Engineered Extracellular Vesicles/Exosomes as a New Tool against Neurodegenerative Diseases

 ,
,  ,
,  and
and 
Abstract
1. Introduction
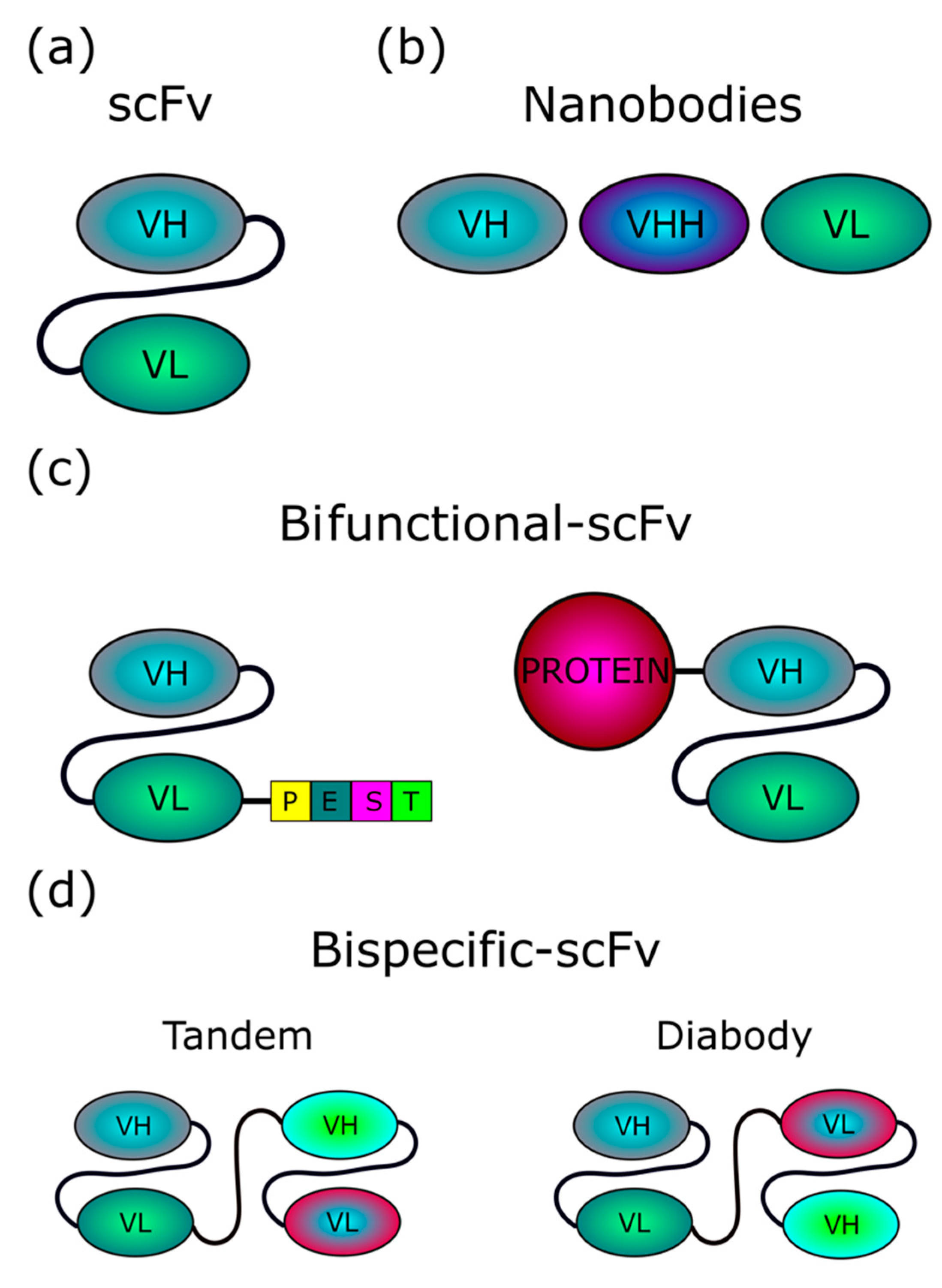
1.1. Intrabodies
1.2. Extracellular Vesicles
2. Genetic Basis of Neurodegenerative Diseases
2.1. Alzheimer’s Disease
2.2. Parkinson’s Disease
2.3. Huntington’s Disease
2.4. Amyotrophic Lateral Sclerosis
2.5. Prion Disorders
3. Intrabodies against ND Targets
3.1. Antibody Targets for Alzheimer’s Disease
3.2. Antibody Targets for Parkinson’s Disease
3.3. Antibody Targets for Huntington’s Disease
3.4. Antibody Targets for Amyotrophic Lateral Sclerosis
3.5. Targets for Prion Disorders
4. Current Strategies for Intrabody Delivery into Cells
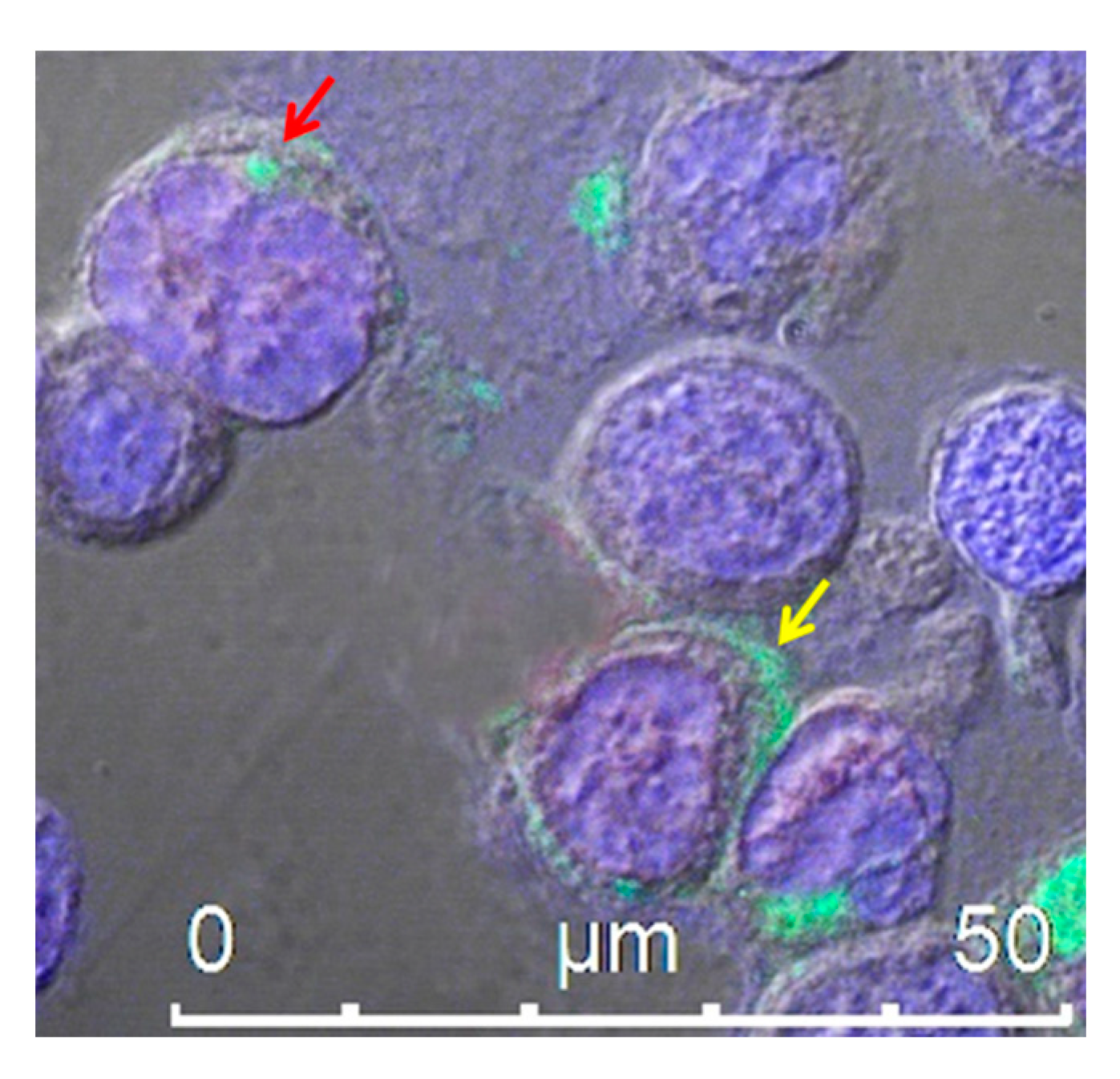
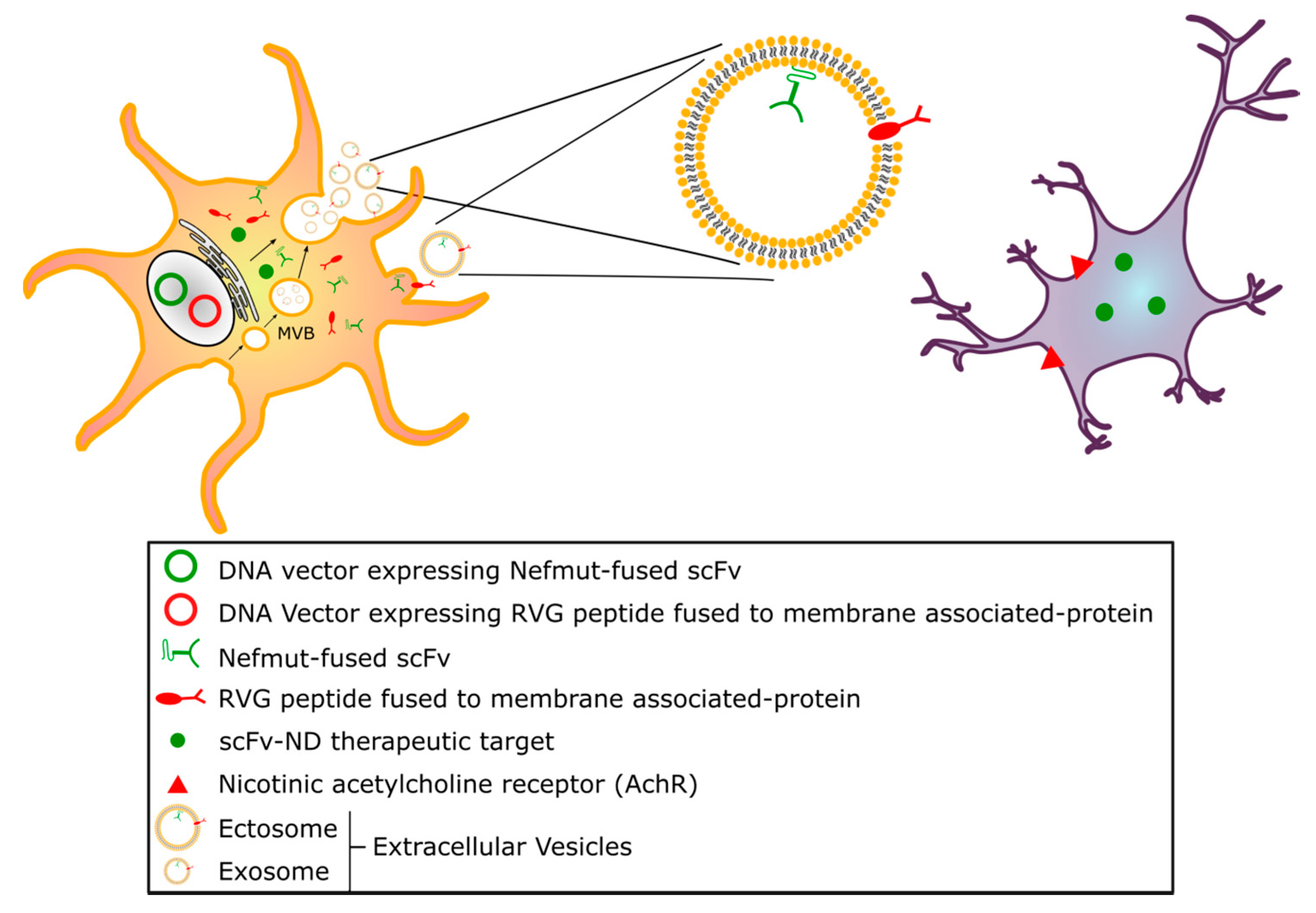
4.1. Extracellular Vesicles/Exosomes as a Tool for scFv Delivery
4.2. Endogenously Engineered Exosomes: Potential and Further Development
4.3. Delivery using Gene Therapy
5. Conclusions
Funding
Conflicts of Interest
References
- La Spada, A.R.; Paulson, H.L.; Fischbeck, K.H. Trinucleotide repeat expansion in neurological disease. Ann. Neurol. 1994, 36, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological Alterations in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2011, 1. [Google Scholar] [CrossRef] [PubMed]
- Ravits, J.M.; La Spada, A.R. ALS motor phenotype heterogeneity, focality, and spread: Deconstructing motor neuron degeneration. Neurology 2009, 73, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.-Y.; Zhu, Q.; Marasco, W.A. Intracellular antibodies (intrabodies) and their therapeutic potential. In Therapeutic Antibodies; Springer: Berlin/Heidelberg, Germany, 2008; pp. 343–373. [Google Scholar]
- Huston, J.S.; Margolies, M.N.; Haber, E. Antibody binding sites. Adv. Protein Chem. 1996, 49, 329–450. [Google Scholar] [CrossRef] [PubMed]
- Marschall, A.L.J.; Dübel, S. Antibodies inside of a cell can change its outside: Can intrabodies provide a new therapeutic paradigm? Comput. Struct. Biotechnol. J. 2016, 14, 304–308. [Google Scholar] [CrossRef]
- Sudol, K.L.; Mastrangelo, M.A.; Narrow, W.C.; Frazer, M.E.; Levites, Y.R.; Golde, T.E.; Federoff, H.J.; Bowers, W.J. Generating differentially targeted amyloid-β specific intrabodies as a passive vaccination strategy for Alzheimer’s disease. Mol. Ther. 2009, 17, 2031–2040. [Google Scholar] [CrossRef]
- Ryan, D.A.; Mastrangelo, M.A.; Narrow, W.C.; Sullivan, M.A.; Federoff, H.J.; Bowers, W.J. AB-directed single-chain antibody delivery via a serotype-1 AAV vector improves learning behavior and pathology in alzheimer’s disease mice. Mol. Ther. 2010, 18, 1471–1481. [Google Scholar] [CrossRef]
- Joshi, S.N.; Butler, D.C.; Messer, A. Fusion to a highly charged proteasomal retargeting sequence increases soluble cytoplasmic expression and efficacy of diverse anti-synuclein intrabodies. In Proceedings of the MAbs; Taylor & Francis: Abingdon, UK, 2012; Volume 4, pp. 686–693. [Google Scholar]
- Kvam, E.; Sierks, M.R.; Shoemaker, C.B.; Messer, A. Physico-chemical determinants of soluble intrabody expression in mammalian cell cytoplasm. Protein Eng. Des. Sel. 2010, 23, 489–498. [Google Scholar] [CrossRef]
- Spencer, B.; Williams, S.; Rockenstein, E.; Valera, E.; Xin, W.; Mante, M.; Florio, J.; Adame, A.; Masliah, E.; Sierks, M.R. α-synuclein conformational antibodies fused to penetratin are effective in models of Lewy body disease. Ann. Clin. Transl. Neurol. 2016, 3, 588–606. [Google Scholar] [CrossRef]
- Valera, E.; Spencer, B.; Fields, J.A.; Trinh, I.; Adame, A.; Mante, M.; Rockenstein, E.; Desplats, P.; Masliah, E. Combination of alpha-synuclein immunotherapy with anti-inflammatory treatment in a transgenic mouse model of multiple system atrophy. Acta Neuropathol. Commun. 2017, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Emadi, S.; Kasturirangan, S.; Wang, M.S.; Schulz, P.; Sierks, M.R. Detecting Morphologically Distinct Oligomeric Forms of α-Synuclein. J. Biol. Chem. 2009, 284, 11048–11058. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Emadi, S.; Sierks, M.R.; Messer, A. A human single-chain Fv intrabody blocks aberrant cellular effects of overexpressed α-synuclein. Mol. Ther. 2004, 10, 1023–1031. [Google Scholar] [CrossRef]
- Butler, D.C.; Joshi, S.N.; De Genst, E.; Baghel, A.S.; Dobson, C.M.; Messer, A. Bifunctional anti-non-amyloid component α-Synuclein nanobodies are protective in situ. PLoS ONE 2016, 11, e0165964. [Google Scholar] [CrossRef]
- Lynch, S.M.; Zhou, C.; Messer, A. An scFv intrabody against the nonamyloid component of alpha-synuclein reduces intracellular aggregation and toxicity. J. Mol. Biol. 2008, 377, 136–147. [Google Scholar] [CrossRef]
- Kvam, E.; Nannenga, B.L.; Wang, M.S.; Jia, Z.; Sierks, M.R.; Messer, A. Conformational targeting of fibrillar polyglutamine proteins in live cells escalates aggregation and cytotoxicity. PLoS ONE 2009, 4, e5727. [Google Scholar] [CrossRef]
- Guilliams, T.; El-Turk, F.; Buell, A.K.; O’Day, E.M.; Aprile, F.A.; Esbjörner, E.K.; Vendruscolo, M.; Cremades, N.; Pardon, E.; Wyns, L. Nanobodies raised against monomeric α-synuclein distinguish between fibrils at different maturation stages. J. Mol. Biol. 2013, 425, 2397–2411. [Google Scholar] [CrossRef]
- Chatterjee, D.; Bhatt, M.; Butler, D.; De Genst, E.; Dobson, C.M.; Messer, A.; Kordower, J.H. Proteasome-targeted nanobodies alleviate pathology and functional decline in an α-synuclein-based Parkinson’s disease model. NPJ Parkinson’s Dis. 2018, 4, 1–10. [Google Scholar] [CrossRef]
- Butler, D.C.; Messer, A. Bifunctional anti-huntingtin proteasome-directed intrabodies mediate efficient degradation of mutant huntingtin exon 1 protein fragments. PLoS ONE 2011, 6, e29199. [Google Scholar] [CrossRef]
- Butler, D.C.; Snyder-Keller, A.; De Genst, E.; Messer, A. Differential nuclear localization of complexes may underlie in vivo intrabody efficacy in Huntington’s disease. Protein Eng. Des. Sel. 2014, 27, 359–363. [Google Scholar] [CrossRef]
- De Genst, E.; Chirgadze, D.Y.; Klein, F.A.C.; Butler, D.C.; Matak-Vinković, D.; Trottier, Y.; Huston, J.S.; Messer, A.; Dobson, C.M. Structure of a Single-Chain Fv Bound to the 17 N-Terminal Residues of Huntingtin Provides Insights into Pathogenic Amyloid Formation and Suppression. J. Mol. Biol. 2015, 427, 2166–2178. [Google Scholar] [CrossRef] [PubMed]
- Hathorn, T.; Snyder-Keller, A.; Messer, A. Nicotinamide improves motor deficits and upregulates PGC-1α and BDNF gene expression in a mouse model of Huntington’s disease. Neurobiol. Dis. 2011, 41, 43–50. [Google Scholar] [CrossRef]
- Lecerf, J.-M.; Shirley, T.L.; Zhu, Q.; Kazantsev, A.; Amersdorfer, P.; Housman, D.E.; Messer, A.; Huston, J.S. Human single-chain Fv intrabodies counteract in situ huntingtin aggregation in cellular models of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 4764–4769. [Google Scholar] [CrossRef]
- McLear, J.A.; Lebrecht, D.; Messer, A.; Wolfgang, W.J. Combinational approach of intrabody with enhanced Hsp70 expression addresses multiple pathologies in a fly model of Huntington’s disease. FASEB J. 2008, 22, 2003–2011. [Google Scholar] [CrossRef]
- Miller, T.W.; Zhou, C.; Gines, S.; MacDonald, M.E.; Mazarakis, N.D.; Bates, G.P.; Huston, J.S.; Messer, A. A human single-chain Fv intrabody preferentially targets amino-terminal huntingtin fragments in striatal models of Huntington’s disease. Neurobiol. Dis. 2005, 19, 47–56. [Google Scholar] [CrossRef]
- Murphy, R.C.; Messer, A. A single-chain Fv intrabody provides functional protection against the effects of mutant protein in an organotypic slice culture model of Huntington’s disease. Mol. Brain Res. 2004, 121, 141–145. [Google Scholar] [CrossRef]
- Snyder-Keller, A.; McLear, J.A.; Hathorn, T.; Messer, A. Early or late-stage anti-N-terminal Huntingtin intrabody gene therapy reduces pathological features in B6.HDR6/1 mice. J. Neuropathol. Exp. Neurol. 2010, 69, 1078–1085. [Google Scholar] [CrossRef]
- Wolfgang, W.J.; Miller, T.W.; Webster, J.M.; Huston, J.S.; Thompson, L.M.; Marsh, J.L.; Messer, A. Suppression of Huntington’s disease pathology in Drosophila by human single-chain Fv antibodies. Proc. Natl. Acad. Sci. USA 2005, 102, 11563–11568. [Google Scholar] [CrossRef]
- Colby, D.W.; Chu, Y.; Cassady, J.P.; Duennwald, M.; Zazulak, H.; Webster, J.M.; Messer, A.; Lindquist, S.; Ingram, V.M.; Wittrup, K.D. Potent inhibition of huntingtin aggregation and cytotoxicity by a disulfide bond-free single-domain intracellular antibody. Proc. Natl. Acad. Sci. USA 2004, 101, 17616–17621. [Google Scholar] [CrossRef]
- Colby, D.W.; Garg, P.; Holden, T.; Chao, G.; Webster, J.M.; Messer, A.; Ingram, V.M.; Wittrup, K.D. Development of a Human Light Chain Variable Domain (VL) Intracellular Antibody Specific for the Amino Terminus of Huntingtin via Yeast Surface Display. J. Mol. Biol. 2004, 342, 901–912. [Google Scholar] [CrossRef]
- Southwell, A.L.; Khoshnan, A.; Dunn, D.E.; Bugg, C.W.; Lo, D.C.; Patterson, P.H. Intrabodies binding the proline-rich domains of mutant huntingtin increase its turnover and reduce neurotoxicity. J. Neurosci. 2008, 28, 9013–9020. [Google Scholar] [CrossRef] [PubMed]
- Southwell, A.L.; Ko, J.; Patterson, P.H. Intrabody Gene Therapy Ameliorates Motor, Cognitive, and Neuropathological Symptoms in Multiple Mouse Models of Huntington’s Disease. J. Neurosci. 2009, 29, 13589–13602. [Google Scholar] [CrossRef]
- Southwell, A.L.; Bugg, C.W.; Kaltenbach, L.S.; Dunn, D.; Butland, S.; Weiss, A.; Paganetti, P.; Lo, D.C.; Patterson, P.H. Perturbation with Intrabodies Reveals That Calpain Cleavage Is Required for Degradation of Huntingtin Exon 1. PLoS ONE 2011, 6, e16676. [Google Scholar] [CrossRef] [PubMed]
- Khoshnan, A.; Ko, J.; Patterson, P.H. Effects of intracellular expression of anti-huntingtin antibodies of various specificities on mutant huntingtin aggregation and toxicity. Proc. Natl. Acad. Sci. USA 2002, 99, 1002–1007. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.; Ou, S.; Patterson, P.H. New anti-huntingtin monoclonal antibodies: Implications for huntingtin conformation and its binding proteins. Brain Res. Bull. 2001, 56, 319–329. [Google Scholar] [CrossRef]
- Wang, C.-E.; Zhou, H.; McGuire, J.R.; Cerullo, V.; Lee, B.; Li, S.-H.; Li, X.-J. Suppression of neuropil aggregates and neurological symptoms by an intracellular antibody implicates the cytoplasmic toxicity of mutant huntingtin. J. Cell Biol. 2008, 181, 803–816. [Google Scholar] [CrossRef]
- Amaro, I.A.; Henderson, L.A. An Intrabody Drug (rAAV6-INT41) Reduces the Binding of N-Terminal Huntingtin Fragment(s) to DNA to Basal Levels in PC12 Cells and Delays Cognitive Loss in the R6/2 Animal Model. J. Neurodegener. Dis. 2016, 2016, 7120753. [Google Scholar] [CrossRef]
- Ghadge, G.D.; Kay, B.K.; Drigotas, C.; Roos, R.P. Single chain variable fragment antibodies directed against SOD1 ameliorate disease in mutant SOD1 transgenic mice. Neurobiol. Dis. 2019, 121, 131–137. [Google Scholar] [CrossRef]
- Ghadge, G.D.; Pavlovic, J.D.; Koduvayur, S.P.; Kay, B.K.; Roos, R.P. Single chain variable fragment antibodies block aggregation and toxicity induced by familial ALS-linked mutant forms of SOD1. Neurobiol. Dis. 2013, 56, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Kriz, J.; Gravel, M.; Soucy, G.; Bareil, C.; Gravel, C.; Julien, J.-P. Adeno-associated Virus–mediated Delivery of a Recombinant Single-chain Antibody Against Misfolded Superoxide Dismutase for Treatment of Amyotrophic Lateral Sclerosis. Mol. Ther. 2014, 22, 498–510. [Google Scholar] [CrossRef]
- Cardinale, A.; Filesi, I.; Vetrugno, V.; Pocchiari, M.; Sy, M.-S.; Biocca, S. Trapping prion protein in the endoplasmic reticulum impairs PrPC maturation and prevents PrPSc accumulation. J. Biol. Chem. 2005, 280, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Vetrugno, V.; Cardinale, A.; Filesi, I.; Mattei, S.; Sy, M.-S.; Pocchiari, M.; Biocca, S. KDEL-tagged anti-prion intrabodies impair PrP lysosomal degradation and inhibit scrapie infectivity. Biochem. Biophys. Res. Commun. 2005, 338, 1791–1797. [Google Scholar] [CrossRef] [PubMed]
- Abskharon, R.N.N.; Soror, S.H.; Pardon, E.; El Hassan, H.; Legname, G.; Steyaert, J.; Wohlkonig, A. Crystallization and preliminary X-ray diffraction analysis of a specific VHH domain against mouse prion protein. Acta Cryst. F 2010, 66, 1644–1646. [Google Scholar] [CrossRef] [PubMed]
- Abskharon, R.N.N.; Giachin, G.; Wohlkonig, A.; Soror, S.H.; Pardon, E.; Legname, G.; Steyaert, J. Probing the N-Terminal β-Sheet Conversion in the Crystal Structure of the Human Prion Protein Bound to a Nanobody. J. Am. Chem. Soc. 2014, 136, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Schorey, J.S.; Cheng, Y.; Singh, P.P.; Smith, V.L. Exosomes and other extracellular vesicles in host-pathogen interactions. EMBO Rep. 2015, 16, 24–43. [Google Scholar] [CrossRef]
- Fröhlich, D.; Kuo, W.P.; Frühbeis, C.; Sun, J.-J.; Zehendner, C.M.; Luhmann, H.J.; Pinto, S.; Toedling, J.; Trotter, J.; Krämer-Albers, E.-M. Multifaceted effects of oligodendroglial exosomes on neurons: Impact on neuronal firing rate, signal transduction and gene regulation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369. [Google Scholar] [CrossRef]
- Janas, A.M.; Sapoń, K.; Janas, T.; Stowell, M.H.B.; Janas, T. Exosomes and other extracellular vesicles in neural cells and neurodegenerative diseases. Biochim. Biophys. Acta (BBA) - Biomembr. 2016, 1858, 1139–1151. [Google Scholar] [CrossRef]
- Jan, A.T.; Malik, M.A.; Rahman, S.; Yeo, H.R.; Lee, E.J.; Abdullah, T.S.; Choi, I. Perspective Insights of Exosomes in Neurodegenerative Diseases: A Critical Appraisal. Front. Aging Neurosci. 2017, 9, 317. [Google Scholar] [CrossRef]
- Ugalde, C.L.; Finkelstein, D.I.; Lawson, V.A.; Hill, A.F. Pathogenic mechanisms of prion protein, amyloid-β and α-synuclein misfolding: The prion concept and neurotoxicity of protein oligomers. J. Neurochem. 2016, 139, 162–180. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, A.; Ekavali. A review on Alzheimer’s disease pathophysiology and its management: An update. Pharmacol. Rep. 2015, 67, 195–203. [Google Scholar] [CrossRef]
- Mendez, M.F. Early-Onset Alzheimer Disease. Neurol. Clin. 2017, 35, 263–281. [Google Scholar] [CrossRef] [PubMed]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Nikolac Perkovic, M.; Pivac, N. Genetic Markers of Alzheimer’s Disease. Adv. Exp. Med. Biol. 2019, 1192, 27–52. [Google Scholar] [CrossRef] [PubMed]
- Tysnes, O.-B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural. Transm. (Vienna) 2017, 124, 901–905. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the α-Synuclein Gene Identified in Families with Parkinson’s Disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Sato, H.; Arawaka, S.; Hara, S.; Fukushima, S.; Koga, K.; Koyama, S.; Kato, T. Authentically phosphorylated α-synuclein at Ser129 accelerates neurodegeneration in a rat model of familial Parkinson’s disease. J. Neurosci. 2011, 31, 16884–16894. [Google Scholar] [CrossRef]
- Karampetsou, M.; Ardah, M.T.; Semitekolou, M.; Polissidis, A.; Samiotaki, M.; Kalomoiri, M.; Majbour, N.; Xanthou, G.; El-Agnaf, O.M.A.; Vekrellis, K. Phosphorylated exogenous alpha-synuclein fibrils exacerbate pathology and induce neuronal dysfunction in mice. Sci. Rep. 2017, 7, 16533. [Google Scholar] [CrossRef]
- Zeng, X.-S.; Geng, W.-S.; Jia, J.-J.; Chen, L.; Zhang, P.-P. Cellular and Molecular Basis of Neurodegeneration in Parkinson Disease. Front. Aging Neurosci. 2018, 10, 109. [Google Scholar] [CrossRef]
- McGinty, R.J.; Mirkin, S.M. Cis- and Trans-Modifiers of Repeat Expansions: Blending Model Systems with Human Genetics. Trends Genet. 2018, 34, 448–465. [Google Scholar] [CrossRef]
- Caron, N.S.; Wright, G.E.; Hayden, M.R. Huntington Disease. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Juenemann, K.; Weisse, C.; Reichmann, D.; Kaether, C.; Calkhoven, C.F.; Schilling, G. Modulation of mutant huntingtin N-terminal cleavage and its effect on aggregation and cell death. Neurotox. Res. 2011, 20, 120–133. [Google Scholar] [CrossRef] [PubMed]
- Swinnen, B.; Robberecht, W. The phenotypic variability of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2014, 10, 661–670. [Google Scholar] [CrossRef]
- Abel, O.; Powell, J.F.; Andersen, P.M.; Al-Chalabi, A. ALSoD: A user-friendly online bioinformatics tool for amyotrophic lateral sclerosis genetics. Hum. Mutat. 2012, 33, 1345–1351. [Google Scholar] [CrossRef] [PubMed]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; Van Den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 1–19. [Google Scholar] [CrossRef]
- Peralta, O.A.; Eyestone, W.H. Quantitative and qualitative analysis of cellular prion protein (PrP(C)) expression in bovine somatic tissues. Prion 2009, 3, 161–170. [Google Scholar] [CrossRef]
- Aguzzi, A.; Haass, C. Games played by rogue proteins in prion disorders and Alzheimer’s disease. Science 2003, 302, 814–818. [Google Scholar] [CrossRef]
- Prusiner, S.B. Neurodegenerative diseases and prion, Shattuck lecture. N. Engl. J. Med. 2001, 344, 1516–1526. [Google Scholar] [CrossRef]
- Huang, L.-K.; Chao, S.-P.; Hu, C.-J. Clinical trials of new drugs for Alzheimer disease. J. Biomed. Sci. 2020, 27, 18. [Google Scholar] [CrossRef]
- Mullard, A. Anti-amyloid failures stack up as Alzheimer antibody flops. Nat. Rev. Drug Discov. 2019, 18, 327. [Google Scholar] [CrossRef]
- Foster, J.K.; Verdile, G.; Bates, K.A.; Martins, R.N. Immunization in Alzheimer’s disease: Naïve hope or realistic clinical potential? Mol. Psychiatry 2009, 14, 239–251. [Google Scholar] [CrossRef]
- Habicht, G.; Haupt, C.; Friedrich, R.P.; Hortschansky, P.; Sachse, C.; Meinhardt, J.; Wieligmann, K.; Gellermann, G.P.; Brodhun, M.; Götz, J. Directed selection of a conformational antibody domain that prevents mature amyloid fibril formation by stabilizing Aβ protofibrils. Proc. Natl. Acad. Sci. USA 2007, 104, 19232–19237. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; McAllister, C.; Lyubchenko, Y.; Sierks, M.R. Proteolytic antibody light chains alter beta-amyloid aggregation and prevent cytotoxicity. Biochemistry 2004, 43, 9999–10007. [Google Scholar] [CrossRef]
- Liu, R.; Yuan, B.; Emadi, S.; Zameer, A.; Schulz, P.; McAllister, C.; Lyubchenko, Y.; Goud, G.; Sierks, M.R. Single chain variable fragments against beta-amyloid (Abeta) can inhibit Abeta aggregation and prevent abeta-induced neurotoxicity. Biochemistry 2004, 43, 6959–6967. [Google Scholar] [CrossRef] [PubMed]
- Paganetti, P.; Calanca, V.; Galli, C.; Stefani, M.; Molinari, M. beta-site specific intrabodies to decrease and prevent generation of Alzheimer’s Abeta peptide. J. Cell Biol. 2005, 168, 863–868. [Google Scholar] [CrossRef]
- Miller, T.W.; Messer, A. Intrabody applications in neurological disorders: Progress and future prospects. Mol. Ther. 2005, 12, 394–401. [Google Scholar] [CrossRef]
- He, P.; Xin, W.; Schulz, P.; Sierks, M.R. Bispecific Antibody Fragment Targeting APP and Inducing α-Site Cleavage Restores Neuronal Health in an Alzheimer’s Mouse Model. Mol. Neurobiol. 2019, 56, 7420–7432. [Google Scholar] [CrossRef]
- Periquet, M.; Fulga, T.; Myllykangas, L.; Schlossmacher, M.G.; Feany, M.B. Aggregated alpha-synuclein mediates dopaminergic neurotoxicity in vivo. J. Neurosci. 2007, 27, 3338–3346. [Google Scholar] [CrossRef]
- Giasson, B.I.; Murray, I.V.; Trojanowski, J.Q.; Lee, V.M. A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. J. Biol. Chem. 2001, 276, 2380–2386. [Google Scholar] [CrossRef]
- Eliezer, D. The mysterious C-terminal tail of alpha-synuclein: Nanobody’s guess. J. Mol. Biol. 2013, 425, 2393–2396. [Google Scholar] [CrossRef][Green Version]
- El-Turk, F.; Newby, F.N.; De Genst, E.; Guilliams, T.; Sprules, T.; Mittermaier, A.; Dobson, C.M.; Vendruscolo, M. Structural effects of two camelid nanobodies directed to distinct C-terminal epitopes on α-synuclein. Biochemistry 2016, 55, 3116–3122. [Google Scholar] [CrossRef]
- Bhatt, M.A.; Messer, A.; Kordower, J.H. Can intrabodies serve as neuroprotective therapies for Parkinson’s disease? Beginning thoughts. J. Parkinson’s Dis. 2013, 3, 581–591. [Google Scholar] [CrossRef] [PubMed]
- De Genst, E.; Messer, A.; Dobson, C.M. Antibodies and protein misfolding: From structural research tools to therapeutic strategies. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2014, 1844, 1907–1919. [Google Scholar] [CrossRef]
- Gupta, S.; Colby, D.W. Protein misfolding detected early in pathogenesis of transgenic mouse model of Huntington disease using amyloid seeding assay. J. Biol. Chem. 2012, 287, 9982–9989. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.W.; Messer, A. Gene therapy for CNS diseases using Intrabodies. In Gene Therapy of the Central Nervous System: From Bench to Bedside; Academic Press: Amsterdam, The Netherlands; Boston, MA, USA, 2006; pp. 133–150. [Google Scholar]
- Cattaneo, E.; Zuccato, C.; Tartari, M. Normal huntingtin function: An alternative approach to Huntington’s disease. Nat. Rev. Neurosci. 2005, 6, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Bortvedt, S.F.; McLear, J.A.; Messer, A.; Ahern-Rindell, A.J.; Wolfgang, W.J. Cystamine and intrabody co-treatment confers additional benefits in a fly model of Huntington’s disease. Neurobiol. Dis. 2010, 40, 130–134. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: Roles in disease and aging. Cell Res. 2014, 24, 92–104. [Google Scholar] [CrossRef]
- Bauer, P.O.; Goswami, A.; Wong, H.K.; Okuno, M.; Kurosawa, M.; Yamada, M.; Miyazaki, H.; Matsumoto, G.; Kino, Y.; Nagai, Y.; et al. Harnessing chaperone-mediated autophagy for the selective degradation of mutant huntingtin protein. Nat. Biotechnol. 2010, 28, 256–263. [Google Scholar] [CrossRef]
- Popiel, H.A.; Burke, J.R.; Strittmatter, W.J.; Oishi, S.; Fujii, N.; Takeuchi, T.; Toda, T.; Wada, K.; Nagai, Y. The Aggregation Inhibitor Peptide QBP1 as a Therapeutic Molecule for the Polyglutamine Neurodegenerative Diseases. J. Amino Acids 2011, 2011, 265084. [Google Scholar] [CrossRef]
- Qin, Z.-H.; Wang, Y.; Sapp, E.; Cuiffo, B.; Wanker, E.; Hayden, M.R.; Kegel, K.B.; Aronin, N.; DiFiglia, M. Huntingtin bodies sequester vesicle-associated proteins by a polyproline-dependent interaction. J. Neurosci. 2004, 24, 269–281. [Google Scholar] [CrossRef]
- Rockabrand, E.; Slepko, N.; Pantalone, A.; Nukala, V.N.; Kazantsev, A.; Marsh, J.L.; Sullivan, P.G.; Steffan, J.S.; Sensi, S.L.; Thompson, L.M. The first 17 amino acids of Huntingtin modulate its sub-cellular localization, aggregation and effects on calcium homeostasis. Hum. Mol. Genet. 2007, 16, 61–77. [Google Scholar] [CrossRef]
- Pozzi, S.; Thammisetty, S.S.; Codron, P.; Rahimian, R.; Plourde, K.V.; Soucy, G.; Bareil, C.; Phaneuf, D.; Kriz, J.; Gravel, C.; et al. Virus-mediated delivery of antibody targeting TAR DNA-binding protein-43 mitigates associated neuropathology. J. Clin. Invest. 2019, 129, 1581–1595. [Google Scholar] [CrossRef]
- Luginbühl, B.; Kanyo, Z.; Jones, R.M.; Fletterick, R.J.; Prusiner, S.B.; Cohen, F.E.; Williamson, R.A.; Burton, D.R.; Plückthun, A. Directed Evolution of an Anti-prion Protein scFv Fragment to an Affinity of 1 pM and its Structural Interpretation. J. Mol. Biol. 2006, 363, 75–97. [Google Scholar] [CrossRef]
- Ha, D.; Yang, N.; Nadithe, V. Exosomes as therapeutic drug carriers and delivery vehicles across biological membranes: Current perspectives and future challenges. Acta Pharm. Sin. B 2016, 6, 287–296. [Google Scholar] [CrossRef]
- McNamara, R.P.; Costantini, L.M.; Myers, T.A.; Schouest, B.; Maness, N.J.; Griffith, J.D.; Damania, B.A.; MacLean, A.G.; Dittmer, D.P. Nef Secretion into Extracellular Vesicles or Exosomes Is Conserved across Human and Simian Immunodeficiency Viruses. MBio 2018, 9. [Google Scholar] [CrossRef]
- Lattanzi, L.; Federico, M. A strategy of antigen incorporation into exosomes: Comparing cross-presentation levels of antigens delivered by engineered exosomes and by lentiviral virus-like particles. Vaccine 2012, 30, 7229–7237. [Google Scholar] [CrossRef]
- D’Aloja, P.; Santarcangelo, A.C.; Arold, S.; Baur, A.; Federico, M. Genetic and functional analysis of the human immunodeficiency virus (HIV) type 1-inhibiting F12-HIVnef allele. J. Gen. Virol. 2001, 82, 2735–2745. [Google Scholar] [CrossRef]
- Ferrantelli, F.; Arenaccio, C.; Manfredi, F.; Olivetta, E.; Chiozzini, C.; Leone, P.; Percario, Z.; Ascione, A.; Flego, M.; Di Bonito, P.; et al. The Intracellular Delivery of Anti-HPV16 E7 scFvs Through Engineered Extracellular Vesicles Inhibits the Proliferation of HPV-Infected Cells. Int. J. Nanomed. 2019, 14, 8755–8768. [Google Scholar] [CrossRef]
- Di Bonito, P.; Chiozzini, C.; Arenaccio, C.; Anticoli, S.; Manfredi, F.; Olivetta, E.; Ferrantelli, F.; Falcone, E.; Ruggieri, A.; Federico, M. Antitumor HPV E7-specific CTL activity elicited by in vivo engineered exosomes produced through DNA inoculation. Int. J. Nanomed. 2017, 12, 4579–4591. [Google Scholar] [CrossRef]
- Kumar, P.; Wu, H.; McBride, J.L.; Jung, K.-E.; Kim, M.H.; Davidson, B.L.; Lee, S.K.; Shankar, P.; Manjunath, N. Transvascular delivery of small interfering RNA to the central nervous system. Nature 2007, 448, 39–43. [Google Scholar] [CrossRef]
- Cui, G.; Guo, H.; Li, H.; Zhai, Y.; Gong, Z.; Wu, J.; Liu, J.; Dong, Y.; Hou, S.; Liu, J. RVG-modified exosomes derived from mesenchymal stem cells rescue memory deficits by regulating inflammatory responses in a mouse model of Alzheimer’s disease. Immun. Ageing 2019, 16, 10. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J.A. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]
- Mullard, A. FDA approves SMA gene therapy. Nat. Rev. Drug Discov. 2019, 18, 488. [Google Scholar] [CrossRef]
- Zhang, H.; Yang, B.; Mu, X.; Ahmed, S.S.; Su, Q.; He, R.; Wang, H.; Mueller, C.; Sena-Esteves, M.; Brown, R.; et al. Several rAAV vectors efficiently cross the blood-brain barrier and transduce neurons and astrocytes in the neonatal mouse central nervous system. Mol. Ther. 2011, 19, 1440–1448. [Google Scholar] [CrossRef]
- Merkel, S.F.; Andrews, A.M.; Lutton, E.M.; Mu, D.; Hudry, E.; Hyman, B.T.; Maguire, C.A.; Ramirez, S.H. Trafficking of adeno-associated virus vectors across a model of the blood-brain barrier; a comparative study of transcytosis and transduction using primary human brain endothelial cells. J. Neurochem. 2017, 140, 216–230. [Google Scholar] [CrossRef]
- Naso, M.F.; Tomkowicz, B.; Perry, W.L.; Strohl, W.R. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs 2017, 31, 317–334. [Google Scholar] [CrossRef]
- Klein, R.L.; Muir, D.F.; King, M.A.; Peel, A.L.; Zolotukhin, S.; Möller, J.C.; Krüttgen, A.; Heymach, J.V.; Muzyczka, N.; Meyer, E.M. Long-term actions of vector-derived nerve growth factor or brain-derived neurotrophic factor on choline acetyltransferase and Trk receptor levels in the adult rat basal forebrain. Neuroscience 1999, 90, 815–821. [Google Scholar] [CrossRef]
- Rivera, V.M.; Gao, G.; Grant, R.L.; Schnell, M.A.; Zoltick, P.W.; Rozamus, L.W.; Clackson, T.; Wilson, J.M. Long-term pharmacologically regulated expression of erythropoietin in primates following AAV-mediated gene transfer. Blood 2005, 105, 1424–1430. [Google Scholar] [CrossRef]
- Leone, P.; Shera, D.; McPhee, S.W.J.; Francis, J.S.; Kolodny, E.H.; Bilaniuk, L.T.; Wang, D.-J.; Assadi, M.; Goldfarb, O.; Goldman, H.W.; et al. Long-Term Follow-Up After Gene Therapy for Canavan Disease. Sci. Transl. Med. 2012, 4, 165ra163. [Google Scholar] [CrossRef]
- Challis, R.C.; Ravindra Kumar, S.; Chan, K.Y.; Challis, C.; Beadle, K.; Jang, M.J.; Kim, H.M.; Rajendran, P.S.; Tompkins, J.D.; Shivkumar, K.; et al. Systemic AAV vectors for widespread and targeted gene delivery in rodents. Nat. Protoc. 2019, 14, 379–414. [Google Scholar] [CrossRef]
- Huang, Q.; Chan, K.Y.; Tobey, I.G.; Chan, Y.A.; Poterba, T.; Boutros, C.L.; Balazs, A.B.; Daneman, R.; Bloom, J.M.; Seed, C.; et al. Delivering genes across the blood-brain barrier: LY6A, a novel cellular receptor for AAV-PHP.B capsids. PLoS ONE 2019. [Google Scholar] [CrossRef]
- Hudry, E.; Vandenberghe, L.H. Therapeutic AAV Gene Transfer to the Nervous System: A Clinical Reality. Neuron 2019, 101, 839–862. [Google Scholar] [CrossRef]
- Stott, S.R.W.; Hayat, S.; Carnwath, T.; Garas, S.; Sleeman, J.P.; Barker, R.A. CD24 expression does not affect dopamine neuronal survival in a mouse model of Parkinson’s disease. PLoS ONE 2017, 12, e0171748. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Alzheimer’s Disease Target: β-amyloid Protein | ||||
| Antigen | Species | Intrabody | Experimental Phase | Reference(s) |
| Aβ1–42 | scFv | scFvAβIB | Preclinical in mice | [8] |
| Aβ | scFv | Aβ-scFv | Preclinical in mice | [9] |
| Parkinson’s Disease Target: α-synuclein Protein | ||||
| Antigen | Species | Intrabody | Experimental Phase | Reference(s) |
| Oligomeric α-synuclein | Human scFv | D5 | Preclinical in mice | [10,11,12,13] |
| Oligomeric α-synuclein | Human scFv | 10H | Preclinical in mice | [10,12,14] |
| Pan-specific α-synuclein | Human scFv | D10 | Preclinical in mice | [10,12,15] |
| Hydrophobic non-amyloid component (NAC) of α-synuclein, AA53–95 | Human nanobody (VH) | VH14 (NAC14) | Preclinical in rats | [10,16,17] |
| α-synuclein AA53–87 | Human scFv | NAC32 | In vitro in cell culture | [17] |
| Fibrillar α-synuclein | Human scFv | 6E | In vitro in cell culture | [11,18] |
| α-synuclein, AA118–131 | Camelid nanobody (VHH) | Nbsyn2 | In vitro in cell culture | [16,19] |
| α-synuclein, AA137-140 | Camelid nanobody (VHH) | Nbsyn87 | Preclinical in rats | [16,19,20] |
| Huntington’s Disease Target: Huntingtin (HTT) Protein | ||||
| Antigen | Species | Intrabody | Experimental Phase | Reference(s) |
| HTT-N17 (PolyQ N-term AA1-17) | Human scFv | scFvC4 | Preclinical in mice | [11,21,22,23,24,25,26,27,28,29,30] |
| HTT-N20 (PolyQ N-term, AA1-20) | Human VL | VL12.3 | Preclinical in mice | [22,31,32,33,34,35] |
| HTT-polyQ | Mouse scFv | MW1 | In vitro in cell culture | [36,37] |
| HTT-polyQ | Mouse scFv | MW2 | In vitro in cell culture | [36,37] |
| HTT-PRR | Mouse scFv | MW7 | Ex vivo in brain tissue | [33,36,37] |
| HTT-PRR | Human VL | Happ1 | Preclinical in mice | [33,34] |
| HTT-PRR | Human VL | Happ3 | Ex vivo in brain tissue | [33] |
| HTT-Exon1 | Mouse scFv | EM48 | Preclinical in mice | [38] |
| HTT-PRR | Human scFv | INT41 | Preclinical in mice | [39] |
| Fibrillar mHTT | Human scFv | 6E | In vitro in cell culture | [18,21] |
| Amyotrophic Lateral Sclerosis (ALS) Disease Target: SOD1 Protein | ||||
| Antigen | Species | Intrabody | Experimental Phase | Reference(s) |
| SOD1 | Human scFv | B1 | Preclinical in mice | [40,41] |
| SOD1 | Human scFv | B12 | Preclinical in mice | [40,41] |
| G93A human SOD1 | Mouse scFv | D3H5 | Preclinical in mice | [42] |
| Prion Disorder Target: Prion Protein (PrP) | ||||
| Antigen | Species | Intrabody | Experimental Phase | Reference(s) |
| Cellular PrP | Mouse scFv | 8H4 | Preclinical in mice | [43,44] |
| Cellular PrP | Mouse scFv | 8F9 | In vitro in cell culture | [43] |
| Cellular PrP | Camelid nanobody (VHH) | Nb_PrP_01 | In vitro in crystallography studies | [45,46] |
| Cellular PrP AA123–125, 164–170, and 174–185 | Camelid nanobody (VHH) | Nb484 | In vitro in crystallography studies | [46] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrantelli, F.; Chiozzini, C.; Leone, P.; Manfredi, F.; Federico, M. Engineered Extracellular Vesicles/Exosomes as a New Tool against Neurodegenerative Diseases. Pharmaceutics 2020, 12, 529. https://doi.org/10.3390/pharmaceutics12060529
Ferrantelli F, Chiozzini C, Leone P, Manfredi F, Federico M. Engineered Extracellular Vesicles/Exosomes as a New Tool against Neurodegenerative Diseases. Pharmaceutics. 2020; 12(6):529. https://doi.org/10.3390/pharmaceutics12060529
Chicago/Turabian StyleFerrantelli, Flavia, Chiara Chiozzini, Patrizia Leone, Francesco Manfredi, and Maurizio Federico. 2020. "Engineered Extracellular Vesicles/Exosomes as a New Tool against Neurodegenerative Diseases" Pharmaceutics 12, no. 6: 529. https://doi.org/10.3390/pharmaceutics12060529
APA StyleFerrantelli, F., Chiozzini, C., Leone, P., Manfredi, F., & Federico, M. (2020). Engineered Extracellular Vesicles/Exosomes as a New Tool against Neurodegenerative Diseases. Pharmaceutics, 12(6), 529. https://doi.org/10.3390/pharmaceutics12060529