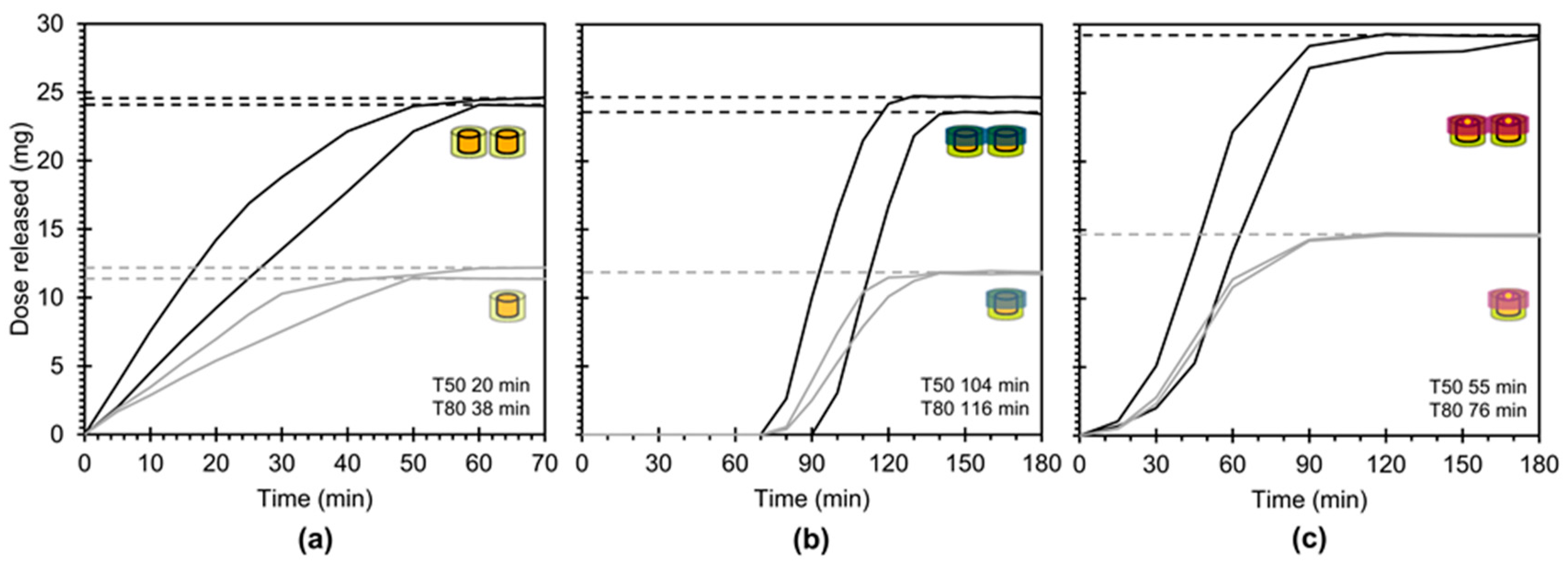
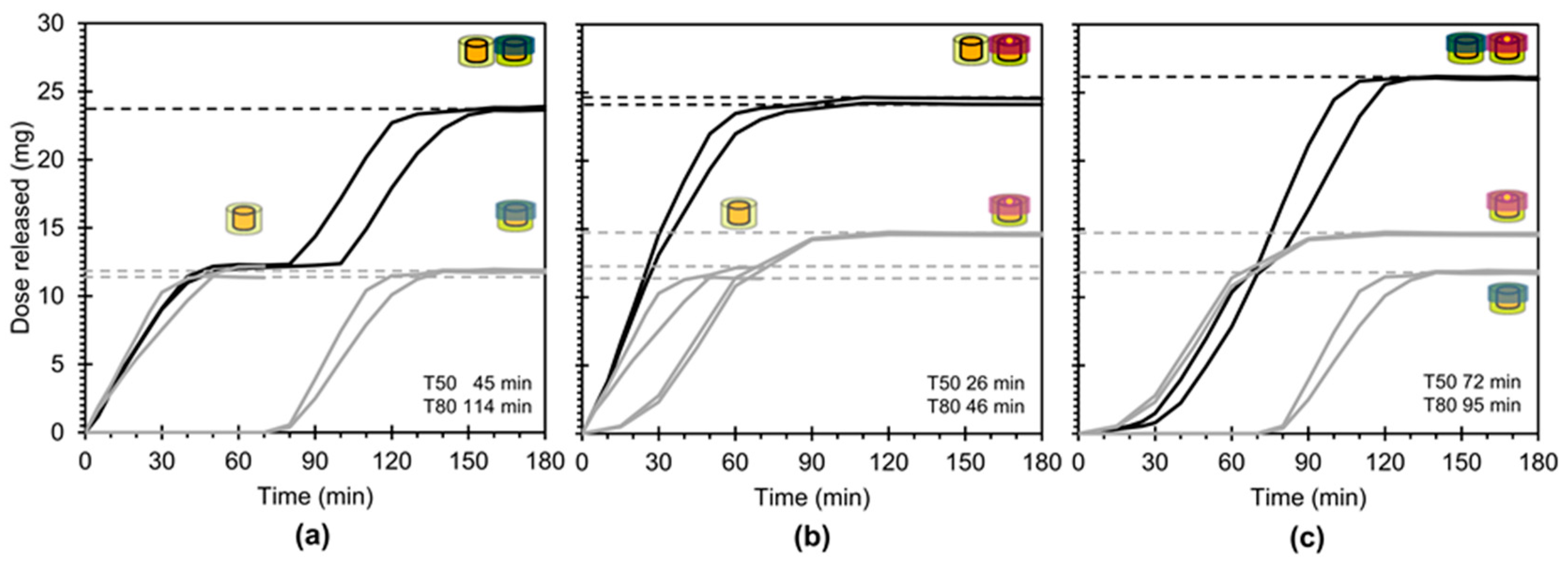
3.4. In Vitro Drug Release
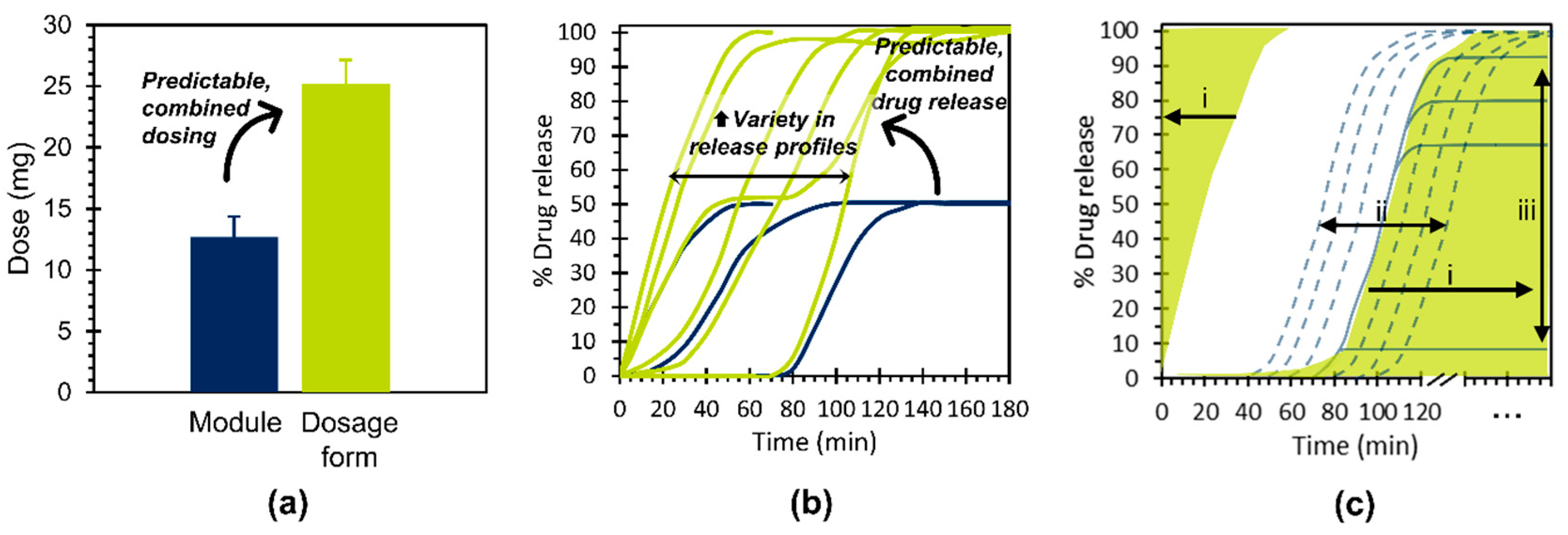
4. Discussion
4.1. Key Study Outcomes and Concept Potential
- Independently achieve predictable, combined dosing and predictable, combined drug release kinetics for dosage forms based on their constituent modules and,
- Contribute to the enhanced product variety necessary for the provision of individualized therapy.
4.2. Realization Challenges
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Florence, A.T.; Lee, V.H. Personalised medicines: More tailored drugs, more tailored delivery. Int. J. Pharm. 2011, 415, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, R.; Aksu, B.; Cuine, A.; Danhof, M.; Takac, M.J.; Linden, H.H.; Link, A.; Muchitsch, E.M.; Wilson, C.G.; Ohrngren, P.; et al. Towards a European strategy for medicines research (2014–2020): The EUFEPS position paper on Horizon 2020. Eur. J. Pharm. Sci. 2012, 47, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Govender, R.; Abrahmsen-Alami, S.; Larsson, A.; Folestad, S. Therapy for the individual: Towards patient integration into the manufacturing and provision of pharmaceuticals. Eur. J. Pharm. Biopharm. 2020, 149, 58–76. [Google Scholar] [CrossRef] [PubMed]
- Lewis, L.D. Personalized drug therapy; the genome, the chip and the physician. Br. J. Clin. Pharmacol. 2005, 60, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Meyer, U.A. Pharmacogenetics—Five decades of therapeutic lessons from genetic diversity. Nat. Rev. Genet. 2004, 5, 669–676. [Google Scholar] [CrossRef]
- Stegemann, S. Towards better understanding of patient centric drug product development in an increasingly older patient population. Int. J. Pharm. 2016, 512, 334–342. [Google Scholar] [CrossRef]
- Thummel, K.E.; Lin, Y.S. Sources of interindividual variability. Methods Mol. Biol. 2014, 1113, 363–415. [Google Scholar] [CrossRef]
- Topol, E.J. Individualized medicine from prewomb to tomb. Cell 2014, 157, 241–253. [Google Scholar] [CrossRef]
- Trusheim, M.R.; Berndt, E.R.; Douglas, F.L. Stratified medicine: Strategic and economic implications of combining drugs and clinical biomarkers. Nat. Rev. Drug Discov. 2007, 6, 287–293. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Paving the Way for Personalised Medicine: FDA’s Role in a New Era of Medical Product Development; U.S. Food and Drug Administration: Silver Spring, MD, USA, 2013; pp. 1–62.
- Deng, J.; Vozmediano, V.; Rodriguez, M.; Cavallari, L.H.; Schmidt, S. Genotype-guided dosing of warfarin through modeling and simulation. Eur. J. Pharm. Sci. 2017, 109, S9–S14. [Google Scholar] [CrossRef]
- Ferrendelli, J.A. Concerns with antiepileptic drug initiation: Safety, tolerability, and efficacy. Epilepsia 2001, 42 (Suppl. 4), 28–30. [Google Scholar] [CrossRef] [PubMed]
- Nidanapu, R.P.; Rajan, S.; Mahadevan, S.; Gitanjali, B. Tablet Splitting of Antiepileptic Drugs in Pediatric Epilepsy: Potential Effect on Plasma Drug Concentrations. Paediatr. Drugs 2016, 18, 451–463. [Google Scholar] [CrossRef]
- Pouplin, T.; Phuong, P.N.; Toi, P.V.; Nguyen Pouplin, J.; Farrar, J. Isoniazid, pyrazinamide and rifampicin content variation in split fixed-dose combination tablets. PLoS ONE 2014, 9, e102047. [Google Scholar] [CrossRef] [PubMed]
- Wening, K.; Breitkreutz, J. Oral drug delivery in personalized medicine: Unmet needs and novel approaches. Int. J. Pharm. 2011, 404, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hens, B.; Corsetti, M.; Spiller, R.; Marciani, L.; Vanuytsel, T.; Tack, J.; Talattof, A.; Amidon, G.L.; Koziolek, M.; Weitschies, W.; et al. Exploring gastrointestinal variables affecting drug and formulation behavior: Methodologies, challenges and opportunities. Int. J. Pharm. 2017, 519, 79–97. [Google Scholar] [CrossRef]
- Khaled, S.A.; Burley, J.C.; Alexander, M.R.; Yang, J.; Roberts, C.J. 3D printing of five-in-one dose combination polypill with defined immediate and sustained release profiles. J. Control. Release 2015, 217, 308–314. [Google Scholar] [CrossRef]
- McConnell, E.L.; Fadda, H.M.; Basit, A.W. Gut instincts: Explorations in intestinal physiology and drug delivery. Int. J. Pharm. 2008, 364, 213–226. [Google Scholar] [CrossRef]
- Bhatia, S.; Kumar, B.; Mittal, S. Oral Chronotherapeutics: Future of Drug Delivery Systems. Int. J. Sci. Study 2014, 2, 55–58. [Google Scholar]
- Effinger, A.; O’Driscoll, C.M.; McAllister, M.; Fotaki, N. Impact of gastrointestinal disease states on oral drug absorption—Implications for formulation design—A PEARRL review. J. Pharm. Pharmacol. 2019, 71, 674–698. [Google Scholar] [CrossRef]
- Trattner, A.; Hvam, L.; Forza, C.; Herbert-Hansen, Z.N.L. Product complexity and operational performance: A systematic literature review. CIRP J. Manuf. Sci. Technol. 2019, 25, 69–83. [Google Scholar] [CrossRef]
- Wilson, M.W. Manufacturing Platforms for Patient-Centric Drug Products. In Developing Drug Products in an Ageing Society; Stegemann, S., Ed.; Springer: Cham, Switzerland, 2016; Volume 24, pp. 447–483. [Google Scholar] [CrossRef]
- Srai, J.S.; Harrington, T.; Alinaghian, L.; Phillips, M. Evaluating the potential for the continuous processing of pharmaceutical products—A supply network perspective. Chem. Eng. Process. 2015, 97, 248–258. [Google Scholar] [CrossRef]
- Fogliatto, F.S.; da Silveira, G.J.C.; Borenstein, D. The mass customization decade: An updated review of the literature. Int. J. Prod. Econ. 2012, 138, 14–25. [Google Scholar] [CrossRef]
- Hu, S.J. Evolving Paradigms of Manufacturing: From Mass Production to Mass Customization and Personalization. Procedia CIRP 2013, 7, 3–8. [Google Scholar] [CrossRef]
- Mourtzis, D. Challenges and future perspectives for the life cycle of manufacturing networks in the mass customisation era. Logist. Res. 2016, 9, 1–20. [Google Scholar] [CrossRef]
- Pillar, F. Mass Customization: Reflections on the State of the Concept. Int. J. Flex. Manuf. Syst. 2004, 16, 313–334. [Google Scholar] [CrossRef]
- Zhang, M.; Guo, H.; Huo, B.; Zhao, X.; Huang, J. Linking supply chain quality integration with mass customization and product modularity. Int. J. Prod. Econ. 2019, 207, 227–235. [Google Scholar] [CrossRef]
- Um, J.; Lyons, A.; Lam, H.K.S.; Cheng, T.C.E.; Dominguez-Pery, C. Product variety management and supply chain performance: A capability perspective on their relationships and competitiveness implications. Int. J. Prod. Econ. 2017, 187, 15–26. [Google Scholar] [CrossRef]
- Salvador, F.; Forza, C.; Rungtusanathan, M. Modularity, product variety, production volume, and component sourcing: Theorizing beyond generic prescriptions. J. Oper. Manag. 2002, 20, 549–575. [Google Scholar] [CrossRef]
- ElMaraghy, H.; Schuh, G.; ElMaraghy, W.; Piller, F.; Schönsleben, P.; Tseng, M.; Bernard, A. Product variety management. CIRP Ann. 2013, 62, 629–652. [Google Scholar] [CrossRef]
- Srai, J.S.; Badman, C.; Krumme, M.; Futran, M.; Johnston, C. Future Supply Chains Enabled by Continuous Processing—Opportunities Challenges May 20–21 2014 Continuous Manufacturing Symposium. J. Pharm. Sci. 2015, 104, 840–849. [Google Scholar] [CrossRef]
- Srai, J.S.; Harrington, T.S.; Tiwari, M.K. Characteristics of redistributed manufacturing systems: A comparative study of emerging industry supply networks. Int. J. Prod. Res. 2016, 54, 6936–6955. [Google Scholar] [CrossRef]
- Roscoe, S.; Blome, C. Understanding the emergence of redistributed manufacturing: An ambidexterity perspective. Prod. Plan. Control 2019, 30, 496–509. [Google Scholar] [CrossRef]
- Aquino, R.P.; Barile, S.; Grasso, A.; Saviano, M. Envisioning smart and sustainable healthcare: 3D printing technologies for personalized medication. Futures 2018, 103, 35–50. [Google Scholar] [CrossRef]
- Guo, F.; Gershenson, J.K. Discovering relationships between modularity and cost. J. Intell. Manuf. 2007, 18, 143–157. [Google Scholar] [CrossRef]
- Gershenson, J.K.; Prasad, G.J.; Zhang, Y. Product modularity: Definitions and benefits. J. Eng. Des. 2003, 14, 295–313. [Google Scholar] [CrossRef]
- Allen, K.R.; Carlson-Skalak, S. Defining product architecture during conceptual design. In Proceedings of the ASME Design Engineering Technical Conference, Atlanta, GA, USA, 13–16 September 1998. [Google Scholar]
- Varum, F.J.; Merchant, H.A.; Basit, A.W. Oral modified-release formulations in motion: The relationship between gastrointestinal transit and drug absorption. Int. J. Pharm. 2010, 395, 26–36. [Google Scholar] [CrossRef]
- Petrovick, G.F.; Kleinebudde, P.; Breitkreutz, J. Orodispersible tablets containing taste-masked solid lipid pellets with metformin hydrochloride: Influence of process parameters on tablet properties. Eur. J. Pharm. Biopharm. 2018, 122, 137–145. [Google Scholar] [CrossRef]
- Dalskov Mosgaard, M.; Strindberg, S.; Abid, Z.; Singh Petersen, R.; Hojlund Eklund Thamdrup, L.; Joukainen Andersen, A.; Sylvest Keller, S.; Mullertz, A.; Hagner Nielsen, L.; Boisen, A. Ex vivo intestinal perfusion model for investigating mucoadhesion of microcontainers. Int. J. Pharm. 2019, 570, 118658. [Google Scholar] [CrossRef]
- Correa, S.; Dreaden, E.C.; Gu, L.; Hammond, P.T. Engineering nanolayered particles for modular drug delivery. J. Control. Release 2016, 240, 364–386. [Google Scholar] [CrossRef]
- Demiri, V.; Stranzinger, S.; Rinner, P.; Piller, M.; Sacher, S.; Lingitz, J.; Khinast, J.; Salar-Behzadi, S. Gluing Pills Technology: A novel route to multilayer tablet manufacturing. Int. J. Pharm. 2018, 548, 672–681. [Google Scholar] [CrossRef]
- Melocchi, A.; Uboldi, M.; Maroni, A.; Foppoli, A.; Palugan, L.; Zema, L.; Gazzaniga, A. 3D printing by fused deposition modeling of single- and multi-compartment hollow systems for oral delivery—A review. Int. J. Pharm. 2020, 579, 119155. [Google Scholar] [CrossRef] [PubMed]
- Melocchi, A.; Uboldi, M.; Parietti, F.; Cerea, M.; Foppoli, A.; Palugan, L.; Gazzaniga, A.; Maroni, A.; Zema, L. Lego-Inspired Capsular Devices for the Development of Personalized Dietary Supplements: Proof of Concept With Multimodal Release of Caffeine. J. Pharm. Sci. 2020, 109, 1990–1999. [Google Scholar] [CrossRef] [PubMed]
- Mitra, B.; Thool, P.; Meruva, S.; Aycinena, J.A.; Li, J.; Patel, J.; Patel, K.; Agarwal, A.; Karki, S.; Bowen, W. Decoding the small size challenges of mini-tablets for enhanced dose flexibility and micro-dosing. Int. J. Pharm. 2020, 574, 118905. [Google Scholar] [CrossRef] [PubMed]
- Niwa, K.; Takaya, T.; Morimoto, T.; Takada, K. Preparation and evaluation of a time-controlled release capsule made of ethylcellulose for colon delivery of drugs. J. Drug Target. 1995, 3, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, P.R.; Bernardi, L.S.; Strusi, O.L.; Mercuri, S.; Segatto Silva, M.A.; Colombo, P.; Sonvico, F. Assembled modules technology for site-specific prolonged delivery of norfloxacin. Int. J. Pharm. 2011, 405, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Patwekar, S.L.; Baramade, M.K. Controlled Release Approach to Novel Multiparticulate Drug Delivery System. Int. J. Pharm. Pharm. Sci. 2012, 4, 757–763. [Google Scholar]
- Pereira, B.C.; Isreb, A.; Isreb, M.; Forbes, R.T.; Oga, E.F.; Alhnan, M.A. Additive Manufacturing of a Point-of-Care “Polypill:” Fabrication of Concept Capsules of Complex Geometry with Bespoke Release against Cardiovascular Disease. Adv. Healthc. Mater. 2020, 9, 2000236. [Google Scholar] [CrossRef]
- Rahmani, S.; Park, T.H.; Dishman, A.F.; Lahann, J. Multimodal delivery of irinotecan from microparticles with two distinct compartments. J. Control. Release 2013, 172, 239–245. [Google Scholar] [CrossRef]
- Sadia, M.; Isreb, A.; Abbadi, I.; Isreb, M.; Aziz, D.; Selo, A.; Timmins, P.; Alhnan, M.A. From ‘fixed dose combinations’ to ‘a dynamic dose combiner’: 3D printed bi-layer antihypertensive tablets. Eur. J. Pharm. Sci. 2018, 123, 484–494. [Google Scholar] [CrossRef]
- Tagami, T.; Nagata, N.; Hayashi, N.; Ogawa, E.; Fukushige, K.; Sakai, N.; Ozeki, T. Defined drug release from 3D-printed composite tablets consisting of drug-loaded polyvinylalcohol and a water-soluble or water-insoluble polymer filler. Int. J. Pharm. 2018, 543, 361–367. [Google Scholar] [CrossRef]
- Tan, Y.J.N.; Yong, W.P.; Kochhar, J.S.; Khanolkar, J.; Yao, X.; Sun, Y.; Ao, C.K.; Soh, S. On-demand fully customizable drug tablets via 3D printing technology for personalized medicine. J. Control. Release 2020, 322, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Huang, L.; Zhang, Y.; Meng, F.; Donoso, M.; Haskell, R.; Luo, L. Tunable Two-Compartment On-Demand Sustained Drug Release Based on Lipid Gels. J. Pharm. Sci. 2020, 109, 1059–1067. [Google Scholar] [CrossRef] [PubMed]
- Awad, A.; Fina, F.; Trenfield, S.J.; Patel, P.; Goyanes, A.; Gaisford, S.; Basit, A.W. 3D Printed Pellets (Miniprintlets): A Novel, Multi-Drug, Controlled Release Platform Technology. Pharmaceutics 2019, 11, 148. [Google Scholar] [CrossRef]
- Cid, A.G.; Sonvico, F.; Bettini, R.; Colombo, P.; Gonzo, E.; Jimenez-Kairuz, A.F.; Bermudez, J.M. Evaluation of the Drug Release Kinetics in Assembled Modular Systems Based on the Dome Matrix Technology. J. Pharm. Sci. 2020, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Fuenmayor, E.; O’Donnell, C.; Gately, N.; Doran, P.; Devine, D.M.; Lyons, J.G.; McConville, C.; Major, I. Mass-customization of oral tablets via the combination of 3D printing and injection molding. Int. J. Pharm. 2019, 569, 118611. [Google Scholar] [CrossRef]
- Jamroz, W.; Kurek, M.; Szafraniec-Szczesny, J.; Czech, A.; Gawlak, K.; Knapik-Kowalczuk, J.; Leszczynski, B.; Wrobel, A.; Paluch, M.; Jachowicz, R. Speed it up, slow it down...An issue of bicalutamide release from 3D printed tablets. Eur. J. Pharm. Sci. 2020, 143, 105169. [Google Scholar] [CrossRef]
- Marucci, M.; Andersson, H.; Hjartstam, J.; Stevenson, G.; Baderstedt, J.; Stading, M.; Larsson, A.; von Corswant, C. New insights on how to adjust the release profile from coated pellets by varying the molecular weight of ethyl cellulose in the coating film. Int. J. Pharm. 2013, 458, 218–223. [Google Scholar] [CrossRef]
- Xu, X.; Zhao, J.; Wang, M.; Wang, L.; Yang, J. 3D Printed Polyvinyl Alcohol Tablets with Multiple Release Profiles. Sci. Rep. 2019, 9, 12487. [Google Scholar] [CrossRef]
- Khaled, S.A.; Burley, J.C.; Alexander, M.R.; Yang, J.; Roberts, C.J. 3D printing of tablets containing multiple drugs with defined release profiles. Int. J. Pharm. 2015, 494, 643–650. [Google Scholar] [CrossRef]
- Laukamp, E.J.; Knop, K.; Thommes, M.; Breitkreutz, J. Micropellet-loaded rods with dose-independent sustained release properties for individual dosing via the Solid Dosage Pen. Int. J. Pharm. 2016, 499, 271–279. [Google Scholar] [CrossRef]
- Smeets, A.; Re, I.L.; Clasen, C.; Van den Mooter, G. Fixed dose combinations for cardiovascular treatment via coaxial electrospraying: Coated amorphous solid dispersion particles. Int. J. Pharm. 2020, 577, 118949. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.C.; Isreb, A.; Forbes, R.T.; Dores, F.; Habashy, R.; Petit, J.B.; Alhnan, M.A.; Oga, E.F. ‘Temporary Plasticiser’: A novel solution to fabricate 3D printed patient-centred cardiovascular ‘Polypill’ architectures. Eur. J. Pharm. Biopharm. 2019, 135, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Genina, N.; Boetker, J.P.; Colombo, S.; Harmankaya, N.; Rantanen, J.; Bohr, A. Anti-tuberculosis drug combination for controlled oral delivery using 3D printed compartmental dosage forms: From drug product design to in vivo testing. J. Control. Release 2017, 268, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Garcia, R.; Prada, M.; Bolas-Fernandez, F.; Ballesteros, M.P.; Serrano, D.R. Oral Fixed-Dose Combination Pharmaceutical Products: Industrial Manufacturing Versus Personalized 3D Printing. Pharm. Res. 2020, 37, 132. [Google Scholar] [CrossRef]
- Siiskonen, M.; Folestad, S.; Malmqvist, J. Applying Function-Means Tree Modelling to Personalized Medicines. In DS91, Proceedings of the NordDesign, Linköping, Sweden, 14–17 August 2018; Ekströmer, P., Schütte, S., Ölvander, J., Eds.; Linköping University: Linköping, Sweden, 2018. [Google Scholar]
- Salvador, F. Toward a Product System Modularity Construct: Literature Review and Reconceptualization. IEEE Trans. Eng. Manag. 2007, 54, 219–240. [Google Scholar] [CrossRef]
- Govender, R.; Abrahmsen-Alami, S.; Folestad, S.; Larsson, A. High Content Solid Dispersions for Dose Window Extension: A Basis for Design Flexibility in Fused Deposition Modelling. Pharm. Res. 2019, 37, 9. [Google Scholar] [CrossRef]
- Shen, J.; Liang, X.; Lei, H. Measurements, Thermodynamic Modeling, and a Hydrogen Bonding Study on the Solubilities of Metoprolol Succinate in Organic Solvents. Molecules 2018, 23, 2469. [Google Scholar] [CrossRef]
- Knopp, M.M.; Tajber, L.; Tian, Y.; Olesen, N.E.; Jones, D.S.; Kozyra, A.; Lobmann, K.; Paluch, K.; Brennan, C.M.; Holm, R.; et al. Comparative Study of Different Methods for the Prediction of Drug-Polymer Solubility. Mol. Pharm. 2015, 12, 3408–3419. [Google Scholar] [CrossRef]
- Quinten, T.; Andrews, G.P.; De Beer, T.; Saerens, L.; Bouquet, W.; Jones, D.S.; Hornsby, P.; Remon, J.P.; Vervaet, C. Preparation and evaluation of sustained-release matrix tablets based on metoprolol and an acrylic carrier using injection moulding. AAPS PharmSciTech 2012, 13, 1197–1211. [Google Scholar] [CrossRef]
- Kolter, K.; Karl, M.; Nalawade, N.; Rottmann, N. Extrusion Compendium: Hot-Melt Extrusion with BASF Pharma Polymers, 2nd ed.; BASF SE Pharma Ingredients and Services: Ludwigshafen, Germany, 2012; pp. 123–125. [Google Scholar]
- Forster, A.; Hempenstall, J.; Tucker, I.; Rades, T. Selection of excipients for melt extrusion with two poorly water-soluble drugs by solubility parameter calculation and thermal analysis. Int. J. Pharm. 2001, 226, 147–161. [Google Scholar] [CrossRef]
- Bochmann, E.S.; Ustuner, E.E.; Gryczke, A.; Wagner, K.G. Predicting melt rheology for hot-melt extrusion by means of a simple Tg-measurement. Eur. J. Pharm. Biopharm. 2017, 119, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.-Y.; Chung, Y.-Y.; Cheah, X.-Z.; Tan, E.Y.-L.; Quah, J. The characterization and dissolution performances of spray dried solid dispersion of ketoprofen in hydrophilic carriers. Asian J. Pharm. Sci. 2015, 10, 372–385. [Google Scholar] [CrossRef]
- Alshafiee, M.; Aljammal, M.K.; Markl, D.; Ward, A.; Walton, K.; Blunt, L.; Korde, S.; Pagire, S.K.; Kelly, A.L.; Paradkar, A.; et al. Hot-melt extrusion process impact on polymer choice of glyburide solid dispersions: The effect of wettability and dissolution. Int. J. Pharm. 2019, 559, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Fahier, J.; Muschert, S.; Fayard, B.; Velghe, C.; Byrne, G.; Doucet, J.; Siepmann, F.; Siepmann, J. Importance of air bubbles in the core of coated pellets: Synchrotron X-ray microtomography allows for new insights. J. Control. Release 2016, 237, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Körner, A.; Piculell, L.; Iselau, F.; Wittgren, B.; Larsson, A. Influence of different polymer types on the overall release mechanism in hydrophilic matrix tablets. Molecules 2009, 14, 2699–2716. [Google Scholar] [CrossRef]
- Burns, S.J.; Higginbottom, S.; Whelan, I.; Bowtle, W.J.; Rosenberg, E.; Corness, D.; Hay, G.; Attwood, D.; Barnwell, S.G. Formulation strategies designed to maintain the biphasic release characteristics of liquid-filled capsules. Int. J. Pharm. 1996, 141, 9–16. [Google Scholar] [CrossRef]
- Smith, D.M.; Kapoor, Y.; Klinzing, G.R.; Procopio, A.T. Pharmaceutical 3D printing: Design and qualification of a single step print and fill capsule. Int. J. Pharm. 2018, 544, 21–30. [Google Scholar] [CrossRef]
- Khaled, S.A.; Burley, J.C.; Alexander, M.R.; Roberts, C.J. Desktop 3D printing of controlled release pharmaceutical bilayer tablets. Int. J. Pharm. 2014, 461, 105–111. [Google Scholar] [CrossRef]
- Maroni, A.; Melocchi, A.; Parietti, F.; Foppoli, A.; Zema, L.; Gazzaniga, A. 3D printed multi-compartment capsular devices for two-pulse oral drug delivery. J. Control. Release 2017, 268, 10–18. [Google Scholar] [CrossRef]
- Matijašić, G.; Gretić, M.; Vinčić, J.; Poropat, A.; Cuculić, L.; Rahelić, T. Design and 3D printing of multi-compartmental PVA capsules for drug delivery. J. Drug Deliv. Sci. Technol. 2019, 52, 677–686. [Google Scholar] [CrossRef]
- Nober, C.; Manini, G.; Carlier, E.; Raquez, J.M.; Benali, S.; Dubois, P.; Amighi, K.; Goole, J. Feasibility study into the potential use of fused-deposition modeling to manufacture 3D-printed enteric capsules in compounding pharmacies. Int. J. Pharm. 2019, 569, 118581. [Google Scholar] [CrossRef] [PubMed]
- Hartman Kok, P.J.; Vonk, P.; Kossen, N.W. A particulate pulse-release system and mathematical description with the Maxwell-Stefan theory. J. Control. Release 2000, 66, 293–306. [Google Scholar] [CrossRef]
- Goyanes, A.; Buanz, A.B.; Basit, A.W.; Gaisford, S. Fused-filament 3D printing (3DP) for fabrication of tablets. Int. J. Pharm. 2014, 476, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Goyanes, A.; Robles Martinez, P.; Buanz, A.; Basit, A.W.; Gaisford, S. Effect of geometry on drug release from 3D printed tablets. Int. J. Pharm. 2015, 494, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Dai, J.; Chang, X.; Yang, M.; Shen, R.; Shan, L.; Qian, Y.; Gao, C. Model drug as pore former for controlled release of water-soluble metoprolol succinate from ethylcellulose-coated pellets without lag phase: Opportunities and challenges. AAPS PharmSciTech 2015, 16, 35–44. [Google Scholar] [CrossRef]
- Chaerunisaa, A.Y.; Ali, R.; Korber, M.; Bodmeier, R. Quantification of porogen effect on the drug release from single- and multi-layered ethylcellulose coated pellets containing single or combined drugs. Int. J. Pharm. 2020, 577, 119050. [Google Scholar] [CrossRef]
- Gebäck, T.; Marucci, M.; Boissier, C.; Arnehed, J.; Heintz, A. Investigation of the Effect of the Tortuous Pore Structure on Water Diffusion through a Polymer Film Using Lattice Boltzmann Simulations. J. Phys. Chem. B 2015, 119, 5220–5227. [Google Scholar] [CrossRef]
- U.S. Department of Health and Human Services Food and Drug Administration. Size, Shape, and Other Physical Attributes of Generic Tablets and Capsules Guidance for Industry. 2015. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/size-shape-and-other-physical-attributes-generic-tablets-and-capsules (accessed on 22 November 2019).
- Siiskonen, M.; Malmqvist, J.; Folestad, S. Integrated product and manufacturing system platforms supporting the design of personalized medicines. J. Manuf. Syst. 2020, 56, 281–295. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Submodular Component | Mean Mass (mg) ± SD | % RSD | n | Dose or Release Rate Determining |
| PLA cups | 29.72 ± 0.32 | 1.1 | 50 | dose and release |
| Core (normal fill) | 28.82 ± 1.04 | 3.6 | 20 | dose |
| Core (overfill) | 33.97 ± 1.42 | 4.2 | 10 | dose |
| Key Feature | Length (mm) ± SD | % RSD | n | Dose or Release Rate Determining |
| Core diameter | 3.81 ± 0.11 | 2.8 | 48 | release |
| MV2 lid thickness | 0.57 ± 0.04 | 6.4 | 16 | release |
| MV3 orifice diameter (for 0.4 mm orifice) | 0.39 ± 0.01 | 2.2 | 15 | release |
| Key Descriptor | Conventional Modular Concepts | Proposed Modular Concept |
|---|---|---|
| Purpose | promotes process flexibility | promotes process flexibility and product variety |
| Production context | mass production—“economies of scale” | mass customization—“economies of scope” |
| Product archetype(s) | fixed assembly of components into products | reconfigurable assembly of components into products |
| Product variant(s) | typically achieved by iterative modifications of single initial product design | achieved by selection from multiple initial product designs |
| Impact on product variety-volume complexity | variety OR affordability achievable | variety AND affordability achievable |
| Impact on product design | no. of module variants > no. of product variants | no. of module variants < no. of product variants |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Govender, R.; Abrahmsén-Alami, S.; Larsson, A.; Borde, A.; Liljeblad, A.; Folestad, S. Independent Tailoring of Dose and Drug Release via a Modularized Product Design Concept for Mass Customization. Pharmaceutics 2020, 12, 771. https://doi.org/10.3390/pharmaceutics12080771
Govender R, Abrahmsén-Alami S, Larsson A, Borde A, Liljeblad A, Folestad S. Independent Tailoring of Dose and Drug Release via a Modularized Product Design Concept for Mass Customization. Pharmaceutics. 2020; 12(8):771. https://doi.org/10.3390/pharmaceutics12080771
Chicago/Turabian StyleGovender, Rydvikha, Susanna Abrahmsén-Alami, Anette Larsson, Anders Borde, Alexander Liljeblad, and Staffan Folestad. 2020. "Independent Tailoring of Dose and Drug Release via a Modularized Product Design Concept for Mass Customization" Pharmaceutics 12, no. 8: 771. https://doi.org/10.3390/pharmaceutics12080771
APA StyleGovender, R., Abrahmsén-Alami, S., Larsson, A., Borde, A., Liljeblad, A., & Folestad, S. (2020). Independent Tailoring of Dose and Drug Release via a Modularized Product Design Concept for Mass Customization. Pharmaceutics, 12(8), 771. https://doi.org/10.3390/pharmaceutics12080771