Effect of Rumex Acetosa Extract, a Herbal Drug, on the Absorption of Fexofenadine

 , , ,
, , , 
Abstract
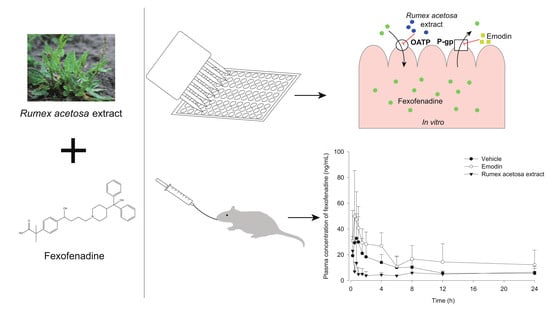
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. R. acetosa Extract
2.3. Cell Culture
2.4. Cytotoxicity Assay
2.5. P-gp Inhibition Test of Anthraquinones and R. acetosa Extract
2.6. Fexofenadine Uptake Test Using OATP1A2/SLCO1A2 Transfected HEK293 Cells
2.7. LC-MS/MS Analysis
2.8. Animal Study
2.8.1. Animals
2.8.2. Pharmacokinetic Study
2.8.3. Sample Preparation
2.9. Physicochemical Interaction Study
2.10. Statistical Analysis
3. Results
3.1. Cytotoxicity Assay
3.2. P-gp Inhibition Test of Anthraquinones and R. acetosa Extract
3.3. Fexofenadine Uptake Test with OATP1A2/SLCO1A2 Transfected HEK293 Cells
3.4. Pharmacokinetic Study
3.5. Physicochemical Interaction Study
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dean, M.; Hamon, Y.; Chimini, G. The human ATP-binding cassette (ABC) transporter superfamily. J. Lipid Res. 2001, 42, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Anderle, P.; Niederer, E.; Rubas, W.; Hilgendorf, C.; Spahn-Langguth, H.; Wunderli-Allenspach, H.; Merkle, H.P.; Langguth, P. P-Glycoprotein (P-gp) mediated efflux in Caco-2 cell monolayers: The influence of culturing conditions and drug exposure on P-gp expression levels. J. Pharm. Sci. 1998, 87, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Fardel, O.; Lecureur, V.; Guillouzo, A. The P-glycoprotein multidrug transporter. Gen. Pharm. 1996, 27, 1283–1291. [Google Scholar] [CrossRef]
- Chan, L.M.; Lowes, S.; Hirst, B.H. The ABCs of drug transport in intestine and liver: Efflux proteins limiting drug absorption and bioavailability. Eur. J. Pharm. Sci. 2004, 21, 25–51. [Google Scholar] [CrossRef]
- Talks, K.L.; Turley, H.; Gatter, K.C.; Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J.; Harris, A.L. The expression and distribution of the hypoxia-inducible factors HIF-1alpha and HIF-2alpha in normal human tissues, cancers, and tumor-associated macrophages. Am. J. Pathol 2000, 157, 411–421. [Google Scholar] [CrossRef]
- Comerford, K.M.; Wallace, T.J.; Karhausen, J.; Louis, N.A.; Montalto, M.C.; Colgan, S.P. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res. 2002, 62, 3387–3394. [Google Scholar]
- Kim, R.B. Drugs as P-glycoprotein substrates, inhibitors, and inducers. Drug Metab. Rev. 2002, 34, 47–54. [Google Scholar] [CrossRef]
- Palmeira, A.; Sousa, E.; Vasconcelos, M.H.; Pinto, M.M. Three decades of P-gp: Skimming through several generations and scaffolds. Curr. Med. Che 2012, 19, 1946–2025. [Google Scholar] [CrossRef]
- Shitara, Y.; Maeda, K.; Ikejiri, K.; Yoshida, K.; Horie, T.; Sugiyama, Y. Clinical significance of organic anion transporting polypeptides (OATPs) in drug disposition: Their roles in hepatic clearance and intestinal absorption. Biopharm. Drug Dispos. 2013, 34, 45–78. [Google Scholar] [CrossRef]
- Yu, J.; Zhou, Z.; Tay-Sontheimer, J.; Levy, R.H.; Ragueneau-Majlessi, I. Intestinal drug interactions mediated by OATPs: A systematic review of preclinical and clinical findings. J. Pharm. Sci. 2017, 106, 2312–2325. [Google Scholar] [CrossRef]
- Agbabiaka, T.B.; Spencer, N.H.; Khanom, S.; Goodman, C. Prevalence of drug-herb and drug-supplement interactions in older adults: A cross-sectional survey. Br. J. Gen. Pr. 2018, 68, e711–e717. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, D.W.; Kelly, J.P.; Rosenberg, L.; Anderson, T.E.; Mitchell, A.A. Recent patterns of medication use in the ambulatory adult population of the United States: The Slone survey. JAMA 2002, 287, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Durr, D.; Stieger, B.; Kullak-Ublick, G.A.; Rentsch, K.M.; Steinert, H.C.; Meier, P.J.; Fattinger, K. St John’s Wort induces intestinal P-glycoprotein/MDR1 and intestinal and hepatic CYP3A4. Clin. Pharm. 2000, 68, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Gescher, K.; Hensel, A.; Hafezi, W.; Derksen, A.; Kuhn, J. Oligomeric proanthocyanidins from Rumex acetosa L. inhibit the attachment of herpes simplex virus type-1. Antivir. Res. 2011, 89, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Vasas, A.; Orban-Gyapai, O.; Hohmann, J. The genus Rumex: Review of traditional uses, phytochemistry and pharmacology. J. Ethnopharmacol. 2015, 175, 198–228. [Google Scholar] [CrossRef] [PubMed]
- Kucekova, Z.; Mlcek, J.; Humpolicek, P.; Rop, O.; Valasek, P.; Saha, P. Phenolic compounds from Allium schoenoprasum, Tragopogon pratensis and Rumex acetosa and their antiproliferative effects. Molecules 2011, 16, 9207–9217. [Google Scholar] [CrossRef]
- Bae, J.Y.; Lee, Y.S.; Han, S.Y.; Jeong, E.J.; Lee, M.K.; Kong, J.Y.; Lee, D.H.; Cho, K.J.; Lee, H.S.; Ahn, M.J. A comparison between water and ethanol extracts of Rumex acetosa for protective effects on gastric ulcers in mice. Biomol. Ther. (Seoul) 2012, 20, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Min, H.; Niu, M.; Zhang, W.; Yan, J.; Li, J.; Tan, X.; Li, B.; Su, M.; Di, B.; Yan, F. Emodin reverses leukemia multidrug resistance by competitive inhibition and downregulation of P-glycoprotein. PLoS ONE 2017, 12, e0187971. [Google Scholar] [CrossRef]
- Feng, Y.; Huang, S.L.; Dou, W.; Zhang, S.; Chen, J.H.; Shen, Y.; Shen, J.H.; Leng, Y. Emodin, a natural product, selectively inhibits 11beta-hydroxysteroid dehydrogenase type 1 and ameliorates metabolic disorder in diet-induced obese mice. Br. J. Pharm. 2010, 161, 113–126. [Google Scholar] [CrossRef]
- Hsu, S.C.; Chung, J.G. Anticancer potential of emodin. BioMedicine (Taipei) 2012, 2, 108–116. [Google Scholar] [CrossRef]
- Simpson, K.; Jarvis, B. Fexofenadine: A review of its use in the management of seasonal allergic rhinitis and chronic idiopathic urticaria. Drugs 2000, 59, 301–321. [Google Scholar] [CrossRef] [PubMed]
- Tahara, H.; Kusuhara, H.; Fuse, E.; Sugiyama, Y. P-glycoprotein plays a major role in the efflux of fexofenadine in the small intestine and blood-brain barrier, but only a limited role in its biliary excretion. Drug Metab. Dispos. 2005, 33, 963–968. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Fuse, K.; Okudaira, K.; Nishigaki, R.; Maeda, K.; Kusuhara, H.; Sugiyama, Y. Contribution of OATP (organic anion-transporting polypeptide) family transporters to the hepatic uptake of fexofenadine in humans. Drug Metab. Dispos. 2005, 33, 1477–1481. [Google Scholar] [CrossRef] [PubMed]
- Molimard, M.; Diquet, B.; Benedetti, M.S. Comparison of pharmacokinetics and metabolism of desloratadine, fexofenadine, levocetirizine and mizolastine in humans. Fundam. Clin. Pharm. 2004, 18, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Prescribing Information for Allegra® (fexofenadine hydrochloride). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2003/20872se8-003,20625se8-010_allegra_lbl.pdf (accessed on 16 March 2020).
- Huang, J.; Guo, L.; Tan, R.; Wei, M.; Zhang, J.; Zhao, Y.; Gong, L.; Huang, Z.; Qiu, X. Interactions between emodin and efflux transporters on rat enterocyte by a validated ussing chamber technique. Front. Pharm. 2018, 9, 646. [Google Scholar] [CrossRef]
- Ullah, H.M.A.; Kim, J.; Rehman, N.U.; Kim, H.J.; Ahn, M.J.; Chung, H.J. A simple and sensitive liquid chromatography with tandem mass spectrometric method for the simultaneous determination of anthraquinone glycosides and their aglycones in rat plasma: Application to a pharmacokinetic study of Rumex acetosa extract. Pharmaceutics 2018, 10, 100. [Google Scholar] [CrossRef]
- Petri, N.; Tannergren, C.; Rungstad, D.; Lennernas, H. Transport characteristics of fexofenadine in the Caco-2 cell model. Pharm. Res. 2004, 21, 1398–1404. [Google Scholar] [CrossRef]
- Rebello, S.; Zhao, S.; Hariry, S.; Dahlke, M.; Alexander, N.; Vapurcuyan, A.; Hanna, I.; Jarugula, V. Intestinal OATP1A2 inhibition as a potential mechanism for the effect of grapefruit juice on aliskiren pharmacokinetics in healthy subjects. Eur. J. Clin. Pharm. 2012, 68, 697–708. [Google Scholar] [CrossRef]
- Yamane, N.; Tozuka, Z.; Sugiyama, Y.; Tanimoto, T.; Yamazaki, A.; Kumagai, Y. Microdose clinical trial: Quantitative determination of fexofenadine in human plasma using liquid chromatography/electrospray ionization tandem mass spectrometry. J. Chromatogr B Anal. Technol Biomed. Life Sci 2007, 858, 118–128. [Google Scholar] [CrossRef]
- Bharathi, V.D.; Radharani, K.; Jagadeesh, B.; Ramulu, G.; Bhushan, I.; Naidu, A.; Mullangi, R. LC–MS–MS assay for simultaneous quantification of fexofenadine and pseudoephedrine in human plasma. Chromatographia 2008, 67, 461–466. [Google Scholar] [CrossRef]
- Li, X.; Hu, J.; Wang, B.; Sheng, L.; Liu, Z.; Yang, S.; Li, Y. Inhibitory effects of herbal constituents on P-glycoprotein in vitro and in vivo: Herb-drug interactions mediated via P-gp. Toxicol. Appl. Pharm. 2014, 275, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Kamath, A.V.; Yao, M.; Zhang, Y.; Chong, S. Effect of fruit juices on the oral bioavailability of fexofenadine in rats. J. Pharm. Sci. 2005, 94, 233–239. [Google Scholar] [CrossRef] [PubMed]
- İşleyen, E.A.Ö.; Özden, T.; Özilhan, S.; Toptan, S. Quantitative determination of fexofenadine in human plasma by HPLC-MS. Chromatographia 2007, 66, 109–113. [Google Scholar] [CrossRef]
- Dai, W.G. In vitro methods to assess drug precipitation. Int. J. Pharm. 2010, 393, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Usami, T.; Katoh, M.; Nadai, M. Effects of thylakoid-rich spinach extract on the pharmacokinetics of drugs in rats. Biol. Pharm. Bull. 2019, 42, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Stippler, E.; Kopp, S.; Dressman, J.B. Comparison of US pharmacopeia simulated intestinal fluid TS (without pancreatin) and phosphate standard buffer pH 6.8, TS of the international pharmacopoeia with respect to their use in in vitro dissolution testing. Dissolution Technol. 2004, 11, 6–10. [Google Scholar] [CrossRef]
- Singh, B.; Saini, G.; Vyas, M.; Verma, S.; Thakur, S. Optimized chronomodulated dual release bilayer tablets of fexofenadine and montelukast: Quality by design, development, and in vitro evaluation. Future J. Pharm. Sci. 2019, 5, 5. [Google Scholar] [CrossRef]
- Arefin, P.; Hasan, I.; Reza, M.S. Design, characterization and in vitro evaluation of HPMC K100 M CR loaded fexofenadine HCl microspheres. Springerplus 2016, 5, 691. [Google Scholar] [CrossRef]
- Hansten, P.D.; Levy, R.H. Role of P-glycoprotein and organic anion transporting polypeptides in drug absorption and distribution. Clin. Drug Investig. 2001, 21, 587–596. [Google Scholar] [CrossRef]
- Mallhi, T.H.; Sarriff, A.; Adnan, A.S.; Khan, Y.H.; Qadir, M.I.; Hamzah, A.A.; Khan, A.H. Effect of fruit/vegetable-drug interactions on CYP450, OATP and p-glycoprotein: A systematic review. Trop. J. Pharm. Res. 2015, 14, 1927–1935. [Google Scholar] [CrossRef]
- Bailey, D.G. Fruit juice inhibition of uptake transport: A new type of food-drug interaction. Br. J. Clin. Pharm. 2010, 70, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Cvetkovic, M.; Leake, B.; Fromm, M.F.; Wilkinson, G.R.; Kim, R.B. OATP and P-glycoprotein transporters mediate the cellular uptake and excretion of fexofenadine. Drug Metab. Dispos. 1999, 27, 866–871. [Google Scholar] [PubMed]
- Niemi, M.; Kivisto, K.T.; Hofmann, U.; Schwab, M.; Eichelbaum, M.; Fromm, M.F. Fexofenadine pharmacokinetics are associated with a polymorphism of the SLCO1B1 gene (encoding OATP1B1). Br. J. Clin. Pharm. 2005, 59, 602–604. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Sharma, A.; Shukla, M.; Vaghasiya, K.; Rangaraj, N.; Lal, J. Novel pre-clinical methodologies for pharmacokinetic drug-drug interaction studies: Spotlight on "humanized" animal models. Drug Metab. Rev. 2014, 46, 475–493. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Gibbs, S.T.; Fang, L.; Miller, H.A.; Landowski, C.P.; Shin, H.C.; Lennernas, H.; Zhong, Y.; Amidon, G.L.; Yu, L.X.; et al. Why is it challenging to predict intestinal drug absorption and oral bioavailability in human using rat model. Pharm. Res. 2006, 23, 1675–1686. [Google Scholar] [CrossRef]
- Mandery, K.; Bujok, K.; Schmidt, I.; Keiser, M.; Siegmund, W.; Balk, B.; Konig, J.; Fromm, M.F.; Glaeser, H. Influence of the flavonoids apigenin, kaempferol, and quercetin on the function of organic anion transporting polypeptides 1A2 and 2B1. Biochem. Pharm. 2010, 80, 1746–1753. [Google Scholar] [CrossRef]
- Masumoto, K.; Quan, Z.; Ishiuchi, K.i.; Matsumoto, T.; Watanabe, J.; Makino, T. Drug interaction between shoseiryuto extract or catechins and fexofenadine through organic-anion-transporting polypeptide 1A2 in vitro. Pharmacogn. Mag. 2019, 15, 304–308. [Google Scholar] [CrossRef]
- Bicker, J.; Petereit, F.; Hensel, A. Proanthocyanidins and a phloroglucinol derivative from Rumex acetosa L. Fitoterapia 2009, 80, 483–495. [Google Scholar] [CrossRef]
- Roth, M.; Timmermann, B.N.; Hagenbuch, B. Interactions of green tea catechins with organic anion-transporting polypeptides. Drug Metab. Dispos. 2011, 39, 920–926. [Google Scholar] [CrossRef]
- Bajad, S.; Bedi, K.L.; Singla, A.K.; Johri, R.K. Piperine inhibits gastric emptying and gastrointestinal transit in rats and mice. Planta Med. 2001, 67, 176–179. [Google Scholar] [CrossRef]
- Hu, M.L.; Rayner, C.K.; Wu, K.L.; Chuah, S.K.; Tai, W.C.; Chou, Y.P.; Chiu, Y.C.; Chiu, K.W.; Hu, T.H. Effect of ginger on gastric motility and symptoms of functional dyspepsia. World J. Gastroenter. 2011, 17, 105–110. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Control (n = 6) | Emodin 11 mg/kg (n = 6) | R. acetosa Extract 2 g/kg (n = 6) |
|---|---|---|---|
| AUC0-24 h (ng∙h/mL) | 222.0 ± 85.5 | 411.9 ± 189.1 * | 132.0 ± 50.5 |
| Cmax (ng/mL) | 36.4 ± 22.8 | 53.4 ± 33.9 | 32.9 ± 28.5 |
| Tmax (h) | 0.75 (0.5–1) | 0.75 (0.5–1) | 0.75 (0.25–8) |
| Solubility | Without R. acetosa Extract(n = 3) | With R. acetosa Extract(n = 3) |
|---|---|---|
| Fexofenadine HCl concentration (mg/mL) | 1.03 ± 0.04 | 0.83 ± 0.10 * |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahn, J.H.; Kim, J.; Rehman, N.U.; Kim, H.-J.; Ahn, M.-J.; Chung, H.J. Effect of Rumex Acetosa Extract, a Herbal Drug, on the Absorption of Fexofenadine. Pharmaceutics 2020, 12, 547. https://doi.org/10.3390/pharmaceutics12060547
Ahn JH, Kim J, Rehman NU, Kim H-J, Ahn M-J, Chung HJ. Effect of Rumex Acetosa Extract, a Herbal Drug, on the Absorption of Fexofenadine. Pharmaceutics. 2020; 12(6):547. https://doi.org/10.3390/pharmaceutics12060547
Chicago/Turabian StyleAhn, Jung Hwan, Junhyeong Kim, Naveed Ur Rehman, Hye-Jin Kim, Mi-Jeong Ahn, and Hye Jin Chung. 2020. "Effect of Rumex Acetosa Extract, a Herbal Drug, on the Absorption of Fexofenadine" Pharmaceutics 12, no. 6: 547. https://doi.org/10.3390/pharmaceutics12060547
APA StyleAhn, J. H., Kim, J., Rehman, N. U., Kim, H.-J., Ahn, M.-J., & Chung, H. J. (2020). Effect of Rumex Acetosa Extract, a Herbal Drug, on the Absorption of Fexofenadine. Pharmaceutics, 12(6), 547. https://doi.org/10.3390/pharmaceutics12060547