Development of 20(S)-Protopanaxadiol-Loaded SNEDDS Preconcentrate Using Comprehensive Phase Diagram for the Enhanced Dissolution and Oral Bioavailability


 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. HPLC Analysis
2.3. Solubility Test
2.4. Particle Size Measurement
2.5. Preparation and Physical Characterization
2.6. Construction of Comprehensive Ternary Phase Diagram
- A = PPD solubility (%) in oil from Figure 2
- B = PPD solubility (%) in surfactant from Figure 2
- C = predetermined composition (%) of oil
- D = predetermined composition (%) of surfactant
- C + D = 100%
- C′ = composition (%) of oil at the calculated E point
- D′ = composition (%) of surfactant at the calculated E point
- C′ + D′ + E = 100%
2.7. Observation of Precipitation
2.8. Scanning Electron Microscopy (SEM) Analysis
2.9. Dissolution Test
2.10. Oral Pharmacokinetic Study in Rats
3. Results
3.1. PPD Solubility in Various Oils and Surfactants
3.2. Physical Characterization of Vehicles of Various Compositions
3.3. Construction of Comprehensive Ternary Phase Diagrams and Characterization of Formulations
3.4. SEM Analysis of Formulations
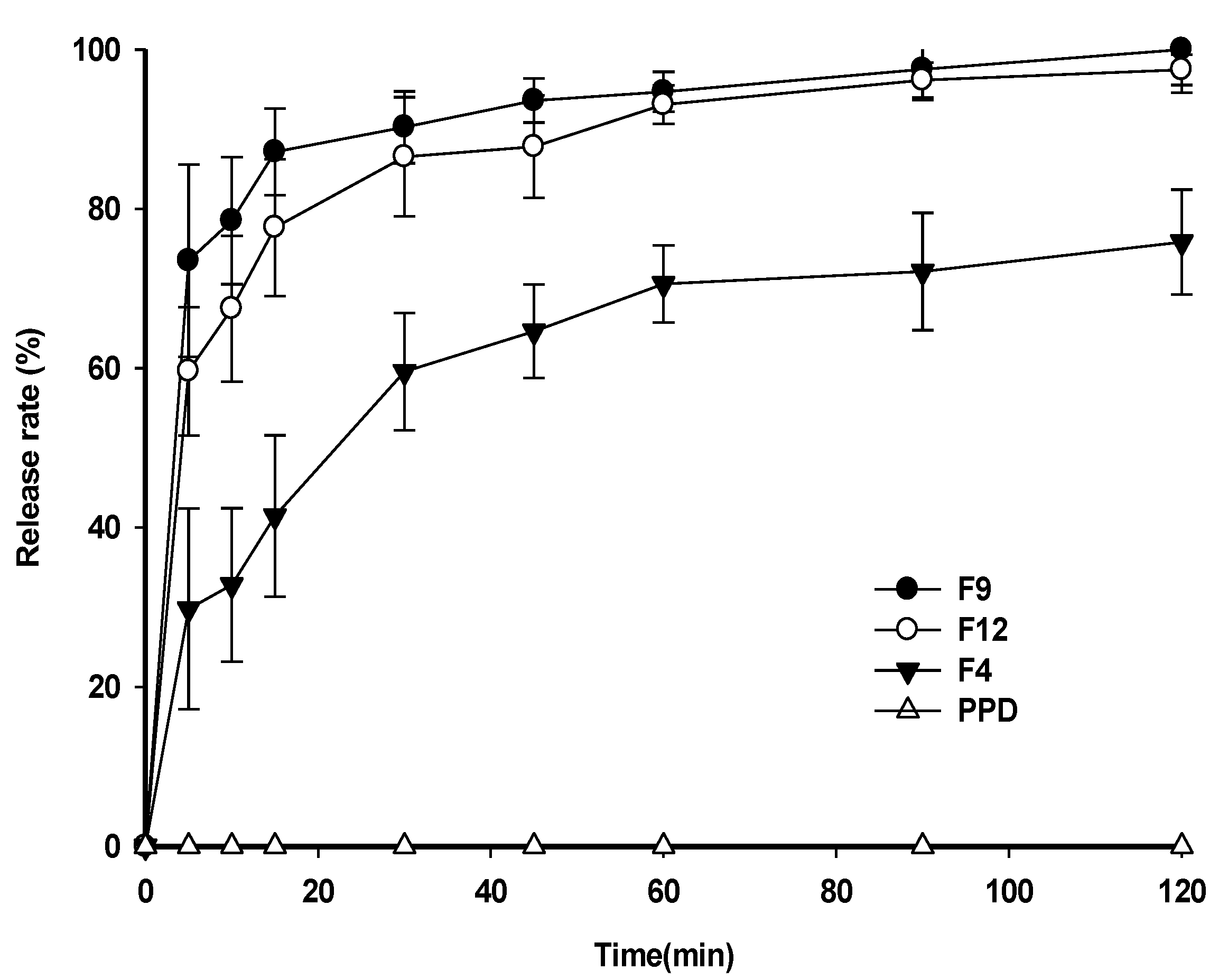
3.5. Dissolution Test
3.6. Oral Pharmacokinetic Study in Rats
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kennedy, O.D.; Scholey, B.A. Ginseng: Potential for the enhancement of cognitive performance and mood. Pharmacol. Biochem. Behav. 2003, 75, 687–700. [Google Scholar] [CrossRef]
- Endale, M.; Im, E.J.; Lee, J.Y.; Kim, S.D.; Yayeh, T.; Song, Y.B.; Kwak, Y.S.; Kim, C.; Kim, S.H.; Roh, S.S.; et al. Korean Red Ginseng Saponin Fraction Rich in Ginsenoside-Rb1, Rc and Rb2 Attenuates the Severity of Mouse Collagen-Induced Arthritis. Mediat. Inflamm. 2014, 2014, 748964. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.Z.; Hu, Y.; Wu, W.Y.; Ye, M.; Guo, D.A. Saponins in the genus Panax L. (Araliaceae): A systematic review of their chemical diversity. Phytochemistry 2014, 106, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Ling, J.; Yu, Y.; Zhu, J.; Li, Y.; Ling, L.; Wang, L.; Xu, C.; Duan, G. A highly sensitive HPLCMS/MS method for quantification of 20(S)-protopanaxadiol in human plasma and its application in phase IIa clinical trial of a novel antidepressant agent. J. Chromatogr. B 2016, 1031, 214–220. [Google Scholar] [CrossRef]
- Han, S.; Lim, T.G.; Kim, J.E.; Yang, H.; Oh, D.K.; Yoon Park, J.H.; Yoon Park, D.; Kim, H.J.; Rhee, Y.K.; Lee, K.W. The ginsenoside derivative 20(S)-protopanaxadiol inhibits solar ultraviolet light-induced matrix metalloproteinase-1 expression. J. Cell. Biochem. 2017, 118, 3756–3764. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.D.; Quan, H.Y.; Zhang, Y.; Kim, S.H.; Chung, S.H. 20(S)-Ginsenoside Rg3-induced apoptosis in HT-29 colon cancer cells is associated with AMPK signaling pathway. Mol. Med. Rep. 2010, 3, 825–831. [Google Scholar]
- Kim, B.J.; Nah, S.Y.; Jeon, J.H.; So, I.; Kim, S.J. Transient receptor potential melastatin 7 channels are involved in ginsenoside Rg3-induced apoptosis in gastric cancer cells. Basic Clin. Pharmacol. Toxicol. 2011, 109, 233–239. [Google Scholar] [CrossRef]
- Jeong, A.; Lee, H.J.; Jeong, S.J.; Lee, H.J.; Lee, E.O.; Bae, H.; Kim, S.H. Compound K inhibits basic fibroblast growth factor-induced angiogenesis via regulation of p38 mitogen activated protein kinase and AKT in human umbilical vein endothelial cells. Biol. Pharm. Bull. 2010, 33, 945–950. [Google Scholar] [CrossRef]
- Benishin, C.G.; Lee, R.; Wang, L.C.; Liu, H.J. Effects of ginsenoside Rb1 on central cholinergic metabolism. Pharmacology 1991, 42, 223–229. [Google Scholar] [CrossRef]
- Zhang, Q.H.; Wu, C.F.; Duan, L.; Yang, J.Y. Protective effects of ginsenoside Rg(3) against cyclophosphamide-induced DNA damage and cell apoptosis in mice. Arch. Toxicol. 2008, 82, 117–123. [Google Scholar] [CrossRef]
- Cai, B.X.; Jin, S.L.; Luo, D.; Lin, X.F.; Gao, J. Ginsenoside Rb1 suppresses ultraviolet radiation-induced apoptosis by inducing DNA repair. Biol. Pharm. Bull. 2009, 32, 837–841. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Ma, L.; Yu, X.; Li, Z.; Guo, Y.; Wang, X. A nanoparticulate drug-delivery system for 20 (S)-protopanaxadiol: Formulation, characterization, increased oral bioavailability and anti-tumor efficacy. Drug Deliv. 2016, 23, 2410–2418. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.T.; Kim, M.H.; Park, J.H.; Lee, J.Y.; Cho, H.J.; Yoon, I.S.; Kim, D.D. Microemulsion-based hydrogels for enhancing epidermal/dermal deposition of topically administered 20 (S)-protopanaxadiol: In vitro and in vivo evaluation studies. J. Ginseng Res. 2018, 42, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Pu, Y.; Wang, B.; Wang, Y.; Dong, T.; Guo, T.; Zhang, T.; Cai, Z. Characterization, Molecular Docking, and In Vitro Dissolution Studies of Solid Dispersions of 20 (S)-Protopanaxadiol. Molecules 2017, 22, 274. [Google Scholar] [CrossRef]
- Kim, R.; Jang, D.J.; Kim, Y.; Yoon, J.H.; Min, K.; Maeng, H.J.; Cho, K. Flurbiprofen-Loaded Solid SNEDDS Preconcentrate for the Enhanced Solubility, In-Vitro Dissolution and Bioavailability in Rats. Pharmaceutics 2018, 10, 247. [Google Scholar] [CrossRef]
- Kim, D.H.; Kim, J.Y.; Kim, R.M.; Maharjan, P.; Ji, Y.G.; Jang, D.J.; Cho, K.H. Orlistat-loaded solid sNeDDs for the enhanced solubility, dissolution, and in vivo performance. Int. J. Nanomed. 2018, 13, 7095. [Google Scholar] [CrossRef]
- Madhav, K.V.; Kishan, V. Self microemulsifying particles of loratadine for improved oral bioavailability: Preparation, characterization and in vivo evaluation. J. Pharm. Investig. 2018, 48, 497–508. [Google Scholar] [CrossRef]
- Singh, D.; Tiwary, A.K.; Bedi, N. Canagliflozin loaded SMEDDS: Formulation optimization for improved solubility, permeability and pharmacokinetic performance. J. Pharm. Investig. 2019, 49, 67–85. [Google Scholar] [CrossRef]
- Patil, S.C.; Tagalpallewar, A.A.; Kokare, C.R. Natural anti-proliferative agent loaded self-microemulsifying nanoparticles for potential therapy in oral squamous carcinoma. J. Pharm. Investig. 2019, 49, 527–541. [Google Scholar] [CrossRef]
- Zhu, W.; Guo, C.; Yu, A.; Gao, Y.; Cao, F.; Zhai, G. Microemulsion-based hydrogel formulation of penciclovir for topical delivery. Int. J. Pharm. 2009, 378, 152–158. [Google Scholar] [CrossRef]
- Kansagra, H.; Mallick, S. Microemulsion-based antifungal gel of luliconazole for dermatophyte infections: Formulation, characterization and efficacy studies. J. Pharm. Investig. 2016, 46, 21–28. [Google Scholar] [CrossRef]
- Jeong, Y.S.; Balla, A.; Chun, K.H.; Chung, S.J.; Maeng, H.J. Physiologically-based pharmacokinetic modeling for drug-drug interactions of procainamide and N-acetylprocainamide with Cimetidine, an inhibitor of rOCT2 and rMATE1, in Rats. Pharmaceutics 2019, 11, 108. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Shu, Y.J.; Cheng, J.Y.; Liang, R.C.; Dian, S.N.; Lv, X.X.; Yang, M.Q.; Huang, S.L.; Chen, G.; Yang, F. Pharmacokinetics and efficiency of brain targeting of ginsenosides Rg1 and Rb1 given as Nao-Qing microemulsion. Drug Dev. Ind. Pharm. 2015, 41, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Mathiyalagan, R.; Subramaniyam, S.; Kim, Y.J.; Kim, Y.C.; Yang, D.C. Ginsenoside compound K-bearing glycol chitosan conjugates: Synthesis, physicochemical characterization, and in vitro biological studies. Carbohydr. Polym. 2014, 112, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Feng, Z.; You, C.; Jin, Y.; Hu, X.; Wang, X.; Han, C. In vitro evaluation of Panax notoginseng Rg1 released from collagen/chitosan-gelatin microsphere scaffolds for angiogenesis. Biomed. Eng. Online 2013, 12, 134. [Google Scholar] [CrossRef]
- Khadka, P.; Ro, J.; Kim, H.; Kim, I.; Kim, J.T.; Kim, H.; Lee, J. Pharmaceutical particle technologies: An approach to improve drug solubility, dissolution and bioavailability. Asian J. Pharm. Sci. 2014, 9, 304–316. [Google Scholar] [CrossRef]
- Singh, A.K.; Chaurasiya, A.; Singh, M.; Upadhyay, S.C.; Mukherjee, R.; Khar, R.K. Exemestane loaded self-microemulsifying drug delivery system (SMEDDS): Development and optimization. Aaps Pharmscitech 2008, 9, 628–634. [Google Scholar] [CrossRef]
- Inugala, S.; Eedara, B.B.; Sunkavalli, S.; Dhurke, R.; Kandadi, P.; Jukanti, R.; Bandari, S. Solid self-nanoemulsifying drug delivery system (S-SNEDDS) of darunavir for improved dissolution and oral bioavailability: In vitro and in vivo evaluation. Eur. J. Pharm. Sci. 2015, 74, 1–10. [Google Scholar] [CrossRef]
- Kyatanwar, A.U.; Jadhav, K.R.; Kadam, V.J. Self micro-emulsifying drug delivery system (SMEDDS). J. Pharm. Res. 2010, 3, 75–83. [Google Scholar]
- Pouton, C.W. Formulation of poorly water-soluble drugs for oral administration: Physicochemical and physiological issues and the lipid formulation classification system. Eur. J. Pharm. Sci. 2006, 29, 278–287. [Google Scholar] [CrossRef]
- Zhang, L.; Xu, C.; Mao, J.; Wang, W.; Han, H.; Pu, Y.; Zhang, T. Formulation and Characterization of Novel Dry Suspension and Dry Emulsion of 20(S)-Protopanaxadiol. AAPS PharmSciTech 2019, 20, 275. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oil | Surfactant | Weight Ratio | Particle Size (nm) | PDI |
|---|---|---|---|---|
| Capmul MCM C8 | Kolliphor EL | 8:2 | Phase separation | N.D. |
| 6:4 | Phase separation | N.D. | ||
| 5:5 | 372.67 ± 45.39 | 0.28 ± 0.01 | ||
| 4:6 | 198.01 ± 14.24 | 0.19 ± 0.02 | ||
| 2:8 | Clear solution | N.D. | ||
| Kolliphor PS80 | 8:2 | Phase separation | N.D. | |
| 6:4 | Phase separation | N.D. | ||
| 5:5 | 352.87 ± 28.07 | 0.34 ± 0.02 | ||
| 4:6 | 277.93 ± 38.30 | 0.23 ± 0.03 | ||
| 2:8 | 164.70 ± 3.83 | 0.37 ± 0.01 | ||
| Kolliphor HS15 | 8:2 | Phase separation | N.D. | |
| 6:4 | Phase separation | N.D. | ||
| 5:5 | Phase separation | N.D. | ||
| 4:6 | 323.23 ± 40.50 | 0.27 ± 0.03 | ||
| 2:8 | Clear solution | N.D. | ||
| Capryol 90 | Kolliphor EL | 8:2 | Phase separation | N.D. |
| 6:4 | 91.63 ± 1.45 | 0.24 ± 0.01 | ||
| 5:5 | 195.67 ± 14.32 | 0.39 ± 0.01 | ||
| 4:6 | 277.00 ± 45.81 | 0.30 ± 0.03 | ||
| 2:8 | 115.97 ± 8.57 | 0.40 ± 0.02 | ||
| Kolliphor PS80 | 8:2 | Phase separation | N.D. | |
| 6:4 | Phase separation | N.D. | ||
| 5:5 | Phase separation | N.D. | ||
| 4:6 | 389.70 ± 7.40 | 0.29 ± 0.03 | ||
| 2:8 | 197.40 ± 57.60 | 0.26 ± 0.01 | ||
| Kolliphor HS15 | 8:2 | Phase separation | N.D. | |
| 6:4 | Phase separation | N.D. | ||
| 5:5 | Phase separation | N.D. | ||
| 4:6 | 270.57 ± 24.79 | 0.32 ± 0.02 | ||
| 2:8 | 182.90 ± 17.56 | 0.23 ± 0.01 |
| Formulation | PPD | Oil | Surfactant | Particle Size (nm) | PDI |
|---|---|---|---|---|---|
| Capmul MCM C8 | Kolliphor EL | ||||
| F1 | 5 | 19 | 76 | Precipitation | N.D. |
| F2 | 10 | 36 | 54 | Precipitation | N.D. |
| Capmul MCM C8 | Kolliphor PS80 | ||||
| F3 | 5 | 19 | 76 | 250.37 ± 23.47 | 0.26 ± 0.01 |
| F4 | 10 | 36 | 54 | Precipitation | N.D. |
| Capmul MCM C8 | Kolliphor HS15 | ||||
| F5 | 5 | 19 | 76 | Precipitation | N.D. |
| Capryol 90 | Kolliphor EL | ||||
| F6 | 5 | 19 | 76 | Precipitation | N.D. |
| F7 | 10 | 36 | 54 | Precipitation | N.D. |
| F8 | 10 | 45 | 45 | Precipitation | N.D. |
| F9 | 10 | 54 | 36 | 125.07 ± 12.56 | 0.23 ± 0.01 |
| Capryol 90 | Kolliphor PS80 | ||||
| F10 | 5 | 19 | 76 | Precipitation | N.D. |
| Capryol 90 | Kolliphor HS15 | ||||
| F11 | 5 | 19 | 76 | Precipitation | N.D. |
| F12 | 10 | 36 | 54 | 247.67 ± 45.79 | 0.21 ± 0.03 |
| Pharmacokinetic Parameters | Raw PPD in Suspension | PSP (F4) | PSP (F9) |
|---|---|---|---|
| Tmax (min) | 150 ± 60 | 150 ± 60 | 160 ± 69 |
| Cmax (μg/mL) | 0.486 ± 0.132 | 0.453 ± 0.180 | 0.942 ± 0.206 * |
| t1/2 (min) | 197 ± 53 | 170 ± 46 | 169 ± 6 |
| AUClast (μg·min/mL) | 179 ± 37 | 186 ± 88 | 324 ± 83 * |
| AUCinf (μg·min/mL) | 180 ± 37 | 188 ± 86 | 325 ± 83 * |
| MRT (min) | 301 ± 41 | 291 ± 63 | 281 ± 49 |
| Relative BA (%) | 104 | 181 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.H.; Kim, Y.C.; Jang, D.-J.; Min, K.A.; Karmacharya, J.; Nguyen, T.-T.-L.; Maeng, H.-J.; Cho, K.H. Development of 20(S)-Protopanaxadiol-Loaded SNEDDS Preconcentrate Using Comprehensive Phase Diagram for the Enhanced Dissolution and Oral Bioavailability. Pharmaceutics 2020, 12, 362. https://doi.org/10.3390/pharmaceutics12040362
Kim YH, Kim YC, Jang D-J, Min KA, Karmacharya J, Nguyen T-T-L, Maeng H-J, Cho KH. Development of 20(S)-Protopanaxadiol-Loaded SNEDDS Preconcentrate Using Comprehensive Phase Diagram for the Enhanced Dissolution and Oral Bioavailability. Pharmaceutics. 2020; 12(4):362. https://doi.org/10.3390/pharmaceutics12040362
Chicago/Turabian StyleKim, Young Hoon, Yu Chul Kim, Dong-Jin Jang, Kyoung Ah Min, Jenisha Karmacharya, Thi-Thao-Linh Nguyen, Han-Joo Maeng, and Kwan Hyung Cho. 2020. "Development of 20(S)-Protopanaxadiol-Loaded SNEDDS Preconcentrate Using Comprehensive Phase Diagram for the Enhanced Dissolution and Oral Bioavailability" Pharmaceutics 12, no. 4: 362. https://doi.org/10.3390/pharmaceutics12040362
APA StyleKim, Y. H., Kim, Y. C., Jang, D.-J., Min, K. A., Karmacharya, J., Nguyen, T.-T.-L., Maeng, H.-J., & Cho, K. H. (2020). Development of 20(S)-Protopanaxadiol-Loaded SNEDDS Preconcentrate Using Comprehensive Phase Diagram for the Enhanced Dissolution and Oral Bioavailability. Pharmaceutics, 12(4), 362. https://doi.org/10.3390/pharmaceutics12040362