Current Status of Supersaturable Self-Emulsifying Drug Delivery Systems



Abstract
1. Introduction
1.1. Aim of This Study
1.2. Conventional Solubilized SEDDSs
2. Su-SEDDSs
2.1. Definition of su-SEDDSs
Confusion between Supersaturated SEDDSs and su-SEDDSs
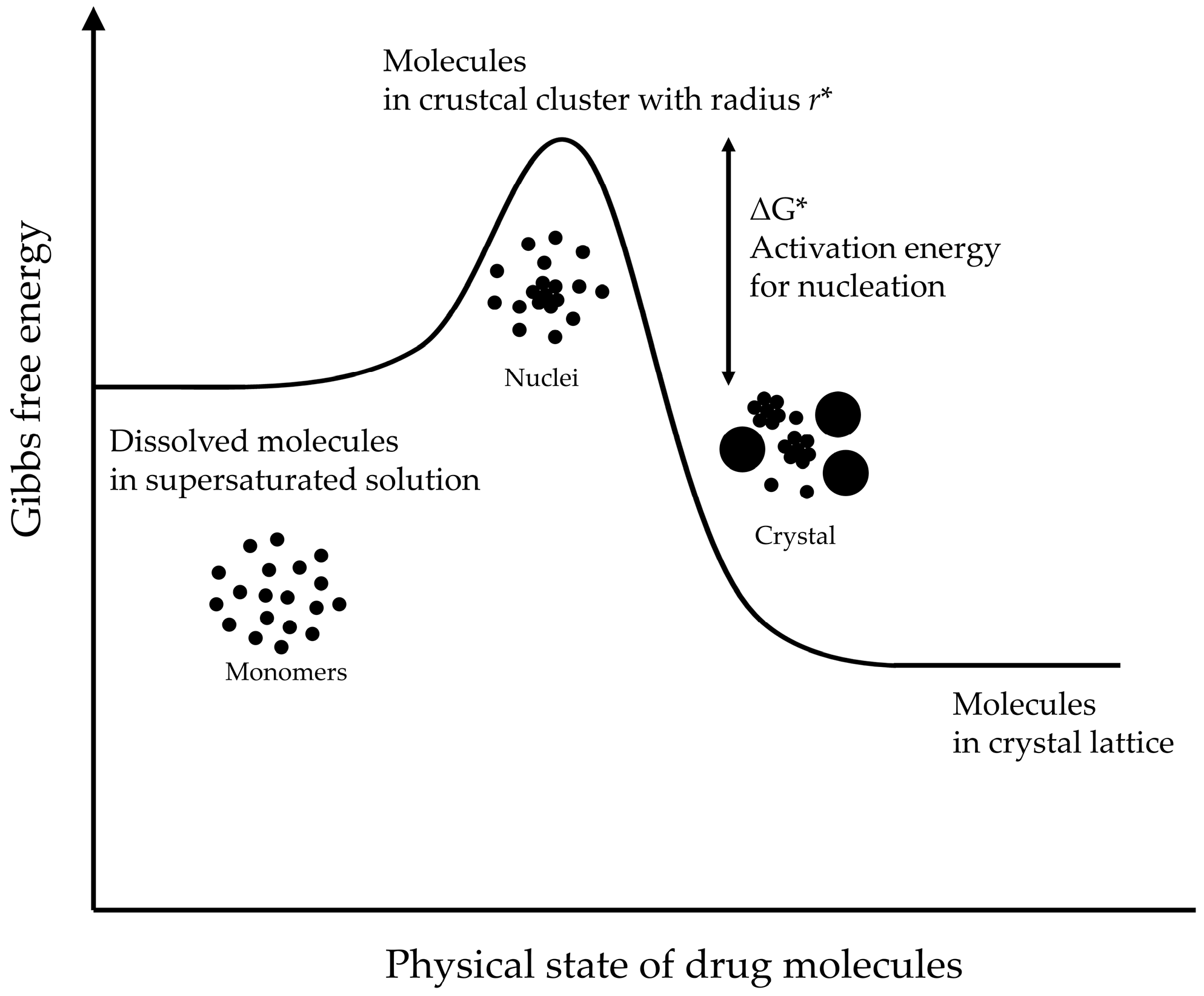
2.2. Understanding the Principles of Drug Precipitation for Successful Development su-SEDDSs
- S < 1 (σ < 0): unsaturated or subsaturated;
- S = 1 (σ = 0): saturated;
- S > 1 (σ > 0): supersaturated.
2.3. In Vivo Drug Absorption from su-SEDDSs
2.3.1. Increase in Absorption by Supersaturation in the GIT
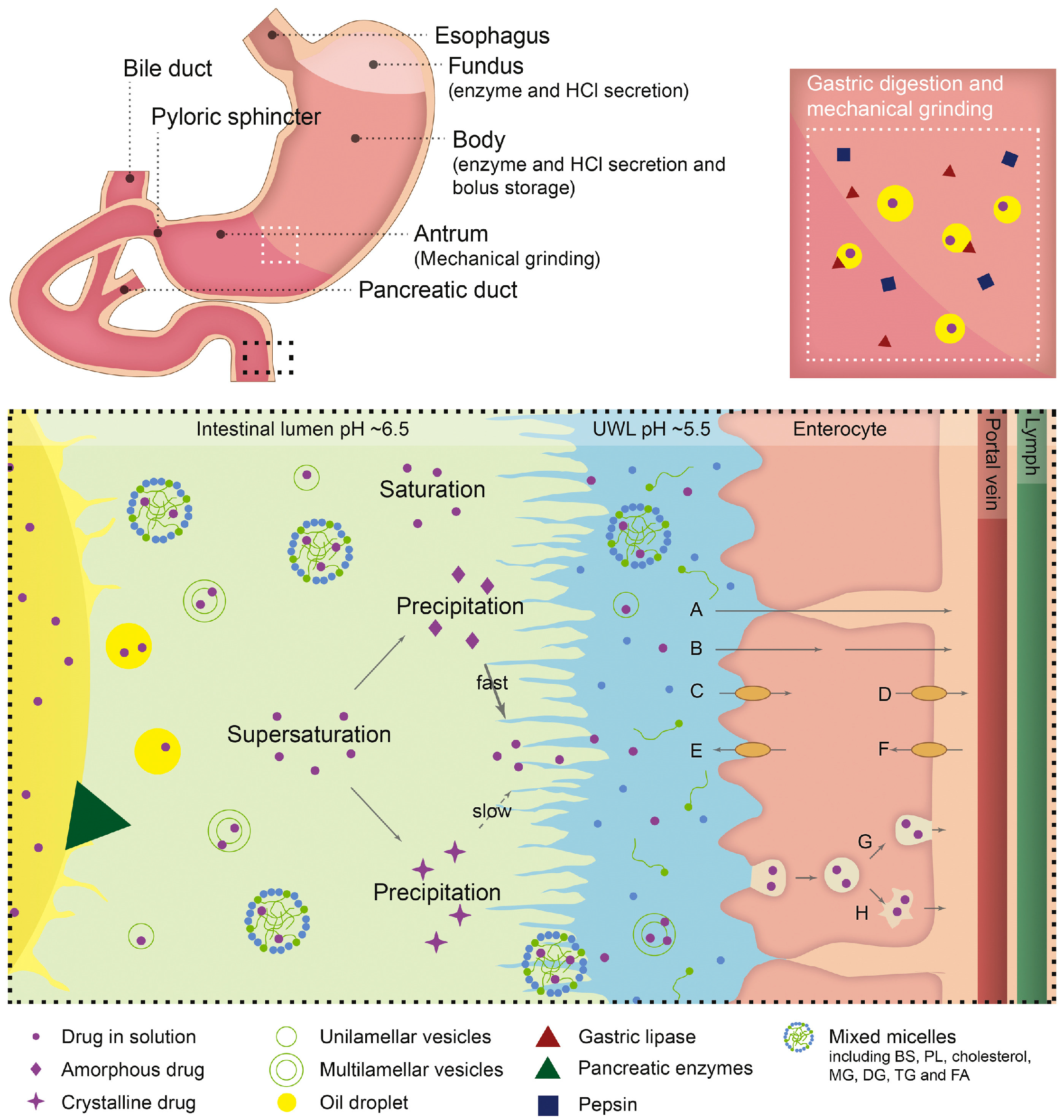
2.3.2. Consideration of GIT Physiological Factors
Effects of GI Physiological pH Changes and Hydrodynamics on Supersaturation
Effect of Food on Supersaturation
Effects of Various Factors on Lipid Digestion and Drug Absorption from su-SEDDSs
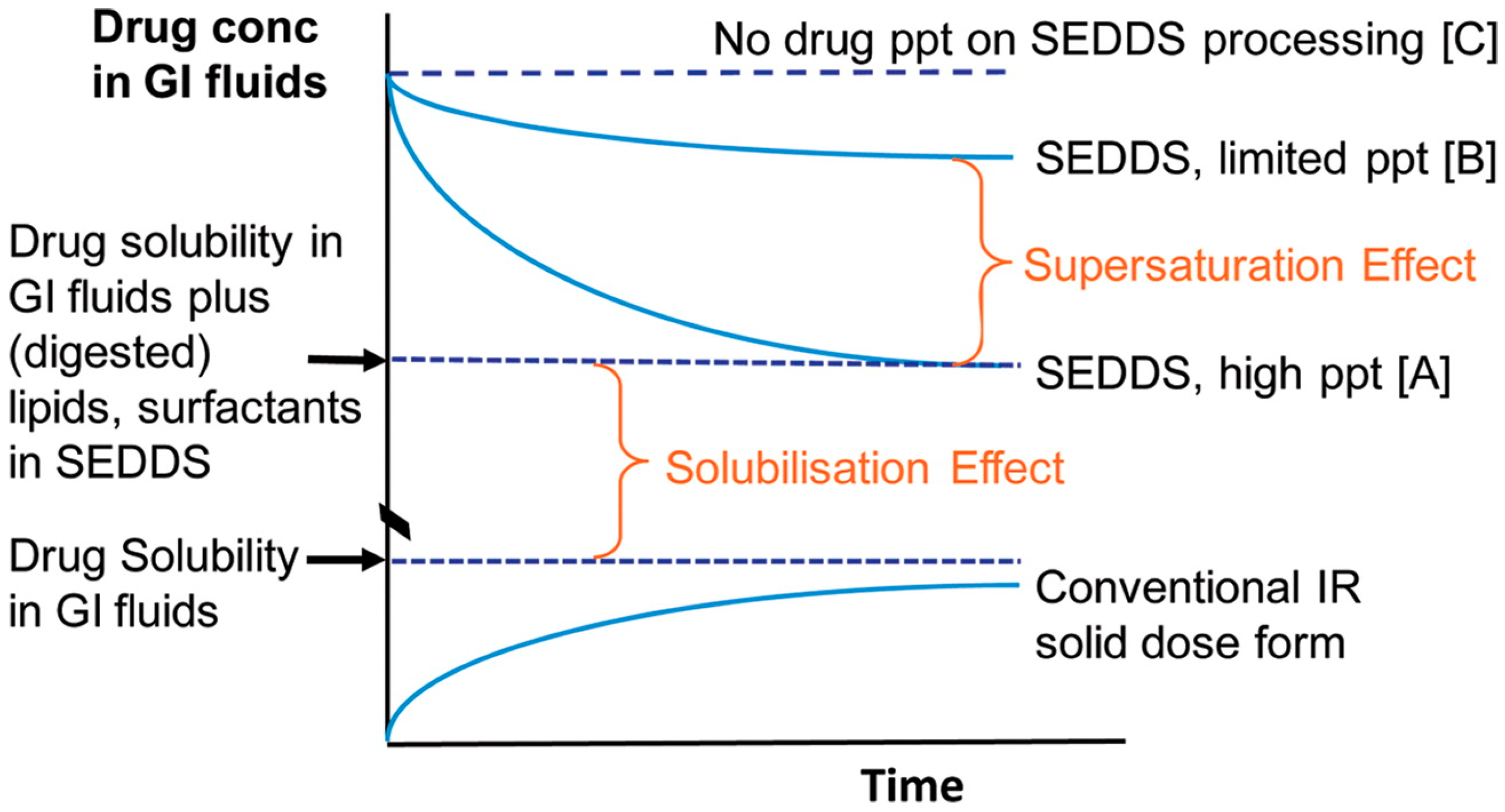
2.3.3. Maintenance of a Supersaturated State for the Improvement of Absorption
2.3.4. New Insight into Precipitation: Considering Increased Absorption by Amorphous Precipitation
2.4. PIs
2.4.1. Mechanisms to Inhibit Drug Precipitation
Hydrogen Bonding
Adsorption Via Hydrophobic Interactions
Reticulate Network Formation and Steric Hindrance
Molecular Rigidity, Molecular Weight, and Viscosity
2.4.2. Classification of PIs
Polymeric PIs
Non-Polymeric PIs
3. Selection and Application of PIs for the Successful Development of su-SEDDSs
3.1. PI Screening in the Preformulation Stage, Early Stages of Drug Development
3.2. Effect of Polymer Solubility in su-SEDDSs on Supersaturation
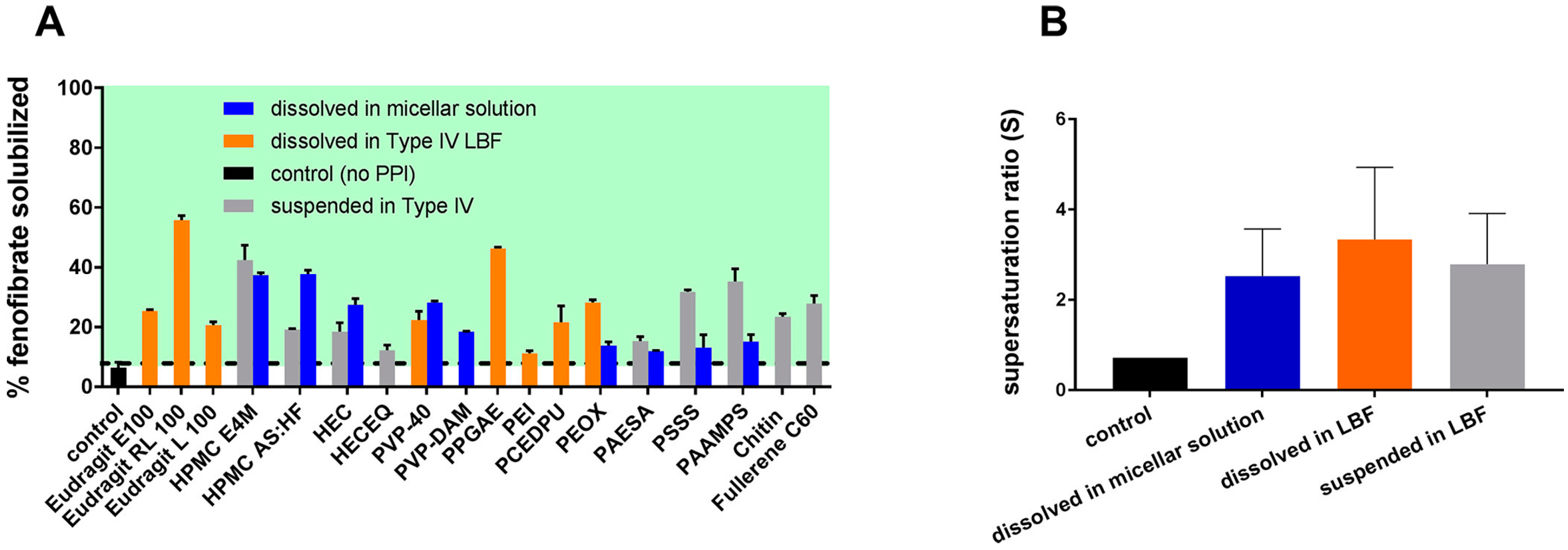
3.3. Cases of PI Application in su-SEDDSs
3.3.1. Dissolving PIs in the Lipid Phase
3.3.2. Suspending PIs in the Lipid Phase
3.3.3. Mixing with Lipids after Dissolving PIs in Soluble Solvent
3.3.4. Blending with Solid SEDDSs
3.3.5. Using Gelatin or HPMC Capsules as PIs
3.4. Potential Effect of PI on Stability, In Vivo Emulsification, and Absorption
3.4.1. Effects of Lipid–PI Interactions on Lipid Digestion
Interactions between PIs and Lipid Components
Interactions between PIs and Lipases
Interactions between PIs and Digested Products
3.5. Pharmaceutical Characterization and Evaluation of su-SEDDSs
3.5.1. Self-Emulsification Properties
3.5.2. Conductivity and Viscosity Measurements
3.5.3. Droplet Size Analysis and Zeta Potential
3.5.4. Phase Separation Test
3.5.5. Spectroscopic Evaluation
3.5.6. Microscopic Evaluation
3.5.7. Small-Angle X-ray Scattering
3.6. Biorelevant Supersaturation Testing
3.6.1. Critical Variables in In Vitro Supersaturation Evaluation
Sink or Non-Sink Conditions
Hydrodynamics
Dissolution Medium
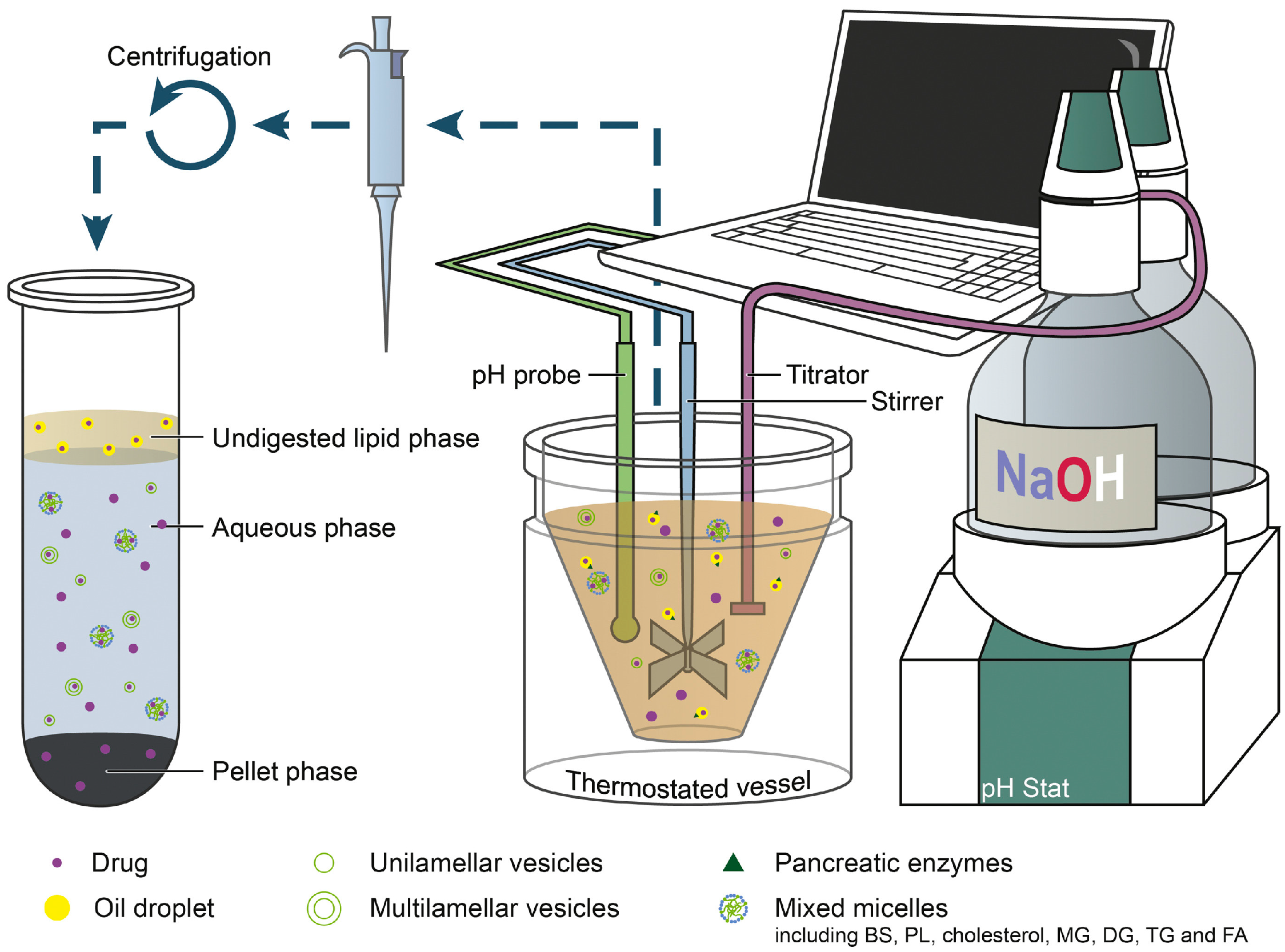
3.6.2. In Vitro Digestion Model
pH-Stat Lipolysis Models
DGM
HTP Lipolysis Model
Real-Time Analysis
Digestion–Permeation Models
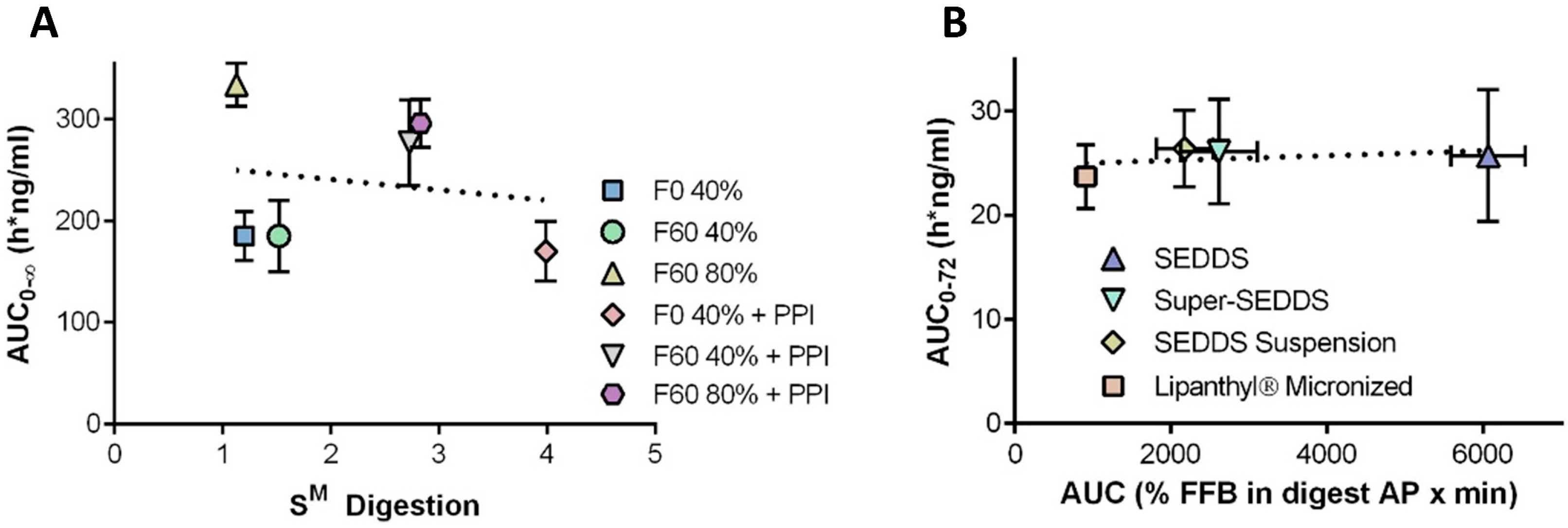
3.6.3. IVIVC Case Studies for su-SEDDSs and Supersaturated SEDDSs
4. Development of su-SEDDSs to Solid Dosage Forms
4.1. Pharmaceutical Excipient for Solidification
4.1.1. Solidification Excipient and Application Cases
Mesoporous Silica
MCC
Polysaccharide-Based Carriers
Polymeric Carriers
Protein-Based Carriers
4.1.2. Influence of Solidification Excipients on the Performance of SEDDSs
4.2. Solidification Process
Influence of the Solidifying Process on the Thermal Stability of Drugs and Excipients
4.3. Applicable Solid Dosage Forms for su-SEDDSs
4.3.1. Powder and Granules
4.3.2. Hard Capsules
4.3.3. Tablets
4.3.4. Suppositories
4.3.5. Implants
4.4. Application of Controlled-Release Technology in su-SEDDSs
4.4.1. Sustained Release
4.4.2. Site-Specific Release
4.5. In Vitro Evaluation of Solid su-SEDDSs
Re-Emulsification and Drug Release from Solid su-SEDDSs
5. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Tang, B.; Cheng, G.; Gu, J.-C.; Xu, C.-H. Development of solid self-emulsifying drug delivery systems: Preparation techniques and dosage forms. Drug Discov. Today 2008, 13, 606–612. [Google Scholar] [CrossRef]
- Herpin, M.J.; Smyth, H.D.C. Super-heated aqueous particle engineering (SHAPE): A novel method for the micronization of poorly water soluble drugs. J. Pharm. Investig. 2018, 48, 135–142. [Google Scholar] [CrossRef]
- Hajjar, B.; Zier, K.-I.; Khalid, N.; Azarmi, S.; Löbenberg, R. Evaluation of a microemulsion-based gel formulation for topical drug delivery of diclofenac sodium. J. Pharm. Investig. 2018, 48, 351–362. [Google Scholar] [CrossRef]
- Singh, D.; Bedi, N.; Tiwary, A.K. Enhancing solubility of poorly aqueous soluble drugs: Critical appraisal of techniques. J. Pharm. Investig. 2018, 48, 509–526. [Google Scholar] [CrossRef]
- Dokania, S.; Joshi, A.K. Self-microemulsifying drug delivery system (SMEDDS)–challenges and road ahead. Drug Deliv. 2015, 22, 675–690. [Google Scholar] [CrossRef]
- Madhav, K.V.; Kishan, V. Self microemulsifying particles of loratadine for improved oral bioavailability: Preparation, characterization and in vivo evaluation. J. Pharm. Investig. 2018, 48, 497–508. [Google Scholar] [CrossRef]
- Chatterjee, B.; Hamed Almurisi, S.; Ahmed Mahdi Dukhan, A.; Mandal, U.K.; Sengupta, P. Controversies with self-emulsifying drug delivery system from pharmacokinetic point of view. Drug Deliv. 2016, 23, 3639–3652. [Google Scholar] [CrossRef]
- Lim, J.H.; Na, Y.G.; Lee, H.K.; Kim, S.J.; Lee, H.J.; Bang, K.H.; Wang, M.; Pyo, Y.C.; Huh, H.W.; Cho, C.W. Effect of surfactant on the preparation and characterization of gemcitabine-loaded particles. J. Pharm. Investig. 2019, 49, 271–278. [Google Scholar] [CrossRef]
- Choi, Y.H.; Han, H.-K. Nanomedicines: Current status and future perspectives in aspect of drug delivery and pharmacokinetics. J. Pharm. Investig. 2018, 48, 43–60. [Google Scholar] [CrossRef]
- Ahsan, M.N.; Verma, P.R.P. Enhancement of in vitro dissolution and pharmacodynamic potential of olanzapine using solid SNEDDS. J. Pharm. Investig. 2018, 48, 269–278. [Google Scholar] [CrossRef]
- Rastogi, V.; Yadav, P.; Verma, N.; Verma, A. Preparation and characterization of transdermal mediated microemulsion delivery of T4 bacteriophages against E.coli bacteria: A novel anti-microbial approach. J. Pharm. Investig. 2018, 48, 393–407. [Google Scholar] [CrossRef]
- Choi, D.H.; Kim, Y.-S.; Kim, D.-D.; Jeong, S.H. QbD based development and evaluation of topical microemulsion-based hydrogel against superficial fungal infections. J. Pharm. Investig. 2019, 49, 87–103. [Google Scholar] [CrossRef]
- McEvoy, C.L.; Trevaskis, N.L.; Feeney, O.M.; Edwards, G.A.; Perlman, M.E.; Ambler, C.M.; Porter, C.J. Correlating in Vitro Solubilization and Supersaturation Profiles with in Vivo Exposure for Lipid Based Formulations of the CETP Inhibitor CP-532,623. Mol. Pharm. 2017, 14, 4525–4538. [Google Scholar] [CrossRef] [PubMed]
- Nikolakakis, I.; Partheniadis, I. Self-emulsifying granules and pellets: Composition and formation mechanisms for instant or controlled release. Pharmaceutics 2017, 9, 50. [Google Scholar] [CrossRef]
- Patil, S.C.; Tagalpallewar, A.A.; Kokare, C.R. Natural anti-proliferative agent loaded self-microemulsifying nanoparticles for potential therapy in oral squamous carcinoma. J. Pharm. Investig. 2019, 49, 527–541. [Google Scholar] [CrossRef]
- Khoo, S.-M.; Humberstone, A.J.; Porter, C.J.; Edwards, G.A.; Charman, W.N. Formulation design and bioavailability assessment of lipidic self-emulsifying formulations of halofantrine. Int. J. Pharm. 1998, 167, 155–164. [Google Scholar] [CrossRef]
- Sek, L.; Boyd, B.J.; Charman, W.N.; Porter, C.J. Examination of the impact of a range of Pluronic surfactants on the in-vitro solubilisation behaviour and oral bioavailability of lipidic formulations of atovaquone. J. Pharm. Pharmacol. 2006, 58, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Feeney, O.M.; Crum, M.F.; McEvoy, C.L.; Trevaskis, N.L.; Williams, H.D.; Pouton, C.W.; Charman, W.N.; Bergström, C.A.; Porter, C.J. 50 years of oral lipid-based formulations: Provenance, progress and future perspectives. Adv. Drug Deliv. Rev. 2016, 101, 167–194. [Google Scholar] [CrossRef] [PubMed]
- Pouton, C.W. Formulation of poorly water-soluble drugs for oral administration: Physicochemical and physiological issues and the lipid formulation classification system. Eur. J. Pharm. Sci. 2006, 29, 278–287. [Google Scholar] [CrossRef]
- Amara, S.; Bourlieu, C.; Humbert, L.; Rainteau, D.; Carrière, F. Variations in gastrointestinal lipases, pH and bile acid levels with food intake, age and diseases: Possible impact on oral lipid-based drug delivery systems. Adv. Drug Deliv. Rev. 2019, 142, 3–15. [Google Scholar] [CrossRef]
- Porter, C.J.; Trevaskis, N.L.; Charman, W.N. Lipids and lipid-based formulations: Optimizing the oral delivery of lipophilic drugs. Nat. Rev. Drug Discov. 2007, 6, 231–248. [Google Scholar] [CrossRef] [PubMed]
- Boyd, B.J.; Bergström, C.A.; Vinarov, Z.; Kuentz, M.; Brouwers, J.; Augustijns, P.; Brandl, M.; Bernkop-Schnürch, A.; Shrestha, N.; Préat, V. Successful oral delivery of poorly water-soluble drugs both depends on the intraluminal behavior of drugs and of appropriate advanced drug delivery systems. Eur. J. Pharm. Sci. 2019, 137, 104967. [Google Scholar] [CrossRef] [PubMed]
- Amidon, G.L.; Lennernäs, H.; Shah, V.P.; Crison, J.R. A theoretical basis for a biopharmaceutic drug classification: The correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Danafar, H.; Rostamizadeh, K.; Hamidi, M. Polylactide/poly(ethylene glycol)/polylactide triblock copolymer micelles as carrier for delivery of hydrophilic and hydrophobic drugs: A comparison study. J. Pharm. Investig. 2018, 48, 381–391. [Google Scholar] [CrossRef]
- Berthelsen, R.; Klitgaard, M.; Rades, T.; Müllertz, A. In vitro digestion models to evaluate lipid based drug delivery systems; present status and current trends. Adv. Drug Deliv. Rev. 2019, 142, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Quan, G.; Niu, B.; Singh, V.; Zhou, Y.; Wu, C.-Y.; Pan, X.; Wu, C. Supersaturable solid self-microemulsifying drug delivery system: Precipitation inhibition and bioavailability enhancement. Int. J. Nanomed. 2017, 12, 8801. [Google Scholar] [CrossRef]
- Gupta, S.; Kesarla, R.; Omri, A. Formulation strategies to improve the bioavailability of poorly absorbed drugs with special emphasis on self-emulsifying systems. ISRN Pharm. 2013, 2013. [Google Scholar] [CrossRef]
- Nardin, I.; Köllner, S. Successful development of oral SEDDS: Screening of excipients from the industrial point of view. Adv. Drug Deliv. Rev. 2019, 142, 128–140. [Google Scholar] [CrossRef]
- Burdock, G.A.; Carabin, I.G. Generally recognized as safe (GRAS): History and description. Toxicol. Lett. 2004, 150, 3–18. [Google Scholar] [CrossRef]
- Joyce, P.; Dening, T.J.; Meola, T.R.; Schultz, H.B.; Holm, R.; Thomas, N.; Prestidge, C.A. Solidification to improve the biopharmaceutical performance of SEDDS: Opportunities and challenges. Adv. Drug Deliv. Rev. 2019, 142, 102–117. [Google Scholar] [CrossRef]
- Gao, P.; Morozowich, W. Development of supersaturatable self-emulsifying drug delivery system formulations for improving the oral absorption of poorly soluble drugs. Expert Opin. Drug Deliv. 2006, 3, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Suys, E.J.; Chalmers, D.K.; Pouton, C.W.; Porter, C.J. Polymeric precipitation inhibitors promote fenofibrate supersaturation and enhance drug absorption from a type IV lipid-based formulation. Mol. Pharm. 2018, 15, 2355–2371. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Ma, X.; Williams Iii, R.O. Polymeric nanomedicines for poorly soluble drugs in oral delivery systems: An update. J. Pharm. Investig. 2018, 48, 61–75. [Google Scholar] [CrossRef]
- Brouwers, J.; Brewster, M.E.; Augustijns, P. Supersaturating drug delivery systems: The answer to solubility-limited oral bioavailability? J. Pharm. Sci. 2009, 98, 2549–2572. [Google Scholar] [CrossRef]
- Thomas, N.; Holm, R.; Müllertz, A.; Rades, T. In vitro and in vivo performance of novel supersaturated self-nanoemulsifying drug delivery systems (super-SNEDDS). J. Control. Release 2012, 160, 25–32. [Google Scholar] [CrossRef]
- Mukherjee, T.; Plakogiannis, F.M. Development and oral bioavailability assessment of a supersaturated self-microemulsifying drug delivery system (SMEDDS) of albendazole. J. Pharm. Pharmacol. 2010, 62, 1112–1120. [Google Scholar] [CrossRef]
- Rao, S.; Tan, A.; Boyd, B.J.; Prestidge, C.A. Synergistic role of self-emulsifying lipids and nanostructured porous silica particles in optimizing the oral delivery of lovastatin. Nanomedicine 2014, 9, 2745–2759. [Google Scholar] [CrossRef]
- Zhou, H.; Wan, J.; Wu, L.; Yi, T.; Liu, W.; Xu, H.; Yang, X. A new strategy for enhancing the oral bioavailability of drugs with poor water-solubility and low liposolubility based on phospholipid complex and supersaturated SEDDS. PLoS ONE 2013, 8, e84530. [Google Scholar] [CrossRef]
- Thomas, N.; Holm, R.; Garmer, M.; Karlsson, J.J.; Müllertz, A.; Rades, T. Supersaturated self-nanoemulsifying drug delivery systems (Super-SNEDDS) enhance the bioavailability of the poorly water-soluble drug simvastatin in dogs. Aaps J. 2013, 15, 219–227. [Google Scholar] [CrossRef]
- Crum, M.F.; Trevaskis, N.L.; Pouton, C.W.; Porter, C.J. Transient supersaturation supports drug absorption from lipid-based formulations for short periods of time, but ongoing solubilization is required for longer absorption periods. Mol. Pharm. 2017, 14, 394–405. [Google Scholar] [CrossRef]
- Zanchetta, B.; Chaud, M.; Santana, M. Self-emulsifying drug delivery systems (SEDDS) in pharmaceutical development. J Adv Chem Eng 2015, 5, 1–7. [Google Scholar]
- Stillhart, C.; Kuentz, M. Trends in the assessment of drug supersaturation and precipitation in vitro using lipid-based delivery systems. J. Pharm. Sci. 2016, 105, 2468–2476. [Google Scholar] [CrossRef] [PubMed]
- Kuentz, M. Drug supersaturation during formulation digestion, including real-time analytical approaches. Adv. Drug Deliv. Rev. 2019, 142, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Sinko, P.J.; Singh, Y. Martin’s Physical Pharmacy and Pharmaceutical Sciences: Physical Chemical and Biopharmaceutical Principles in the Pharmaceutical Sciences, 5th ed.; Lippincott Williams & Wilkins: Baltimore, MD, USA, 2011. [Google Scholar]
- Ye, J.; Wu, H.; Huang, C.; Lin, W.; Zhang, C.; Huang, B.; Lu, B.; Xu, H.; Li, X.; Long, X. Comparisons of in vitro Fick’s first law, lipolysis, and in vivo rat models for oral absorption on BCS II drugs in SNEDDS. Int. J. Nanomed. 2019, 14, 5623. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, T. Physical chemical analysis of percutaneous absorption process from creams and ointments. J. Soc. Cosmet. Chem 1960, 11, 85–97. [Google Scholar]
- Hens, B.; Brouwers, J.; Corsetti, M.; Augustijns, P. Supersaturation and precipitation of posaconazole upon entry in the upper small intestine in humans. J. Pharm. Sci. 2016, 105, 2677–2684. [Google Scholar] [CrossRef]
- Brouwers, J.; Augustijns, P. Resolving intraluminal drug and formulation behavior: Gastrointestinal concentration profiling in humans. Eur. J. Pharm. Sci. 2014, 61, 2–10. [Google Scholar] [CrossRef]
- Kourentas, A.; Vertzoni, M.; Symillides, M.; Hens, B.; Brouwers, J.; Augustijns, P.; Reppas, C. In vitro evaluation of the impact of gastrointestinal transfer on luminal performance of commercially available products of posaconazole and itraconazole using BioGIT. Int. J. Pharm. 2016, 515, 352–358. [Google Scholar] [CrossRef]
- Knoebel, R.W.; Larson, R.A. Pepsi® or Coke®? Influence of acid on dasatinib absorption. J. Oncol. Pharm. Pract. 2018, 24, 156–158. [Google Scholar] [CrossRef]
- Walravens, J.; Brouwers, J.; Spriet, I.; Tack, J.; Annaert, P.; Augustijns, P. Effect of pH and comedication on gastrointestinal absorption of posaconazole. Clin. Pharmacokinet. 2011, 50, 725–734. [Google Scholar] [CrossRef]
- Anby, M.U.; Williams, H.D.; McIntosh, M.; Benameur, H.; Edwards, G.A.; Pouton, C.W.; Porter, C.J. Lipid digestion as a trigger for supersaturation: Evaluation of the impact of supersaturation stabilization on the in vitro and in vivo performance of self-emulsifying drug delivery systems. Mol. Pharm. 2012, 9, 2063–2079. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Rush, B.D.; Pfund, W.P.; Huang, T.; Bauer, J.M.; Morozowich, W.; Kuo, M.S.; Hageman, M.J. Development of a supersaturable SEDDS (S-SEDDS) formulation of paclitaxel with improved oral bioavailability. J. Pharm. Sci. 2003, 92, 2386–2398. [Google Scholar] [CrossRef] [PubMed]
- Taylor, L.S.; Zhang, G.G. Physical chemistry of supersaturated solutions and implications for oral absorption. Adv. Drug Deliv. Rev. 2016, 101, 122–142. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Dai, W.-G. Drug precipitation inhibitors in supersaturable formulations. Int. J. Pharm. 2013, 453, 36–43. [Google Scholar] [CrossRef]
- Bevernage, J.; Brouwers, J.; Brewster, M.E.; Augustijns, P. Evaluation of gastrointestinal drug supersaturation and precipitation: Strategies and issues. Int. J. Pharm. 2013, 453, 25–35. [Google Scholar] [CrossRef]
- Guzmán, H.R.; Tawa, M.; Zhang, Z.; Ratanabanangkoon, P.; Shaw, P.; Gardner, C.R.; Chen, H.; Moreau, J.P.; Almarsson, Ö.; Remenar, J.F. Combined use of crystalline salt forms and precipitation inhibitors to improve oral absorption of celecoxib from solid oral formulations. J. Pharm. Sci. 2007, 96, 2686–2702. [Google Scholar] [CrossRef]
- Mohsin, K.; Long, M.A.; Pouton, C.W. Design of lipid-based formulations for oral administration of poorly water-soluble drugs: Precipitation of drug after dispersion of formulations in aqueous solution. J. Pharm. Sci. 2009, 98, 3582–3595. [Google Scholar] [CrossRef]
- Miller, J.M.; Beig, A.; Carr, R.A.; Webster, G.K.; Dahan, A. The solubility–permeability interplay when using cosolvents for solubilization: Revising the way we use solubility-enabling formulations. Mol. Pharm. 2012, 9, 581–590. [Google Scholar] [CrossRef]
- Warren, D.B.; Benameur, H.; Porter, C.J.; Pouton, C.W. Using polymeric precipitation inhibitors to improve the absorption of poorly water-soluble drugs: A mechanistic basis for utility. J. Drug Target. 2010, 18, 704–731. [Google Scholar] [CrossRef]
- Porter, C.J.; Anby, M.U.; Warren, D.B.; Williams, H.D.; Benameur, H.; Pouton, C.W. Lipid-Based Formulations: Exploring the Link between in Vitro Supersaturation and in Vivo Exposure. Bull. Tech. Gattefosse. 2011, 104, 60–68. [Google Scholar]
- Augustijns, P.; Brewster, M.E. Supersaturating drug delivery systems: Fast is not necessarily good enough. J. Pharm. Sci. 2012, 101, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Six, K.; Daems, T.; de Hoon, J.; Van Hecken, A.; Depre, M.; Bouche, M.-P.; Prinsen, P.; Verreck, G.; Peeters, J.; Brewster, M.E. Clinical study of solid dispersions of itraconazole prepared by hot-stage extrusion. Eur. J. Pharm. Sci. 2005, 24, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Popov, A.; Schopf, L.; Bourassa, J.; Chen, H. Enhanced pulmonary delivery of fluticasone propionate in rodents by mucus-penetrating nanoparticles. Int. J. Pharm. 2016, 502, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Amin, O.M.; Ammar, A.; Eladawy, S.A. Febuxostat loaded β-cyclodextrin based nanosponge tablet: An in vitro and in vivo evaluation. J. Pharm. Investig. 2019. [Google Scholar] [CrossRef]
- Carlert, S.; Pålsson, A.; Hanisch, G.; Von Corswant, C.; Nilsson, C.; Lindfors, L.; Lennernäs, H.; Abrahamsson, B. Predicting intestinal precipitation—A case example for a basic BCS class II drug. Pharm. Res. 2010, 27, 2119–2130. [Google Scholar] [CrossRef]
- Gao, P.; Akrami, A.; Alvarez, F.; Hu, J.; Li, L.; Ma, C.; Surapaneni, S. Characterization and optimization of AMG 517 supersaturatable self-emulsifying drug delivery system (S-SEDDS) for improved oral absorption. J. Pharm. Sci. 2009, 98, 516–528. [Google Scholar] [CrossRef]
- Dash, R.N.; Mohammed, H.; Humaira, T. Design, optimization, and evaluation of ezetimibe solid supersaturatable self-nanoemulsifying drug delivery for enhanced solubility and dissolution. J. Pharm. Investig. 2016, 46, 153–168. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Katare, O.; Singh, B. Development of optimized supersaturable self-nanoemulsifying systems of ezetimibe: Effect of polymers and efflux transporters. Expert Opin. Drug Deliv. 2014, 11, 479–492. [Google Scholar] [CrossRef]
- Sassene, P.J.; Knopp, M.M.; Hesselkilde, J.Z.; Koradia, V.; Larsen, A.; Rades, T.; Müllertz, A. Precipitation of a poorly soluble model drug during in vitro lipolysis: Characterization and dissolution of the precipitate. J. Pharm. Sci. 2010, 99, 4982–4991. [Google Scholar] [CrossRef]
- Stillhart, C.; Dürr, D.; Kuentz, M. Toward an improved understanding of the precipitation behavior of weakly basic drugs from oral lipid-based formulations. J. Pharm. Sci. 2014, 103, 1194–1203. [Google Scholar] [CrossRef]
- Sassene, P.J.; Mosgaard, M.D.; Löbmann, K.; Mu, H.; Larsen, F.H.; Rades, T.; Müllertz, A. Elucidating the molecular interactions occurring during drug precipitation of weak bases from lipid-based formulations: A case study with cinnarizine and a long chain self-nanoemulsifying drug delivery system. Mol. Pharm. 2015, 12, 4067–4076. [Google Scholar] [CrossRef] [PubMed]
- Kaur, N.; Narang, A.; Bansal, A.K. Use of biorelevant dissolution and PBPK modeling to predict oral drug absorption. Eur. J. Pharm. Biopharm. 2018, 129, 222–246. [Google Scholar] [CrossRef] [PubMed]
- Thomas, N.; Holm, R.; Rades, T.; Müllertz, A. Characterising lipid lipolysis and its implication in lipid-based formulation development. Aaps J. 2012, 14, 860–871. [Google Scholar] [CrossRef]
- Khan, J.; Rades, T.; Boyd, B. The precipitation behavior of poorly water-soluble drugs with an emphasis on the digestion of lipid based formulations. Pharm. Res. 2016, 33, 548–562. [Google Scholar] [CrossRef]
- Vetter, T.; Mazzotti, M.; Brozio, J.R. Slowing the growth rate of ibuprofen crystals using the polymeric additive Pluronic F127. Cryst. Growth Des. 2011, 11, 3813–3821. [Google Scholar] [CrossRef]
- Liu, X.; Feng, X.; Williams Iii, R.O.; Zhang, F. Characterization of amorphous solid dispersions. J. Pharm. Investig. 2018, 48, 19–41. [Google Scholar] [CrossRef]
- Do Thi, T.; Van Speybroeck, M.; Barillaro, V.; Martens, J.; Annaert, P.; Augustijns, P.; Van Humbeeck, J.; Vermant, J.; Van den Mooter, G. Formulate-ability of ten compounds with different physicochemical profiles in SMEDDS. Eur. J. Pharm. Sci. 2009, 38, 479–488. [Google Scholar] [CrossRef]
- Gao, P.; Shi, Y. Characterization of supersaturatable formulations for improved absorption of poorly soluble drugs. Aaps J. 2012, 14, 703–713. [Google Scholar] [CrossRef]
- Mu, H.; Holm, R.; Müllertz, A. Lipid-based formulations for oral administration of poorly water-soluble drugs. Int. J. Pharm. 2013, 453, 215–224. [Google Scholar] [CrossRef]
- Li, P.; Hynes, S.R.; Haefele, T.F.; Pudipeddi, M.; Royce, A.E.; Serajuddin, A.T. Development of clinical dosage forms for a poorly water-soluble drug II: Formulation and characterization of a novel solid microemulsion preconcentrate system for oral delivery of a poorly water-soluble drug. J. Pharm. Sci. 2009, 98, 1750–1764. [Google Scholar] [CrossRef]
- Serajuddin, A.; Li, P.; Haefele, T. Development of lipid-based drug delivery systems for poorly water-soluble drugs as viable oral dosage forms—Present status and future prospects. Am Pharm Rev 2008, 11, 34–42. [Google Scholar]
- Chauhan, H.; Hui-Gu, C.; Atef, E. Correlating the behavior of polymers in solution as precipitation inhibitor to its amorphous stabilization ability in solid dispersions. J. Pharm. Sci. 2013, 102, 1924–1935. [Google Scholar] [CrossRef] [PubMed]
- Usui, F.; Maeda, K.; Kusai, A.; Nishimura, K.; Yamamoto, K. Inhibitory effects of water-soluble polymers on precipitation of RS-8359. Int. J. Pharm. 1997, 154, 59–66. [Google Scholar] [CrossRef]
- Vandecruys, R.; Peeters, J.; Verreck, G.; Brewster, M.E. Use of a screening method to determine excipients which optimize the extent and stability of supersaturated drug solutions and application of this system to solid formulation design. Int. J. Pharm. 2007, 342, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Patole, V.C.; Pandit, A.P. Mesalamine-loaded alginate microspheres filled in enteric coated HPMC capsules for local treatment of ulcerative colitis: In vitro and in vivo characterization. J. Pharm. Investig. 2018, 48, 257–267. [Google Scholar] [CrossRef]
- Raghavan, S.; Trividic, A.; Davis, A.; Hadgraft, J. Crystallization of hydrocortisone acetate: Influence of polymers. Int. J. Pharm. 2001, 212, 213–221. [Google Scholar] [CrossRef]
- Miller, D.A.; DiNunzio, J.C.; Yang, W.; McGinity, J.W.; Williams III, R.O. Enhanced in vivo absorption of itraconazole via stabilization of supersaturation following acidic-to-neutral pH transition. Drug Dev. Ind. Pharm. 2008, 34, 890–902. [Google Scholar] [CrossRef]
- Chen, Z.-Q.; Liu, Y.; Zhao, J.-H.; Wang, L.; Feng, N.-P. Improved oral bioavailability of poorly water-soluble indirubin by a supersaturatable self-microemulsifying drug delivery system. Int. J. Nanomed. 2012, 7, 1115. [Google Scholar]
- Gosangari, S.; Dyakonov, T. Enhanced dissolution performance of curcumin with the use of supersaturatable formulations. Pharm. Dev. Technol. 2013, 18, 475–480. [Google Scholar] [CrossRef]
- Douroumis, D.; Fahr, A. Stable carbamazepine colloidal systems using the cosolvent technique. Eur. J. Pharm. Sci. 2007, 30, 367–374. [Google Scholar] [CrossRef]
- Rasenack, N.; Hartenhauer, H.; Müller, B.W. Microcrystals for dissolution rate enhancement of poorly water-soluble drugs. Int. J. Pharm. 2003, 254, 137–145. [Google Scholar] [CrossRef]
- Zimmermann, A.; Millqvist-Fureby, A.; Elema, M.R.; Hansen, T.; Müllertz, A.; Hovgaard, L. Adsorption of pharmaceutical excipients onto microcrystals of siramesine hydrochloride: Effects on physicochemical properties. Eur. J. Pharm. Biopharm. 2009, 71, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.-G. In vitro methods to assess drug precipitation. Int. J. Pharm. 2010, 393, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.D. Kinetics and Mechanisms of Crystal Growth Inhibition of Indomethacin by Model Precipitation Inhibitors. Ph.D. Thesis, University of Kentucky Dissertion, Lexington, KY, USA, 2015. [Google Scholar]
- Plaizier-Vercammen, J.A. Interaction of povidone with aromatic compounds IV: Effects of macromolecule molecular weight, solvent dielectric constant, and ligand solubility on complex formation. J. Pharm. Sci. 1983, 72, 1042–1044. [Google Scholar] [CrossRef] [PubMed]
- Naik, J.B.; Naik, J.B.; Waghulde, M.R.; Waghulde, M.R. Development of vildagliptin loaded Eudragit® microspheres by screening design: In vitro evaluation. J. Pharm. Investig. 2018, 48, 627–637. [Google Scholar] [CrossRef]
- Phaechamud, T.; Phaechamud, T.; Lertsuphotvanit, N.; Lertsuphotvanit, N.; Issarayungyuen, P.; Issarayungyuen, P.; Chantadee, T.; Chantadee, T. Design, fabrication and characterization of xanthan gum/liquid-loaded porous natural rubber film. J. Pharm. Investig. 2019, 49, 149–160. [Google Scholar] [CrossRef]
- Dias, M.; Raghavan, S.; Pellett, M.; Hadgraft, J. The effect of β-cyclodextrins on the permeation of diclofenac from supersaturated solutions. Int. J. Pharm. 2003, 263, 173–181. [Google Scholar] [CrossRef]
- Iervolino, M.; Raghavan, S.; Hadgraft, J. Membrane penetration enhancement of ibuprofen using supersaturation. Int. J. Pharm. 2000, 198, 229–238. [Google Scholar] [CrossRef]
- Brewster, M.E.; Vandecruys, R.; Peeters, J.; Neeskens, P.; Verreck, G.; Loftsson, T. Comparative interaction of 2-hydroxypropyl-β-cyclodextrin and sulfobutylether-β-cyclodextrin with itraconazole: Phase-solubility behavior and stabilization of supersaturated drug solutions. Eur. J. Pharm. Sci. 2008, 34, 94–103. [Google Scholar] [CrossRef]
- Loftsson, T.; Vogensen, S.B.; Brewster, M.E.; Konráðsdóttir, F. Effects of cyclodextrins on drug delivery through biological membranes. J. Pharm. Sci. 2007, 96, 2532–2546. [Google Scholar] [CrossRef]
- Wu, Z.; Tucker, I.G.; Razzak, M.; Yang, L.; McSporran, K.; Medlicott, N.J. Absorption and tissue tolerance of ricobendazole in the presence of hydroxypropyl-β-cyclodextrin following subcutaneous injection in sheep. Int. J. Pharm. 2010, 397, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; Hussain, A.; Hussain, M.S.; Mirza, M.A.; Iqbal, Z. Role of excipients in successful development of self-emulsifying/microemulsifying drug delivery system (SEDDS/SMEDDS). Drug Dev. Ind. Pharm. 2013, 39, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Tiwary, A.K.; Bedi, N. Canagliflozin loaded SMEDDS: Formulation optimization for improved solubility, permeability and pharmacokinetic performance. J. Pharm. Investig. 2019, 49, 67–85. [Google Scholar] [CrossRef]
- Zhang, N.; Zhang, W.; Jin, Y.; Quan, D.-Q. Studies on preparation of carbamazepine (CBZ) supersaturatable self-microemulsifying (S-SMEDDS) formulation and relative bioavailability in beagle dogs. Pharm. Dev. Technol. 2011, 16, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Gao, P.; Gong, Y.; Ping, H. Application of a biphasic test for characterization of in vitro drug release of immediate release formulations of celecoxib and its relevance to in vivo absorption. Mol. Pharm. 2010, 7, 1458–1465. [Google Scholar] [CrossRef] [PubMed]
- Chavan, R.B.; Modi, S.R.; Bansal, A.K. Role of solid carriers in pharmaceutical performance of solid supersaturable SEDDS of celecoxib. Int. J. Pharm. 2015, 495, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Song, W.H.; Yeom, D.W.; Lee, D.H.; Lee, K.M.; Yoo, H.J.; Chae, B.R.; Song, S.H.; Choi, Y.W. In situ intestinal permeability and in vivo oral bioavailability of celecoxib in supersaturating self-emulsifying drug delivery system. Arch. Pharmacal Res. 2014, 37, 626–635. [Google Scholar] [CrossRef]
- Song, W.H.; Park, J.H.; Yeom, D.W.; Ahn, B.K.; Lee, K.M.; Lee, S.G.; Woo, H.S.; Choi, Y.W. Enhanced dissolution of celecoxib by supersaturating self-emulsifying drug delivery system (S-SEDDS) formulation. Arch. Pharmacal Res. 2013, 36, 69–78. [Google Scholar] [CrossRef]
- Jaisamut, P.; Wiwattanawongsa, K.; Graidist, P.; Sangsen, Y.; Wiwattanapatapee, R. Enhanced oral bioavailability of curcumin using a supersaturatable self-microemulsifying system incorporating a hydrophilic polymer; in vitro and in vivo investigations. Aaps Pharmscitech 2018, 19, 730–740. [Google Scholar] [CrossRef]
- Lee, D.R.; Ho, M.J.; Choi, Y.W.; Kang, M.J. A Polyvinylpyrrolidone-Based Supersaturable Self-Emulsifying Drug Delivery System for Enhanced Dissolution of Cyclosporine A. Polymers 2017, 9, 124. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, C.; Zheng, J.; Chen, Z.; Shi, Q.; Liu, H. Development of a solid supersaturatable self-emulsifying drug delivery system of docetaxel with improved dissolution and bioavailability. Biol. Pharm. Bull. 2011, 34, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Baek, I.-h.; Ha, E.-S.; Yoo, J.-W.; Jung, Y.; Kim, M.-S. Design of a gelatin microparticle-containing self-microemulsifying formulation for enhanced oral bioavailability of dutasteride. Drug Des. Dev. Ther. 2015, 9, 3231. [Google Scholar]
- Kim, M.-S.; Ha, E.-S.; Choo, G.-H.; Baek, I.-H. Preparation and in vivo evaluation of a dutasteride-loaded solid-supersaturatable self-microemulsifying drug delivery system. Int. J. Mol. Sci. 2015, 16, 10821–10833. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Yeom, D.W.; Song, Y.S.; Cho, H.R.; Choi, Y.S.; Kang, M.J.; Choi, Y.W. Improved oral absorption of dutasteride via Soluplus®-based supersaturable self-emulsifying drug delivery system (S-SEDDS). Int. J. Pharm. 2015, 478, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Lv, C.; Sun, X.; Wang, J.; Zhao, Z. Preparation of a supersaturatable self-microemulsion as drug delivery system for ellagic acid and evaluation of its antioxidant activities. J. Drug Deliv. Sci. Technol. 2019, 53, 101209. [Google Scholar] [CrossRef]
- Ogino, M.; Yakushiji, K.; Suzuki, H.; Shiokawa, K.; Kikuchi, H.; Seto, Y.; Sato, H.; Onoue, S. Enhanced pharmacokinetic behavior and hepatoprotective function of ginger extract-loaded supersaturable self-emulsifying drug delivery systems. J. Funct. Foods 2018, 40, 156–163. [Google Scholar] [CrossRef]
- Dash, R.N.; Mohammed, H.; Humaira, T.; Reddy, A.V. Solid supersaturatable self-nanoemulsifying drug delivery systems for improved dissolution, absorption and pharmacodynamic effects of glipizide. J. Drug Deliv. Sci. Technol. 2015, 28, 28–36. [Google Scholar] [CrossRef]
- Zadeha, B.S.M.; Salimi, A.; Aminib, R. Novel Super Saturated Self-Emulsifying System for Oral Delivery of Griseofulvin: Design, Preparation and ex-vivo Intestinal Permeability. J. Rep. Pharm. Sci. 2017, 6, 180–190. [Google Scholar]
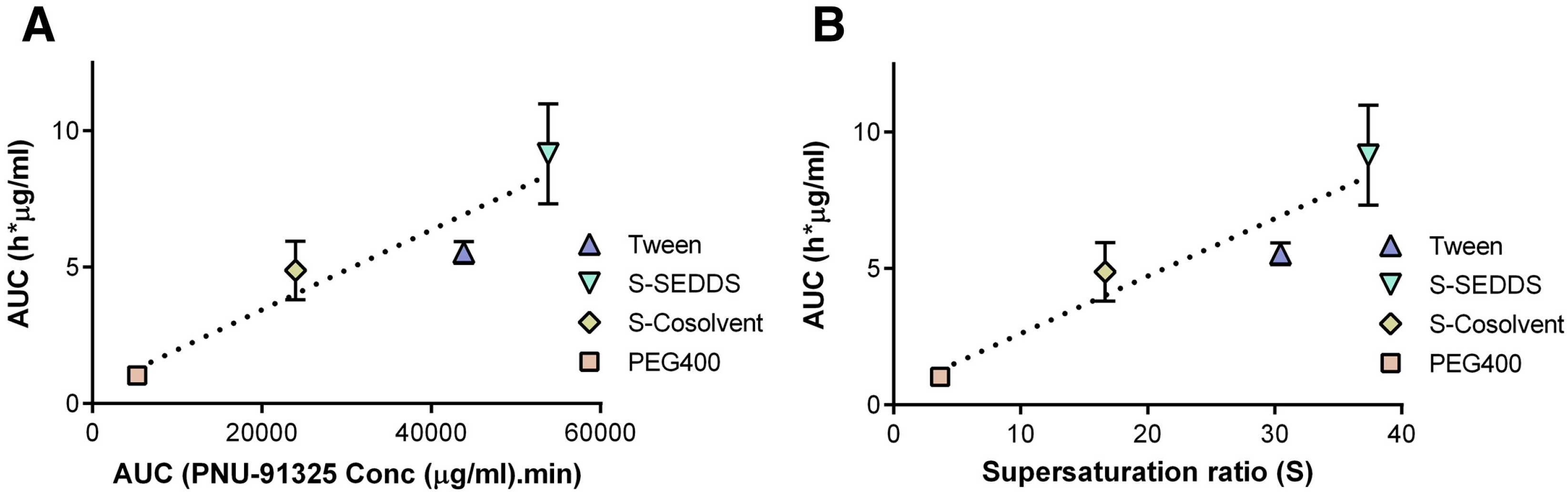
- Gao, P.; Guyton, M.E.; Huang, T.; Bauer, J.M.; Stefanski, K.J.; Lu, Q. Enhanced oral bioavailability of a poorly water soluble drug PNU-91325 by supersaturatable formulations. Drug Dev. Ind. Pharm. 2004, 30, 221–229. [Google Scholar] [CrossRef]
- Lee, J.-H.; Kim, H.H.; Cho, Y.H.; Koo, T.-S.; Lee, G.W. Development and evaluation of raloxifene-hydrochloride-loaded supersaturatable SMEDDS containing an acidifier. Pharmaceutics 2018, 10, 78. [Google Scholar] [CrossRef]
- Singh, G.; Pai, R.S. In vitro and in vivo performance of supersaturable self-nanoemulsifying system of trans-resveratrol. Artif. Cells Nanomed. Biotechnol. 2016, 44, 510–516. [Google Scholar] [CrossRef]
- Wei, Y.; Ye, X.; Shang, X.; Peng, X.; Bao, Q.; Liu, M.; Guo, M.; Li, F. Enhanced oral bioavailability of silybin by a supersaturatable self-emulsifying drug delivery system (S-SEDDS). Colloids Surf. A Physicochem. Eng. Asp. 2012, 396, 22–28. [Google Scholar] [CrossRef]
- Tung, N.-T.; Tran, C.-S.; Nguyen, H.-A.; Nguyen, T.-D.; Chi, S.-C.; Pham, D.-V.; Bui, Q.-D.; Ho, X.-H. Formulation and biopharmaceutical evaluation of supersaturatable self-nanoemulsifying drug delivery systems containing silymarin. Int. J. Pharm. 2019, 555, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.R.; Ho, M.J.; Jung, H.J.; Cho, H.R.; Park, J.S.; Yoon, S.-H.; Choi, Y.S.; Choi, Y.W.; Oh, C.-H.; Kang, M.J. Enhanced dissolution and oral absorption of tacrolimus by supersaturable self-emulsifying drug delivery system. Int. J. Nanomed. 2016, 11, 1109. [Google Scholar]
- Shin, D.J.; Chae, B.R.; Goo, Y.T.; Yoon, H.Y.; Kim, C.H.; Sohn, S.I.; Oh, D.; Lee, A.; Song, S.H.; Choi, Y.W. Improved Dissolution and Oral Bioavailability of Valsartan Using a Solidified Supersaturable Self-Microemulsifying Drug Delivery System Containing Gelucire® 44/14. Pharmaceutics 2019, 11, 58. [Google Scholar] [CrossRef]
- Ouellet, D.; Grossmann, K.F.; Limentani, G.; Nebot, N.; Lan, K.; Knowles, L.; Gordon, M.S.; Sharma, S.; Infante, J.R.; Lorusso, P.M. Effects of particle size, food, and capsule shell composition on the oral bioavailability of dabrafenib, a BRAF inhibitor, in patients with BRAF mutation-positive tumors. J. Pharm. Sci. 2013, 102, 3100–3109. [Google Scholar] [CrossRef]
- Zhang, W.; Li, Y.; Zou, P.; Wu, M.; Zhang, Z.; Zhang, T. The effects of pharmaceutical excipients on gastrointestinal tract metabolic enzymes and transporters—An update. Aaps J. 2016, 18, 830–843. [Google Scholar] [CrossRef]
- Dening, T.J.; Rao, S.; Thomas, N.; Prestidge, C.A. Novel nanostructured solid materials for modulating oral drug delivery from solid-state lipid-based drug delivery systems. Aaps J. 2016, 18, 23–40. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.; Rao, S.; Prestidge, C.A. Transforming lipid-based oral drug delivery systems into solid dosage forms: An overview of solid carriers, physicochemical properties, and biopharmaceutical performance. Pharm. Res. 2013, 30, 2993–3017. [Google Scholar] [CrossRef]
- Reis, P.; Holmberg, K.; Watzke, H.; Leser, M.E.; Miller, R. Lipases at interfaces: A review. Adv. Colloid Interface Sci. 2009, 147, 237–250. [Google Scholar] [CrossRef]
- Joyce, P.; Whitby, C.P.; Prestidge, C.A. Nanostructuring Biomaterials with Specific Activities towards Digestive Enzymes for Controlled Gastrointestinal Absorption of Lipophilic Bioactive Molecules. Adv. Colloid Interface Sci. 2016, 237, 52–75. [Google Scholar] [CrossRef] [PubMed]
- Joyce, P.; Dening, T.J.; Gustafsson, H.; Prestidge, C.A. Modulating the Lipase-Mediated Bioactivity of Particle-Lipid Conjugates Through Changes in Nanostructure and Surface Chemistry. Eur. J. Lipid Sci. Technol. 2017, 119, 1700213. [Google Scholar] [CrossRef]
- Joyce, P.; Whitby, C.P.; Prestidge, C.A. Bioactive Hybrid Particles from Poly(d, l-lactide-co-glycolide) Nanoparticle Stabilized Lipid Droplets. Acs Appl. Mater. Interfaces 2015, 7, 17460–17470. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Li, C.; Huang, Q. Kafirin Nanoparticle-Stabilized Pickering Emulsions as Oral Delivery Vehicles: Physicochemical Stability and in Vitro Digestion Profile. J. Agric. Food Chem. 2015, 63, 10263–10270. [Google Scholar] [CrossRef] [PubMed]
- Tzoumaki, M.V.; Moschakis, T.; Scholten, E.; Biliaderis, C.G. In vitro lipid digestion of chitin nanocrystal stabilized o/w emulsions. Food Funct. 2013, 4, 121–129. [Google Scholar] [CrossRef]
- Li, Y.; Hu, M.; McClements, D.J. Factors affecting lipase digestibility of emulsified lipids using an in vitro digestion model: Proposal for a standardised pH-stat method. Food Chem. 2011, 126, 498–505. [Google Scholar] [CrossRef]
- Lesmes, U.; McClements, D.J. Controlling lipid digestibility: Response of lipid droplets coated by β-lactoglobulin-dextran Maillard conjugates to simulated gastrointestinal conditions. Food Hydrocoll. 2012, 26, 221–230. [Google Scholar] [CrossRef]
- Li, Y.; McClements, D.J. Controlling lipid digestion by encapsulation of protein-stabilized lipid droplets within alginate–chitosan complex coacervates. Food Hydrocoll. 2011, 25, 1025–1033. [Google Scholar] [CrossRef]
- Mun, S.; Decker, E.A.; Park, Y.; Weiss, J.; McClements, D.J. Influence of Interfacial Composition on in Vitro Digestibility of Emulsified Lipids: Potential Mechanism for Chitosan’s Ability to Inhibit Fat Digestion. Food Biophys. 2006, 1, 21–29. [Google Scholar] [CrossRef]
- Reis, P.; Holmberg, K.; Debeche, T.; Folmer, B.; Fauconnot, L.; Watzke, H. Lipase-Catalyzed Reactions at Different Surfaces. Langmuir 2006, 22, 8169–8177. [Google Scholar] [CrossRef]
- Joyce, P.; Kempson, I.; Prestidge, C.A. QCM-D and ToF-SIMS Investigation to Deconvolute the Relationship between Lipid Adsorption and Orientation on Lipase Activity. Langmuir 2015, 31, 10198–10207. [Google Scholar] [CrossRef] [PubMed]
- Joyce, P.; Kempson, I.; Prestidge, C.A. Orientating lipase molecules through surface chemical control for enhanced activity: A QCM-D and ToF-SIMS investigation. Colloids Surf. B Biointerfaces 2016, 142, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Reis, P.; Holmberg, K.; Miller, R.; Leser, M.E.; Raab, T.; Watzke, H.J. Lipase reaction at interfaces as self-limiting processes. Comptes Rendus Chim. 2009, 12, 163–170. [Google Scholar] [CrossRef]
- Reis, P.; Watzke, H.; Leser, M.; Holmberg, K.; Miller, R. Interfacial mechanism of lipolysis as self-regulated process. Biophys. Chem. 2010, 147, 93–103. [Google Scholar] [CrossRef]
- Reis, P.; Holmberg, K.; Miller, R.; Krägel, J.; Grigoriev, D.O.; Leser, M.E.; Watzke, H.J. Competition between Lipases and Monoglycerides at Interfaces. Langmuir 2008, 24, 7400–7407. [Google Scholar] [CrossRef]
- Dening, T.J.; Joyce, P.; Prestidge, C.A. Improving Correlations Between Drug Solubilization and In Vitro Lipolysis by Monitoring the Phase Partitioning of Lipolytic Species for Lipid-Based Formulations. J. Pharm. Sci. 2019, 108, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Gershanik, T.; Benita, S. Self-Dispersing Lipid Formulations for Improving Oral Absorption of Lipophilic Drugs; Elsevier B.V.: Amsterdam, The Netherlands, 2000; Volume 50, pp. 179–188. [Google Scholar]
- Nazzal, S.; Smalyukh, I.I.; Lavrentovich, O.D.; Khan, M.A. Preparation and in vitro characterization of a eutectic based semisolid self-nanoemulsified drug delivery system (SNEDDS) of ubiquinone: Mechanism and progress of emulsion formation. Int. J. Pharm. 2002, 235, 247–265. [Google Scholar] [CrossRef]
- Ujhelyi, Z.; Vecsernyés, M.; Fehér, P.; Kósa, D.; Arany, P.; Nemes, D.; Sinka, D.; Vasvári, G.; Fenyvesi, F.; Váradi, J. Physico-chemical characterization of self-emulsifying drug delivery systems. Drug Discov. Today Technol. 2018, 27, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Pouton, C.W.; Charman, W.N. The potential of oily formulations for drug delivery to the gastro-intestinal tract. Adv. Drug Deliv. Rev. 1997, 25, 1–2. [Google Scholar] [CrossRef]
- Kohli, K.; Chopra, S.; Dhar, D.; Arora, S.; Khar, R.K. Self-emulsifying drug delivery systems: An approach to enhance oral bioavailability. Drug Discov. Today 2010, 15, 958–965. [Google Scholar] [CrossRef]
- Patil, P.; Joshi, P.; Paradkar, A. Effect of formulation variables on preparation and evaluation of gelled self-emulsifying drug delivery system (SEDDS) of ketoprofen. Aaps Pharmscitech 2004, 5, 43–50. [Google Scholar] [CrossRef]
- Yi, T.; Wan, J.; Xu, H.; Yang, X. A new solid self-microemulsifying formulation prepared by spray-drying to improve the oral bioavailability of poorly water soluble drugs. Eur. J. Pharm. Biopharm. 2008, 70, 439–444. [Google Scholar] [CrossRef]
- Patel, M.R.; Hirani, S.N.; Patel, R.B. Microemulsion for nasal delivery of Asenapine maleate in treatment of schizophrenia: Formulation considerations. J. Pharm. Investig. 2018, 48, 301–312. [Google Scholar] [CrossRef]
- Barker, S.A.; Craig, D.Q.M.; Taylor, K.M.G.; Hill, R.M. The study of liposomes by low frequency dielectric spectroscopy. J. Pharm. Pharmacol. Suppl. 1989, 41, 1. [Google Scholar]
- Craig, D.Q.M.; Barker, S.A.; Banning, D.; Booth, S.W. An investigation into the mechanisms of self-emulsification using particle size analysis and low frequency dielectric spectroscopy. Int. J. Pharm. 1995, 114, 103–110. [Google Scholar] [CrossRef]
- Breitkreitz, M.C.; Sabin, G.P.; Polla, G.; Poppi, R.J. Characterization of semi-solid Self-Emulsifying Drug Delivery Systems (SEDDS) of atorvastatin calcium by Raman image spectroscopy and chemometrics. J. Pharm. Biomed. Anal. 2013, 73, 3–12. [Google Scholar] [CrossRef]
- Abdalla, A.; Mäder, K. Preparation and characterization of a self-emulsifying pellet formulation. Eur. J. Pharm. Biopharm. 2007, 66, 220–226. [Google Scholar] [CrossRef]
- Sonawale, P.; Patil, A.; Kamble, A.; Bhutkar, M. Solubility Enhancement of Lipophilic Drugs-Solid Self Micro-Emulsifying Drug Delivery System. Asian J. Pharm. Technol. 2016, 6, 155. [Google Scholar] [CrossRef]
- Bolzinger-Thevenin, M.A.; Grossiord, J.L.; Poelman, M.C. Characterization of a Sucrose Ester Microemulsion by Freeze Fracture Electron Micrograph and Small Angle Neutron Scattering Experiments. Langmuir 1999, 15, 2307–2315. [Google Scholar] [CrossRef]
- Angell, C.A.; Kadiyala, R.K.; MacFarlane, D.R. Glass-forming microemulsions. J. Phys. Chem. 1984, 88, 4593–4596. [Google Scholar] [CrossRef]
- Goddeeris, C.; Goderis, B.; Van den Mooter, G. Lyotropic, liquid crystalline nanostructures of aqueous dilutions of SMEDDS revealed by small-angle X-ray scattering: Impact on solubility and drug release. Eur. J. Pharm. Sci. 2010, 40, 110–117. [Google Scholar] [CrossRef]
- Williams, H.D.; Sassene, P.; Kleberg, K.; Bakala-N’Goma, J.-C.; Calderone, M.; Jannin, V.; Igonin, A.; Partheil, A.; Marchaud, D.; Jule, E.; et al. Toward the Establishment of Standardized In Vitro Tests for Lipid-Based Formulations, Part 1: Method Parameterization and Comparison of In Vitro Digestion Profiles Across a Range of Representative Formulations. J. Pharm. Sci. 2012, 101, 3360–3380. [Google Scholar] [CrossRef]
- Bakala-N’Goma, J.-C.; Williams, H.D.; Sassene, P.J.; Kleberg, K.; Calderone, M.; Jannin, V.; Igonin, A.; Partheil, A.; Marchaud, D.; Jule, E.; et al. Toward the establishment of standardized in vitro tests for lipid-based formulations. 5. lipolysis of representative formulations by gastric lipase. Pharm. Res. 2015, 32, 1279–1287. [Google Scholar] [CrossRef]
- Williams, H.D.; Sassene, P.; Kleberg, K.; Calderone, M.; Igonin, A.; Jule, E.; Vertommen, J.; Blundell, R.; Benameur, H.; Müllertz, A.; et al. Toward the Establishment of Standardized In Vitro Tests for Lipid-Based Formulations, Part 4: Proposing a New Lipid Formulation Performance Classification System. J. Pharm. Sci. 2014, 103, 2441–2455. [Google Scholar] [CrossRef]
- Sassene, P.; Kleberg, K.; Williams, H.D.; Bakala-N’Goma, J.-C.; Carrière, F.; Calderone, M.; Jannin, V.; Igonin, A.; Partheil, A.; Marchaud, D.; et al. Toward the Establishment of Standardized In Vitro Tests for Lipid-Based Formulations, Part 6: Effects of Varying Pancreatin and Calcium Levels. Aaps J. 2014, 16, 1344–1357. [Google Scholar] [CrossRef]
- Williams, H.D.; Sassene, P.; Kleberg, K.; Calderone, M.; Igonin, A.; Jule, E.; Vertommen, J.; Blundell, R.; Benameur, H.; Müllertz, A.; et al. Toward the Establishment of Standardized In Vitro Tests for Lipid-Based Formulations, Part 3: Understanding Supersaturation Versus Precipitation Potential During the In Vitro Digestion of Type I, II, IIIA, IIIB and IV Lipid-Based Formulations. Pharm. Res. 2013, 30, 3059–3076. [Google Scholar] [CrossRef]
- Williams, H.D.; Anby, M.U.; Sassene, P.; Kleberg, K.; Bakala-N’Goma, J.-C.; Calderone, M.; Jannin, V.; Igonin, A.; Partheil, A.; Marchaud, D.; et al. Toward the Establishment of Standardized in Vitro Tests for Lipid-Based Formulations. 2. The Effect of Bile Salt Concentration and Drug Loading on the Performance of Type I, II, IIIA, IIIB, and IV Formulations during in Vitro Digestion. Mol. Pharm. 2012, 9, 3286–3300. [Google Scholar] [CrossRef]
- Fatouros, D.G.; Nielsen, F.S.; Douroumis, D.; Hadjileontiadis, L.J.; Mullertz, A. In vitro–in vivo correlations of self-emulsifying drug delivery systems combining the dynamic lipolysis model and neuro-fuzzy networks. Eur. J. Pharm. Biopharm. 2008, 69, 887–898. [Google Scholar] [CrossRef]
- Birru, W.A.; Warren, D.B.; Headey, S.J.; Benameur, H.; Porter, C.J.H.; Pouton, C.W.; Chalmers, D.K. Computational Models of the Gastrointestinal Environment. 1. The Effect of Digestion on the Phase Behavior of Intestinal Fluids. Mol. Pharm. 2017, 14, 566–579. [Google Scholar] [CrossRef]
- Birru, W.A.; Warren, D.B.; Han, S.; Benameur, H.; Porter, C.J.H.; Pouton, C.W.; Chalmers, D.K. Computational Models of the Gastrointestinal Environment. 2. Phase Behavior and Drug Solubilization Capacity of a Type I Lipid-Based Drug Formulation after Digestion. Mol. Pharm. 2017, 14, 580–592. [Google Scholar] [CrossRef]
- Suys, E.J.A.; Warren, D.B.; Porter, C.J.H.; Benameur, H.; Pouton, C.W.; Chalmers, D.K. Computational Models of the Intestinal Environment. 3. The Impact of Cholesterol Content and pH on Mixed Micelle Colloids. Mol. Pharm. 2017, 14, 3684–3697. [Google Scholar] [CrossRef]
- Bałdyga, J.; Orciuch, W. Some hydrodynamic aspects of precipitation. Powder Technol. 2001, 121, 9–19. [Google Scholar] [CrossRef]
- Manth, T.; Mignon, D.; Offermann, H. Experimental investigation of precipitation reactions under homogeneous mixing conditions. Chem. Eng. Sci. 1996, 51, 2571–2576. [Google Scholar] [CrossRef]
- McAllister, M. Dynamic Dissolution: A Step Closer to Predictive Dissolution Testing? Mol. Pharm. 2010, 7, 1374–1387. [Google Scholar] [CrossRef]
- Jantratid, E.; Janssen, N.; Reppas, C.; Dressman, J.B. Dissolution Media Simulating Conditions in the Proximal Human Gastrointestinal Tract: An Update. Pharm. Res. 2008, 25, 1663–1676. [Google Scholar] [CrossRef]
- Dressman, J.B.; Amidon, G.L.; Reppas, C.; Shah, V.P. Dissolution Testing as a Prognostic Tool for Oral Drug Absorption: Immediate Release Dosage Forms. Pharm. Res. 1998, 15, 11–22. [Google Scholar] [CrossRef]
- Dahan, A.; Hoffman, A. The effect of different lipid based formulations on the oral absorption of lipophilic drugs: The ability of in vitro lipolysis and consecutive ex vivo intestinal permeability data to predict in vivo bioavailability in rats. Eur. J. Pharm. Biopharm. 2007, 67, 96–105. [Google Scholar] [CrossRef]
- Cuiné, J.F.; McEvoy, C.L.; Charman, W.N.; Pouton, C.W.; Edwards, G.A.; Benameur, H.; Porter, C.J.H. Evaluation of the Impact of Surfactant Digestion on the Bioavailability of Danazol after Oral Administration of Lipidic Self-Emulsifying Formulations to Dogs. J. Pharm. Sci. 2008, 97, 995–1012. [Google Scholar] [CrossRef]
- Pafumi, Y.; Lairon, D.; Paulette Lechene de la, P.; Juhel, C.; Storch, J.; Hamosh, M.; Armand, M. Mechanisms of Inhibition of Triacylglycerol Hydrolysis by Human Gastric Lipase. J. Biol. Chem. 2002, 277, 28070–28079. [Google Scholar] [CrossRef]
- Carrière, F. Impact of gastrointestinal lipolysis on oral lipid-based formulations and bioavailability of lipophilic drugs. Biochimie 2016, 125, 297–305. [Google Scholar] [CrossRef]
- Christophersen, P.C.; Christiansen, M.L.; Holm, R.; Kristensen, J.; Jacobsen, J.; Abrahamsson, B.; Müllertz, A. Fed and fasted state gastro-intestinal in vitro lipolysis: In vitro in vivo relations of a conventional tablet, a SNEDDS and a solidified SNEDDS. Eur. J. Pharm. Sci. 2014, 57, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, S.; Chevrier, S.; Ritter, N.; Mahler, B.; Demarne, F.; Carrière, F.; Jannin, V. In vitro gastrointestinal lipolysis of four formulations of piroxicam and cinnarizine with the self emulsifying excipients Labrasol® and Gelucire® 44/14. Pharm. Res. 2009, 26, 1901–1910. [Google Scholar] [CrossRef] [PubMed]
- Klitgaard, M.; Sassene, P.J.; Selen, A.; Müllertz, A.; Berthelsen, R. Studying furosemide solubilization using an in vitro model simulating gastrointestinal digestion and drug solubilization in neonates and young infants. Eur. J. Pharm. Sci. 2017, 109, 191–199. [Google Scholar] [CrossRef]
- Mercuri, A.; Passalacqua, A.; Wickham, M.S.J.; Faulks, R.M.; Craig, D.Q.M.; Barker, S.A. The Effect of Composition and Gastric Conditions on the Self-Emulsification Process of Ibuprofen-Loaded Self-Emulsifying Drug Delivery Systems: A Microscopic and Dynamic Gastric Model Study. Pharm. Res. 2011, 28, 1540–1551. [Google Scholar] [CrossRef]
- Thuenemann, E.C.; Giuseppina, G.M.; Rich, G.T.; Faulks, R.M. Dynamic Gastric Model (DGM). In The Impact of Food Bioactives on Health; Verhoeckx, K., Cotter, P., López-Expósito, I., Kleiveland, C., Lea, T., Mackie, A., Requena, T., Swiatecka, D., Wichers, H., Eds.; Springer: Cham, Switzerland, 2015; pp. 47–59. [Google Scholar] [CrossRef]
- Mosgaard, M.D.; Sassene, P.J.; Mu, H.; Rades, T.; Müllertz, A. High-Throughput Lipolysis in 96-Well Plates for Rapid Screening of Lipid-Based Drug Delivery Systems. J. Pharm. Sci. 2017, 106, 1183–1186. [Google Scholar] [CrossRef]
- Mosgaard, M.D.; Sassene, P.; Mu, H.; Rades, T.; Müllertz, A. Development of a high-throughput in vitro intestinal lipolysis model for rapid screening of lipid-based drug delivery systems. Eur. J. Pharm. Biopharm. 2015, 94, 493–500. [Google Scholar] [CrossRef]
- Keemink, J.; Martensson, E.; Bergstrom, C.A.S.; Medicinska och farmaceutiska, v.; Uppsala, u.; Institutionen för, f.; Farmaceutiska, f. Lipolysis-Permeation Setup for Simultaneous Study of Digestion and Absorption in Vitro. Mol. Pharm. 2019, 16, 921–930. [Google Scholar] [CrossRef]
- Alskär, L.C.; Parrow, A.; Keemink, J.; Johansson, P.; Abrahamsson, B.; Bergström, C.A.S.; Medicinska och farmaceutiska, V.; Uppsala, U.; Institutionen för, F.; Farmaceutiska, F. Effect of lipids on absorption of carvedilol in dogs: Is coadministration of lipids as efficient as a lipid-based formulation? J. Control. Release 2019, 304, 90–100. [Google Scholar] [CrossRef]
- Crum, M.F.; Trevaskis, N.L.; Williams, H.D.; Pouton, C.W.; Porter, C.J.H. A new in vitro lipid digestion—in vivo absorption model to evaluate the mechanisms of drug absorption from lipid-based formulations. Pharm. Res. 2016, 33, 970–982. [Google Scholar] [CrossRef]
- Thomas, N.; Richter, K.; Pedersen, T.B.; Holm, R.; Müllertz, A.; Rades, T. In Vitro Lipolysis Data Does Not Adequately Predict the In Vivo Performance of Lipid-Based Drug Delivery Systems Containing Fenofibrate. Aaps J. 2014, 16, 539–549. [Google Scholar] [CrossRef]
- Monton, C.; Kulvanich, P. Characterization of crosslinked hard gelatin capsules for a structural assembly of elementary osmotic pump delivery system. J. Pharm. Investig. 2019, 49, 655–665. [Google Scholar] [CrossRef]
- Thabet, Y.; Walsh, J.; Breitkreutz, J. Flexible and precise dosing of enalapril maleate for all paediatric age groups utilizing orodispersible minitablets. Int. J. Pharm. 2018, 541, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Mistry, P.; Batchelor, H. Smart Paediatric Drug Development–UK. Evidence of acceptability of oral paediatric medicines: A review. J. Pharm. Pharmacol. 2017, 69, 361–376. [Google Scholar] [CrossRef]
- Liu, F.; Ranmal, S.; Batchelor, H.K.; Orlu-Gul, M.; Ernest, T.B.; Thomas, I.W.; Flanagan, T.; Tuleu, C. Patient-Centered Pharmaceutical Design to Improve Acceptability of Medicines: Similarities and Differences in Paediatric and Geriatric Populations. Drugs 2014, 74, 1871–1889. [Google Scholar] [CrossRef] [PubMed]
- Cole, E.T.; Cadé, D.; Benameur, H. Challenges and opportunities in the encapsulation of liquid and semi-solid formulations into capsules for oral administration. Adv. Drug Deliv. Rev. 2008, 60, 747–756. [Google Scholar] [CrossRef]
- Hong, S.H.; Choi, Y. Mesoporous silica-based nanoplatforms for the delivery of photodynamic therapy agents. J. Pharm. Investig. 2018, 48, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Alabi, C.O.; Singh, I.; Odeku, O.A. Evaluation of natural and pregelatinized forms of three tropical starches as excipients in tramadol tablet formulation. J. Pharm. Investig. 2018, 48, 333–340. [Google Scholar] [CrossRef]
- Okunlola, A.; Ghomorai, T. Development of ibuprofen microspheres using acetylated plantain starches as polymer for sustained release. J. Pharm. Investig. 2018, 48, 551–564. [Google Scholar] [CrossRef]
- Kim, D.W.; Kang, J.H.; Oh, D.H.; Yong, C.S.; Choi, H.-G. Development of novel flurbiprofen-loaded solid self-microemulsifying drug delivery system using gelatin as solid carrier. J. Microencapsul. 2012, 29, 323–330. [Google Scholar] [CrossRef]
- Kalepu, S.; Manthina, M.; Padavala, V. Oral lipid-based drug delivery systems—An overview. Acta Pharm. Sin. B 2013, 3, 361–372. [Google Scholar] [CrossRef]
- Joyce, P.; Tan, A.; Whitby, C.P.; Prestidge, C.A. The Role of Porous Nanostructure in Controlling Lipase-Mediated Digestion of Lipid Loaded into Silica Particles. Langmuir 2014, 30, 2779–2788. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.; Simovic, S.; Davey, A.K.; Rades, T.; Boyd, B.J.; Prestidge, C.A. Silica Nanoparticles To Control the Lipase-Mediated Digestion of Lipid-Based Oral Delivery Systems. Mol. Pharm. 2010, 7, 522–532. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.D.; Speybroeck, M.V.; Augustijns, P.; Porter, C.J.H. Lipid-Based Formulations Solidified Via Adsorption onto the Mesoporous Carrier Neusilin® US2: Effect of Drug Type and Formulation Composition on In Vitro Pharmaceutical Performance. J. Pharm. Sci. 2014, 103, 1734–1746. [Google Scholar] [CrossRef] [PubMed]
- Unger, K.K.; Kumar, D.; Grün, M.; Büchel, G.; Lüdtke, S.; Adam, T.; Schumacher, K.; Renker, S. Synthesis of Spherical Porous Silicas in the Micron and Submicron Size Range: Challenges and Opportunities for Miniaturized High-Resolution Chromatographic and Electrokinetic Separations; Elsevier B.V.: Amsterdam, The Netherlands, 2000; Volume 892, pp. 47–55. [Google Scholar]
- Sauzet, C.; Claeys-Bruno, M.; Nicolas, M.; Kister, J.; Prinderre, P. An innovative floating gastro retentive dosage system: Formulation and in vitro evaluation. Int. J. Pharm. 2009, 378, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.J.; Jung, S.Y.; Song, W.H.; Park, J.S.; Choi, S.-U.; Oh, K.T.; Choi, H.-K.; Choi, Y.W.; Lee, J.; Lee, B.-J.; et al. Immediate release of ibuprofen from Fujicalin®-based fast-dissolving self-emulsifying tablets. Drug Dev. Ind. Pharm. 2011, 37, 1298–1305. [Google Scholar] [CrossRef]
- Quan, G.; Wu, Q.; Zhang, X.; Zhan, Z.; Zhou, C.; Chen, B.; Zhang, Z.; Li, G.; Pan, X.; Wu, C. Enhancing in vitro dissolution and in vivo bioavailability of fenofibrate by solid self-emulsifying matrix combined with SBA-15 mesoporous silica. Colloids Surf. B Biointerfaces 2016, 141, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Almeida, S.R.D.; Tippavajhala, V.K. A Rundown Through Various Methods Used in the Formulation of Solid Self-Emulsifying Drug Delivery Systems (S-SEDDS). Aaps Pharmscitech 2019, 20, 323. [Google Scholar] [CrossRef]
- Jannin, V.; Musakhanian, J.; Marchaud, D. Approaches for the development of solid and semi-solid lipid-based formulations. Adv. Drug Deliv. Rev. 2008, 60, 734–746. [Google Scholar] [CrossRef]
- Vadlamudi, H.C.; Yalavarthi, P.R.; Nagaswaram, T.; Rasheed, A.; Peesa, J.P. In-vitro and pharmacodynamic characterization of solidified self microemulsified system of quetiapine fumarate. J. Pharm. Investig. 2019, 49, 161–172. [Google Scholar] [CrossRef]
- Maniyar, M.G.; Kokare, C.R. Formulation and evaluation of spray dried liposomes of lopinavir for topical application. J. Pharm. Investig. 2019, 49, 259–270. [Google Scholar] [CrossRef]
- Pokharkar, V.; Patil-Gadhe, A.; Kaur, G. Physicochemical and pharmacokinetic evaluation of rosuvastatin loaded nanostructured lipid carriers: Influence of long- and medium-chain fatty acid mixture. J. Pharm. Investig. 2018, 48, 465–476. [Google Scholar] [CrossRef]
- Pasquali, I.; Bettini, R.; Giordano, F. Supercritical fluid technologies: An innovative approach for manipulating the solid-state of pharmaceuticals. Adv. Drug Deliv. Rev. 2008, 60, 399–410. [Google Scholar] [CrossRef]
- Alinaghi, A.; Tan, A.; Rao, S.; Prestidge, C.A. Impact of solidification on the performance of lipid-based colloidal carriers: Oil-based versus self-emulsifying systems. Curr. Drug Deliv. 2015, 12, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Van Speybroeck, M.; Williams, H.D.; Nguyen, T.-H.; Anby, M.U.; Porter, C.J.H.; Augustijns, P. Incomplete Desorption of Liquid Excipients Reduces the in Vitro and in Vivo Performance of Self-Emulsifying Drug Delivery Systems Solidified by Adsorption onto an Inorganic Mesoporous Carrier. Mol. Pharm. 2012, 9, 2750–2760. [Google Scholar] [CrossRef]
- Madagul, J.K.; Parakh, D.R.; Kumar, R.S.; Abhang, R.R. Formulation and evaluation of solid self-microemulsifying drug delivery system of chlorthalidone by spray drying technology. Dry. Technol. 2017, 35, 1433–1449. [Google Scholar] [CrossRef]
- Singh, S.; Singh, S.K.; Vuddanda, P.R.; Srivastava, A.K. A comparison between use of spray and freeze drying techniques for preparation of solid self-microemulsifying formulation of valsartan and in vitro and in vivo evaluation. Biomed Res. Int. 2013, 2013, 909045. [Google Scholar] [CrossRef]
- Yasmin, R.; Tan, A.; Bremmell, K.E.; Prestidge, C.A. Lyophilized Silica Lipid Hybrid (SLH) Carriers for Poorly Water-Soluble Drugs: Physicochemical and In Vitro Pharmaceutical Investigations. J. Pharm. Sci. 2014, 103, 2950–2959. [Google Scholar] [CrossRef]
- Kuncahyo, I.; Choiri, S.; Fudholi, A. Solidification of meloxicam self-nano emulsifying drug delivery system formulation incorporated into soluble and insoluble carriers using freeze drying method. Iop Conf. Ser. Mater. Sci. Eng. 2019, 578, 12051. [Google Scholar] [CrossRef]
- Bertoni, S.; Dolci, L.S.; Albertini, B.; Passerini, N. Spray congealing: A versatile technology for advanced drug-delivery systems. Ther. Deliv. 2018, 9, 833–845. [Google Scholar] [CrossRef]
- Albertini, B.; Sabatino, M.D.; Melegari, C.; Passerini, N. Formulation of spray congealed microparticles with self-emulsifying ability for enhanced glibenclamide dissolution performance. J. Microencapsul. 2015, 32, 181–192. [Google Scholar] [CrossRef]
- Sun, C.; Gui, Y.; Hu, R.; Chen, J.; Wang, B.; Guo, Y.; Lu, W.; Nie, X.; Shen, Q.; Gao, S.; et al. Preparation and Pharmacokinetics Evaluation of Solid Self-Microemulsifying Drug Delivery System (S-SMEDDS) of Osthole. Aaps Pharmscitech 2018, 19, 2301–2310. [Google Scholar] [CrossRef] [PubMed]
- Park, M.J.; Ren, S.; Lee, B.J. In vitro and in vivo comparative study of itraconazole bioavailability when formulated in highly soluble self-emulsifying system and in solid dispersion. Biopharm. Drug Dispos. 2007, 28, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Katteboina, S.; Chandrasekhar, P.V.S.R.; Balaji, S. Approaches for the development of solid self-emulsifying drug delivery systems and dosage forms. Asian J. Pharm. Sci. 2009, 4, 240–253. [Google Scholar]
- Meng, X.; Zu, Y.; Zhao, X.; Li, Q.; Jiang, S.; Sang, M. Characterization and pharmacokinetics of coenzyme Q10 nanoparticles prepared by a rapid expansion of supercritical solution process. Die Pharm. 2012, 67, 161. [Google Scholar]
- Ha, E.-S.; Lee, S.-K.; Choi, D.H.; Jeong, S.H.; Hwang, S.-J.; Kim, M.-S. Application of diethylene glycol monoethyl ether in solubilization of poorly water-soluble drugs. J. Pharm. Investig. 2019. [Google Scholar] [CrossRef]
- Soottitantawat, A.; Yoshii, H.; Furuta, T.; Ohkawara, M.; Linko, P. Microencapsulation by Spray Drying: Influence of Emulsion Size on the Retention of Volatile Compounds. J. Food Sci. 2003, 68, 2256–2262. [Google Scholar] [CrossRef]
- Jang, D.-J.; Jeong, E.J.; Lee, H.-M.; Kim, B.-C.; Lim, S.-J.; Kim, C.-K. Improvement of bioavailability and photostability of amlodipine using redispersible dry emulsion. Eur. J. Pharm. Sci. 2006, 28, 405–411. [Google Scholar] [CrossRef]
- Soottitantawat, A.; Takayama, K.; Okamura, K.; Muranaka, D.; Yoshii, H.; Furuta, T.; Ohkawara, M.; Linko, P. Microencapsulation of l-menthol by spray drying and its release characteristics. Innov. Food Sci. Emerg. Technol. 2005, 6, 163–170. [Google Scholar] [CrossRef]
- Liang, R.; Li, X.; Shi, Y.; Wang, A.; Sun, K.; Liu, W.; Li, Y. Effect of water on exenatide acylation in poly (lactide-co-glycolide) microspheres. Int. J. Pharm. 2013, 454, 344–353. [Google Scholar] [CrossRef]
- Matsaridou, I.; Barmpalexis, P.; Salis, A.; Nikolakakis, I. The Influence of Surfactant HLB and Oil/Surfactant Ratio on the Formation and Properties of Self-emulsifying Pellets and Microemulsion Reconstitution. Aaps Pharmscitech 2012, 13, 1319–1330. [Google Scholar] [CrossRef]
- Nikolakakis, I.; Panagopoulou, A.; Salis, A.; Malamataris, S. Relationships Between the Properties of Self-Emulsifying Pellets and of the Emulsions Used as Massing Liquids for Their Preparation. Aaps Pharmscitech 2015, 16, 129–139. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Agarwal, V.; Siddiqui, A.; Ali, H.; Nazzal, S. Dissolution and powder flow characterization of solid self-emulsified drug delivery system (SEDDS). Int. J. Pharm. 2009, 366, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Cavinato, M.; Franceschinis, E.; Cavallari, S.; Realdon, N.; Santomaso, A. Relationship between particle shape and some process variables in high shear wet granulation using binders of different viscosity. Chem. Eng. J. 2010, 164, 292–298. [Google Scholar] [CrossRef]
- Patel, P.; Pailla, S.R.; Rangaraj, N.; Cheruvu, H.S.; Dodoala, S.; Sampathi, S. Quality by Design Approach for Developing Lipid-Based Nanoformulations of Gliclazide to Improve Oral Bioavailability and Anti-Diabetic Activity. Aaps Pharmscitech 2019, 20, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Kallakunta, V.R.; Bandari, S.; Jukanti, R.; Veerareddy, P.R. Oral self emulsifying powder of lercanidipine hydrochloride: Formulation and evaluation. Powder Technol. 2012, 221, 375–382. [Google Scholar] [CrossRef]
- Agarwal, V.; Alayoubi, A.; Siddiqui, A.; Nazzal, S. Powdered self-emulsified lipid formulations of meloxicam as solid dosage forms for oral administration. Drug Dev. Ind. Pharm. 2013, 39, 1681–1689. [Google Scholar] [CrossRef]
- Comoglu, T.; Dilek Ozyilmaz, E. Orally disintegrating tablets and orally disintegrating mini tablets–novel dosage forms for pediatric use. Pharm. Dev. Technol. 2019, 24, 902–914. [Google Scholar] [CrossRef]
- Nazzal, S.; Khan, M.A. Controlled release of a self-emulsifying formulation from a tablet dosage form: Stability assessment and optimization of some processing parameters. Int. J. Pharm. 2006, 315, 110–121. [Google Scholar] [CrossRef]
- Ayenew, Z.; Paudel, A.; Rombaut, P.; Van den Mooter, G. Effect of compression on non-isothermal crystallization behaviour of amorphous indomethacin. Pharm. Res. 2012, 29, 2489–2498. [Google Scholar] [CrossRef]
- Mah, P.T.; Novakovic, D.; Saarinen, J.; Van Landeghem, S.; Peltonen, L.; Laaksonen, T.; Isomäki, A.; Strachan, C.J. Elucidation of compression-induced surface crystallization in amorphous tablets using sum frequency generation (SFG) microscopy. Pharm. Res. 2017, 34, 957–970. [Google Scholar] [CrossRef]
- Kim, J.Y.; Ku, Y.S. Enhanced absorption of indomethacin after oral or rectal administration of a self-emulsifying system containing indomethacin to rats. Int. J. Pharm. 2000, 194, 81–89. [Google Scholar] [CrossRef]
- Chae, G.S.; Lee, J.S.; Kim, S.H.; Seo, K.S.; Kim, M.S.; Lee, H.B.; Khang, G. Enhancement of the stability of BCNU using self-emulsifying drug delivery systems (SEDDS) and in vitro antitumor activity of self-emulsified BCNU-loaded PLGA wafer. Int. J. Pharm. 2005, 301, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-H.; Park, C.; Oh, G.; Park, J.-B.; Lee, B.-J. New blends of hydroxypropylmethylcellulose and Gelucire 44/14: Physical property and controlled release of drugs with different solubility. J. Pharm. Investig. 2018, 48, 313–321. [Google Scholar] [CrossRef]
- Dawaba, H.M.; Dawaba, A.M. Development and evaluation of extended release ciprofloxacin HCl ocular inserts employing natural and synthetic film forming agents. J. Pharm. Investig. 2019, 49, 245–257. [Google Scholar] [CrossRef]
- Luu, T.D.; Lee, B.-J.; Tran, P.H.L.; Tran, T.T.D. Modified sprouted rice for modulation of curcumin crystallinity and dissolution enhancement by solid dispersion. J. Pharm. Investig. 2019, 49, 127–134. [Google Scholar] [CrossRef]
- Setthacheewakul, S.; Kedjinda, W.; Maneenuan, D.; Wiwattanapatapee, R. Controlled Release of Oral Tetrahydrocurcumin from a Novel Self-Emulsifying Floating Drug Delivery System (SEFDDS). Aaps Pharmscitech 2011, 12, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhang, S.; Cui, S.; Chen, F.; Jia, L.; Wang, S.; Gai, X.; Li, P.; Yang, F.; Pan, W.; et al. Preparation and evaluation of Vinpocetine self-emulsifying pH gradient release pellets. Drug Deliv. 2017, 24, 1598–1604. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, R.; Wu, J.; Shen, Q. Characterization and evaluation of self-microemulsifying sustained-release pellet formulation of puerarin for oral delivery. Int. J. Pharm. 2012, 427, 337–344. [Google Scholar] [CrossRef]
- Tran, P.H.-L.; Tran, T.T.-D.; Piao, Z.Z.; Van Vo, T.; Park, J.B.; Lim, J.; Oh, K.T.; Rhee, Y.-S.; Lee, B.-J. Physical properties and in vivo bioavailability in human volunteers of isradipine using controlled release matrix tablet containing self-emulsifying solid dispersion. Int. J. Pharm. 2013, 450, 79–86. [Google Scholar] [CrossRef]
- Tao, C.; Chen, J.; Huang, A.; Zhang, J.; Lin, B.; Liu, Z.; Zhang, M.; Chen, X.; Zeng, L.; Zhang, L.; et al. Development of solidified self-microemulsifying delivery systems with enhanced stability of sirolimus and extended release. Int. J. Pharm. 2016, 513, 255–261. [Google Scholar] [CrossRef]
- Serratoni, M.; Newton, M.; Booth, S.; Clarke, A. Controlled drug release from pellets containing water-insoluble drugs dissolved in a self-emulsifying system. Eur. J. Pharm. Biopharm. 2007, 65, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.P.; Gan, Y.; Zhang, X.X. Novel gastroretentive sustained-release tablet of tacrolimus based on self-microemulsifying mixture: In vitro evaluation and in vivo bioavailability test. Acta Pharmacol. Sin. 2011, 32, 1294–1302. [Google Scholar] [CrossRef] [PubMed]
- Tung, N.-T.; Nguyen, C.-H.; Nguyen, V.-D.; Nguyen, T.-H.-T.; Nguyen, V.-L.; Tran, C.-S.; Pham, T.-M.-H. Formulation and in vivo imaging evaluation of colonic targeting tablets prepared by a simple dry powder coating technique. J. Pharm. Investig. 2019. [Google Scholar] [CrossRef]
- Huang, Y.; Tian, R.; Hu, W.; Jia, Y.; Zhang, J.; Jiang, H.; Zhang, L. A novel plug-controlled colon-specific pulsatile capsule with tablet of curcumin-loaded SMEDDS. Carbohydr. Polym. 2013, 92, 2218–2223. [Google Scholar] [CrossRef]
- Nikolakakis, I.; Malamataris, S. Self-Emulsifying Pellets: Relations Between Kinetic Parameters of Drug Release and Emulsion Reconstitution—Influence of Formulation Variables. J. Pharm. Sci. 2014, 103, 1453–1465. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| API 1 | BCS Class 2 | Product Name/Company (Strength, mg) | Dosage Form |
|---|---|---|---|
| Cyclosporin | IV | Sandimmune®/Novartis | Soft gelatin capsule |
| - | - | Neoral®/Novartis | Soft gelatin capsule |
| - | - | Gengraf®/AbbVie | Hard gelatin capsule |
| Ritonavir | II | Norvir®/AbbVie | Soft gelatin capsule |
| Saquinavir | IV | Fortovase®/Roche | Soft gelatin capsule |
| Amprenavir | II | Agenerase®/GlaxoSmithKline | Soft gelatin capsule |
| Valproic acid | II | Depakene®/AbbVie | Soft gelatin capsule |
| Calcitriol | II | Rocaltrol®/Roche | Soft gelatin capsule |
| Bexarotene | II | Targretin®/Ligand | Soft gelatin capsule |
| Tretinoin | II | Vesanoid®/Roche | Soft gelatin capsule |
| Isotretinoin | II | Accutane®/Roche | Soft gelatin capsule |
| Tipranavir | II | Aptivus®/Boehringer Ingelheim | Soft gelatin capsule |
| Drug (BCS Class) | Pre-Concentrate | PI | Dosage form (Solidification Method) | Result/Outcome | Solid State of Precipitate 2 | Ref. | |
|---|---|---|---|---|---|---|---|
| Formulation (Drug Conc.) | Substance (Conc.) | PI Addition Method (Appearance) | |||||
| AMG 517 (ND, BCS II or IV) | Capmul MCM, Tween 80, PEG 400 (12.5 mg/450 mg) | HPMC-E5 (5%, w/w) | Suspending in pre-concentrate by vortexing (Suspension) | Liquid filled in hard gelatin capsule | -PI effect: HPMC > PVP -Hydrophobicity dependent PI effect. -In vivo in Cynomolgus monkeys at a dose of 12.5 mg: −30% higher Cmax and AUC, and short Tmax as compared to an aqueous suspension. | HPMC: amorphous PVP or without PI: crystalline | [67] |
| Carbamazepine (BCS II) | Miglyol 812 N, Tween 80, Cremphor EL-35, PEG 400 (25 mg/830 mg) | PVP-K90 (2%, w/w) | Dissolving in pre-concentrate by heating and stirring (Clear solution) | Liquid filled in soft capsule | -PI effect: PVP > HPMC -In vivo in Beagle dog at a dose of 200 mg, 6.7 times higher Cmax, 5.9 times higher AUC as compared to commercial tablet. | ND 1 | [106] |
| Celecoxib (BCS II) | PEG 400, EtOH, Tween 80, Oleic acid, Tromethamine, Water (200 mg/g) | HPMC-E5 (3.8%, w/w) + PVP-12PF (4.7%, w/w) | Suspending in pre-concentrate by vortexing (Suspension) | Liquid filled in hard gelatin capsule | -Highly supersaturated state in the aqueous phase, resulting in high drug concentrations in octanol for biphasic in vitro test dissolution method. -Good in vitro–in vivo correlations (IVIVC) with Human PK as compared to solution and marketed capsule formulation. | ND | [107] |
| Celecoxib (BCS II) | Capryol 90, Tween 20, Transcutol HP (180 mg/mL) | Soluplus (4%, w/v) | Adding in pre-concentrate (ND) | Solid su-SEDDS (Adsorption method, Sylysia 350 fcp) | -Physico-chemical properties (surface area, hydrophobicity) of solid carrier dependent drug dissolution. -In vivo in SD rats, 2.34-fold increase in Cmax and 4.82 fold increase in AUC as compared to drug powder. | ND | [108] |
| Celecoxib (BCS II) | Capryol 90, Tween 20, Tetraglycol (200 mg/mL) | Soluplus (4%, w/v) | Adding in pre-concentrate (ND) | Liquid | -In vivo in SD rats at a dose of 100 mg/kg, 1.32-fold increase in Cmax and 1.35-fold increase in AUC and 0.49-fold decreased in Tmax as compared to conventional SEDDS without PI. -Good correlation between in vitro dissolution, permeation and in vivo PK. | ND | [109] |
| Celecoxib (BCS II) | Capryol 90, Tween 20, Tetraglycol (200 mg/mL) | Soluplus (4%, w/v) | Adding in pre-concentrate (ND) | Liquid | -PI effect: Soluplus > PVP VA64, poloxamer 407, PEG 6000 | ND | [110] |
| Curcumin (BCS IV) | Capryol 90, Labrafac PG, Cremophor EL, Labrasol (40 mg/940 mg) | Eudragit E PO (5%, w/w) | Suspending in pre-concentrate by blending (Suspension) | Liquid filled in hard gelatin capsule | -Concentration dependent PI effect. -In vivo in rabbits at a dose of 50 mg/kg, 1.22- and 53.14-fold increased absorption as compared to the conventional SEDDS without PI and the aqueous suspension, respectively. | Amorphous | [111] |
| Curcumin (BCS IV) | Capryol 90, Labrasol, PEG 400 (50 mg/g) | PVP (10%, w/w) | Suspending in pre-concentrate by vortexing (Suspension) | Liquid filled in hard gelatin capsule | -PI effect: PVP-K30 < PVP-K90 < without PI < HPMC -Concentration dependent PI effect. | ND | [90] |
| Cyclosporine A (BCS II) | Maisine 35-1, Kolliphor RH40, ethanol, and propylene glycol (Drug:Vehicle = 1:4.5 (w/v)) | PVP:Vehicle = 0.3:4.5 (w/v) | Suspending (HPC)or dissolving (Kollidon VA64 and PVP) in pre-concentrate by vortexing (Suspension) | Liquid | -PI effect: without PI = HPC = PVP VA64 < PVP K17 -Concentration-dependent PI effect. -In vitro dialysis test, equivalent concentration profile with that of conventional SEDDS prepared with two times more amount of lipid vehicle. | ND | [112] |
| Danazol (BCS II) | Captex 300, Capmul MCM, Cremophor EL, EtOH (40% or 80% of saturated solubility in formulation) | HPMC E4M (5%, w/w) | Suspending in pre-concentrate (Suspension) | Liquid filled in hard gelatin capsule | -PI effect: Cellulosic PPI > Mesoporous silica, Eudragits, Polyvinylpyrrolidones (PVPs). -In vivo in beagle dogs, PPI to promote drug exposure at moderate drug loads (40% of saturated solubility in the formulation), but not at higher drug loads (80% saturation). | Crystalline | [52] |
| Docetaxel (BCS II) | Labrafac, Cremophor RH40, Transcutol P (40 mg/640 mg) | HPMC K100 (2.5%, w/w) | Dispersing in pre-concentrate (ND) | Solid su-SEDDS (Spray drying, Lactose: pre-concentrate = 6 g:8 g in 100 mL water) | -In vivo in SD rats at a dose of 10 mg/kg, AUC increased by nearly 8.77-fold, 1.45-fold more than those of the powder drug and the conventional SEDDS without PI. | ND | [113] |
| Dutasteride (BCS II) | Capryol 90, Cremophor EL, Transcutol HP (0.5 mg/170.5 mg) | Gelatin (44%, w/w) + Soluplus (14.7%, w/w) | Mixing with pre-concentrate and PI solution (Clear solution) | Solid su-SEDDS (Spray drying, Gelatin) | -PI effect on dissolution and prolonged supersaturated state: Combination of gelatin with Soluplus > Gelucire 44/14, poloxamer 407, sodium lauryl sulfate, Soluplus, Solutol HS15, or TPGS. | ND | [114] |
| Dutasteride (BCS II) | Capryol 90, Cremophor EL, Transcutol HP (100 mg/20.1 g) | HPMC, Soluplus (1:1 w/w ratio compared to pre-concentrate) | Mixing with pre-concentrate (2.01 g) and PI solution (2 g in 400 mL EtOH) dispersed with solid carrier, Aerosil 200 (2 g) | Solid su-SEDDS (Spray drying) | -In vivo in SD rats at a dose of 2 mg/kg, higher oral BA with 6.8- and 5.0-fold for Cmax and AUC, respectively, compared to the physical mixture. | ND | [115] |
| Dutasteride (BCS II) | Capryol 90, Cremophor EL, Transcutol HP (Drug:Vehicle = 1:67.6, w/v) | Soluplus:Vehicle = 10:67.6 (w/v) | Suspending in pre-concentrate by vortexing (Suspension) | Liquid | -In vivo in SD rats at a dose of 2 mg/kg, 3.9-fold greater AUC than that of the drug suspension and 1.3-fold greater than that of conventional SEDDS. The 5.6- and 2.0-fold higher Cmax as compared to drug suspension and SEDDS, respectively. | ND | [116] |
| Ellagic acid (BCS IV) | Ethyl oleate, Tween 80, polyethylene glycol (4 mg/g) | PVP K30 (0.5%, w/w) | Adding in pre-concentrate by vortexing (ND) | Liquid | -Concentration dependent PI effect. -Good correlation between in vitro nucleation inhibition effect of PI and in vivo antioxidant ability. | ND | [117] |
| Ezetimibe (BCS II) | Captex 355, Cremophor RH40, Imwitor 988 (90% saturation solubility level of 90.62 mg/ml) | HPMC-E5 (5%, w/w) | Suspending in pre-concentrate by Cyclo-mixer (Suspension) | Solid su-SEDDS (Adsorption and granulation, MCC and talc) | -In vitro release, improved by 1.17-, 1.69-, and 13.21-fold as compared to solid-SEDDS, commercial product, and the free drug, respectively. | Amorphous | [68] |
| Fenofibrate (BCS II) | Ethyl oleate, Cremophor RH40, Transcutol HP (15%, w/w) | Soluplus:Drug = 1:1 (w/w) | Physical blending with solid su-SEDDS | Solid su-SEDDS (Solvent evaporation, mesoporous silica) | -In vivo in beagle dogs at a dose of 100 mg, 1.4-fold greater AUC than that without Soluplus. | ND | [26] |
| Fenofibrate (BCS II) | Captex 300, Capmul MCM, Cremophor EL, Transcutol HP (40% or 85% of saturated solubility in formulation) | Lipid soluble: Eudragit RL100 (5%, w/w), PPGAE (1%, w/w) Water soluble: HPMC E4M (5%, w/w) | Dissolving or suspending in pre-concentrate by vortexing (Lipid soluble: Clear solution, Water soluble: Suspension) | Liquid | -Polymer specific stabilizing effect -In vitro−in situ model using SD rats, potential utility of PPIs in promoting drug absorption via stabilization of supersaturation. | ND | [32] |
| Ginger extract (ND) | Medium chai triglyceride, Lysolecithin, glycerin (5%, w/w) | HPMC (5%, w/w) | Suspending in pre-concentrate by vortexing (Suspension) | Liquid | -In vivo in SD rats at a dose of 100 mg/kg, three-fold higher BA of 6-gingerol and 8-gingerol than those of unformulated extract treated group. | ND | [118] |
| Glipizide (BCS II) | Captex 355: Solutol HS15: Imwitor 988 (4%, w/v) | HPMC-E5 (5%, w/w) | Suspending in pre-concentrate by Cyclo-mixer (Suspension) | Solid su-SEDDS (Adsorption, calcium carbonate and talc) | -In vivo in Himalayan rabbits at a dose of 1 mg/kg, increase in Cmax (3.4-fold) and AUC (2.7-fold) from solid su-SEDDS as compared with the pure drug. | Amorphous | [119] |
| Griseofulvin (BCS II) | Oleic acid, Labrafil, Tween 20, Labrafac PG (5 mg/16.545 g) | Poloxamer (0.48%, w/w) | Adding in pre-concentrate (ND) | Liquid | -PI effect: Poloxamer > HPMC -In vivo permeability in Wister rats at a dose of 1 mL with a concentration of 0.05 mg/ml, three-fold more permeability through the rat intestine, compared with aqueous suspension. | ND | [120] |
| Indirubin (ND, BCS II or IV) | Maisine 35-1, Cremophor EL, Transcutol P (ND) | PVP-K17 (0.5%, w/w) | Dispersing in pre-concentrate by vortexing (ND) | Liquid | -PI effect: PVP-K17 > PEG 4000 and HPMC -In vivo in SD rats at a dose of 2.58 mg/kg, improved oral absorption and relative BA (129.5%) compared with conventional SEDDS. | ND | [89] |
| Paclitaxel (BCS IV) | EtOH, PEG 400, Cremophor EL, Glyceryl dioleate (57 mg/g) | HPMC-E5LV (5%, w/w) | Suspending in pre-concentrate by hand mixing (Suspension) | Liquid | -In vivo in SD rats at a dose of 10 mg/kg, 10- and 20-fold higher Cmax and 5- and 10-fold higher AUC compared with those of Taxol® formulation and the conventional SEDDS, respectively | ND | [53] |
| PNU-91325 (ND, BCS II or IV) | Cremophor EL, PEG 400, Dimethyl acetamide, Pluronic L44, HPMC, Glycerol monooleate, Glycerol dioleate, Water (4%, w/w) | HPMC-E50LV (20%, w/w) | Suspending in pre-concentrate by vortexing (Suspension) | Liquid filled in hard gelatin capsule | -In vivo in beagle dogs at a dose of 10 mg/kg, Oral BA of −76% compared to that of a PEG 400 (−12%) or tween (−68%) formulations. | ND | [121] |
| Raloxifene HCl (BCS II) | Tween 20, PEG 200, TEC or Capryol 90, Labrasol, PEG 200 (6%, w/v) | HPC-L (3%, w/v) | ND | Liquid filled in hard gelatin capsule | -PI effect: HPC-L > HPMC-More than 50% release from the pH-modified su-SEDDS at pH 2.5 compared with only −5% release of a conventional tablet. | ND | [122] |
| Resveratrol (BCS II) | Lauroglycol FCC, Transcutol P (100 mg/450 mg) | HPMC-E15LV (5%, w/w) | Suspending in pre-concentrate by vortexing (Suspension) | Liquid | -In vivo in Wistar rats at a dose of 20 mg/kg, 1.33-fold increased AUC of the su-SEDDS than conventional SEDDS without PI. | Amorphous | [123] |
| Silybin (BCS II) | Labrafac CC, Cremophor RH40, Labrasol (40 mg/1090 mg) | HPMC-E50LV (5%, w/w) | Suspending in pre-concentrate by vortexing (Suspension) | Liquid | -In vivo in SD rats at a dose of 533 mg/kg, three-fold increased AUC than those of the conventional SEDDS without HPMC. | Amorphous | [124] |
| Silymarin (BCS II) | Labrafil M 1944 CS, Kolliphor® RH 40, Transcutol HP (15.6% as milk thistle powder, w/w) | Poloxamer 407 (10% w/w) | Dissolving in pre-concentrate by heating and magnetic stirring (Poloxamer 407: Clear solution)Suspending in pre-concentrate by milling (Other PIs: Suspension) | Liquid | -PI effect: Poloxamer 407 > HPβCD, HPMCP, Eudragit L100. -Concentration dependent PI effect. -In vivo in Rabbits at a dose of 28 mg/kg as silybin, 760% BA of su-SEDDS versus Legalon®, commercial product. | Crystalline | [125] |
| Tacrolimus (BCS II) | Capmul MCM, Cremophor EL, and Transcutol P (5.9%, w/w) | Soluplus (5.9%, w/w) | Suspending in pre-concentrate by vortexing using magnetic stirrer (Suspension) | Liquid | -PI effect: Soluplus > HPMC, PVP. -Concentration dependent PI effect -In vivo in SD rats at a dose of 5 mg/kg, Similar or higher AUC and Cmax of su-SEDDS containing one-quarter the amount of vehicle compared to conventional SEDDS. | ND | [126] |
| Valsartan (BCS III) | Capmul MCM, Tween 80, Gelucire 44/14, Water (80 mg/190 mg) | Poloxamer 407 (5.3%, w/w) | Adding in pre-concentrate (ND) | Solid su-SEDDS (Kneading and granulation by sieving, HPC and Florite® PS-10) | -Concentration dependent PI effect. -In vivo in SD rats at a dose of 10 mg/kg. approximately 177%–198% AUC versus raw drug and Diovan®, commercial product. | ND | [127] |
| Process | Physical State of Pre-Concentrate [213] | Solvent Use 1 | Description/Advantages (A) and Disadvantages (D) [30,131,212] | Max Loading Capacity (%, w/w) 2 [30,213] | Suitable Drug Dose/Potency [30] | Self-Emulsification Rate [30] | Ref. | ||
|---|---|---|---|---|---|---|---|---|---|
| O | X | Drug | Lipid Vehicle | ||||||
| Physical adsorption | L 3 | √ | Adsorption onto solid carrier using physical blending with a mixer -A: Easy and Low cost, Easy scale-up -D: Low adsorption efficiency (large amount of solid carrier) and exudation of liquid lipid by tablet compaction | 10 | 80 | Low dose, high potency | ++ | [68,108,119] | |
| Granulation/pelletization followed by drying | L 3, SS 4, S 5 | √ | Similar with conventional wet granulation -A: Easy and low cost, Excellent powder property, Flexible applicable to various solid dosage forms (including controlled release system), Easy scale-up -D: Destabilization by high temperature for drying | - | - | Low dose, high potency | ++ | [5,127,211] | |
| Spray drying | L 4, SS 4, S 5 | √ | Evaporation of solvent from atomized SEDDS with solid carrier -A: Narrow particle size distribution, Relatively easy scale-up -D: Destabilization or loss of drug and/or volatile component by high temperature for drying | 50 | 60 | Low dose, high potency | +++ | [113,114,115,215,220] | |
| Freeze drying or Spray freeze-drying | L 3, SS 4, S 5 | √ | Lyophilization of solvent (mainly aqueous) from frozen SEDDS with solid carrier -A: Prevent destabilization or loss of drug and/or volatile by heat -D: Destabilization of drug by freezing and lyophilization, Low process efficiency, particle aggregation and poor re-dispersibility | 50 | 60 | Low dose, high potency | +++ | [221,222,223] | |
| Spray congealing | SS 4, S 5 | √ | Spraying of molten formula with or without solid carrier into a cooling chamber -A: Prevent destabilization or loss of drug and/or volatile by heat, Solvent free -D: Destabilization of drug by freezing, Bad flow property, Limitations on the use of liquid lipids, particle aggregation and poor re-dispersibility | 30 | 99 | Low-medium dose | ++ | [224,225] | |
| Melt granulation or Melt extrusion and spheronization | SS 4, S 5 | √ | High shear mixing at high temperature -A: High process efficiency, Solvent free -D: Destabilization or loss of drug and/or volatile component by high temperature for drying | 60–80 | 50 | Low-medium dose | ++ | [226,227] | |
| Supercritical fluid (SCF) | L 3, SS 4, S 5 | √ | √ | Adsorption and/or coating onto solid carrier using high diffusivity of SCF -A: Mild condition, High loading efficiency -D: High cost for development of commercial scale equipment | 20 | 99 | Low-medium dose | +++ | [204,228,229] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, H.; Ha, E.-S.; Kim, M.-S. Current Status of Supersaturable Self-Emulsifying Drug Delivery Systems. Pharmaceutics 2020, 12, 365. https://doi.org/10.3390/pharmaceutics12040365
Park H, Ha E-S, Kim M-S. Current Status of Supersaturable Self-Emulsifying Drug Delivery Systems. Pharmaceutics. 2020; 12(4):365. https://doi.org/10.3390/pharmaceutics12040365
Chicago/Turabian StylePark, Heejun, Eun-Sol Ha, and Min-Soo Kim. 2020. "Current Status of Supersaturable Self-Emulsifying Drug Delivery Systems" Pharmaceutics 12, no. 4: 365. https://doi.org/10.3390/pharmaceutics12040365
APA StylePark, H., Ha, E.-S., & Kim, M.-S. (2020). Current Status of Supersaturable Self-Emulsifying Drug Delivery Systems. Pharmaceutics, 12(4), 365. https://doi.org/10.3390/pharmaceutics12040365