Co-crystals, Salts or Mixtures of Both? The Case of Tenofovir Alafenamide Fumarates


 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Characterization Methods
2.2.1. Attenuated Total Reflection Fourier-Transform Infrared (ATR-FTIR) Measurements
2.2.2. Solution Nuclear Magnetic Resonance (NMR) Spectroscopy
2.2.3. Solid-State Nuclear Magnetic Resonance Analysis
2.2.4. DSC Measurements
2.2.5. p-XRD Measurements
2.2.6. X-Ray Single Crystal Analysis
2.3. Synthesis and Characterization of Tenofovir Alafenamide Derivatives
2.3.1. Synthesis of Tenofovir Alafenamide (TA)
2.3.2. Synthesis of Tenofovir Alafenamide Hydrochloride (TA HCl)
2.3.3. Characterization of Tenofovir Alafenamide Hemifumarate (TA HF)
2.3.4. Characterization of Tenofovir Alafenamide Monofumarate Form I (TA MF1)
2.3.5. Synthesis of Tenofovir Alafenamide Monofumarate Form II (TA MF2)
2.3.6. Synthesis of Tenofovir Alafenamide Monofumarate Form III (TA MF3)
3. Results
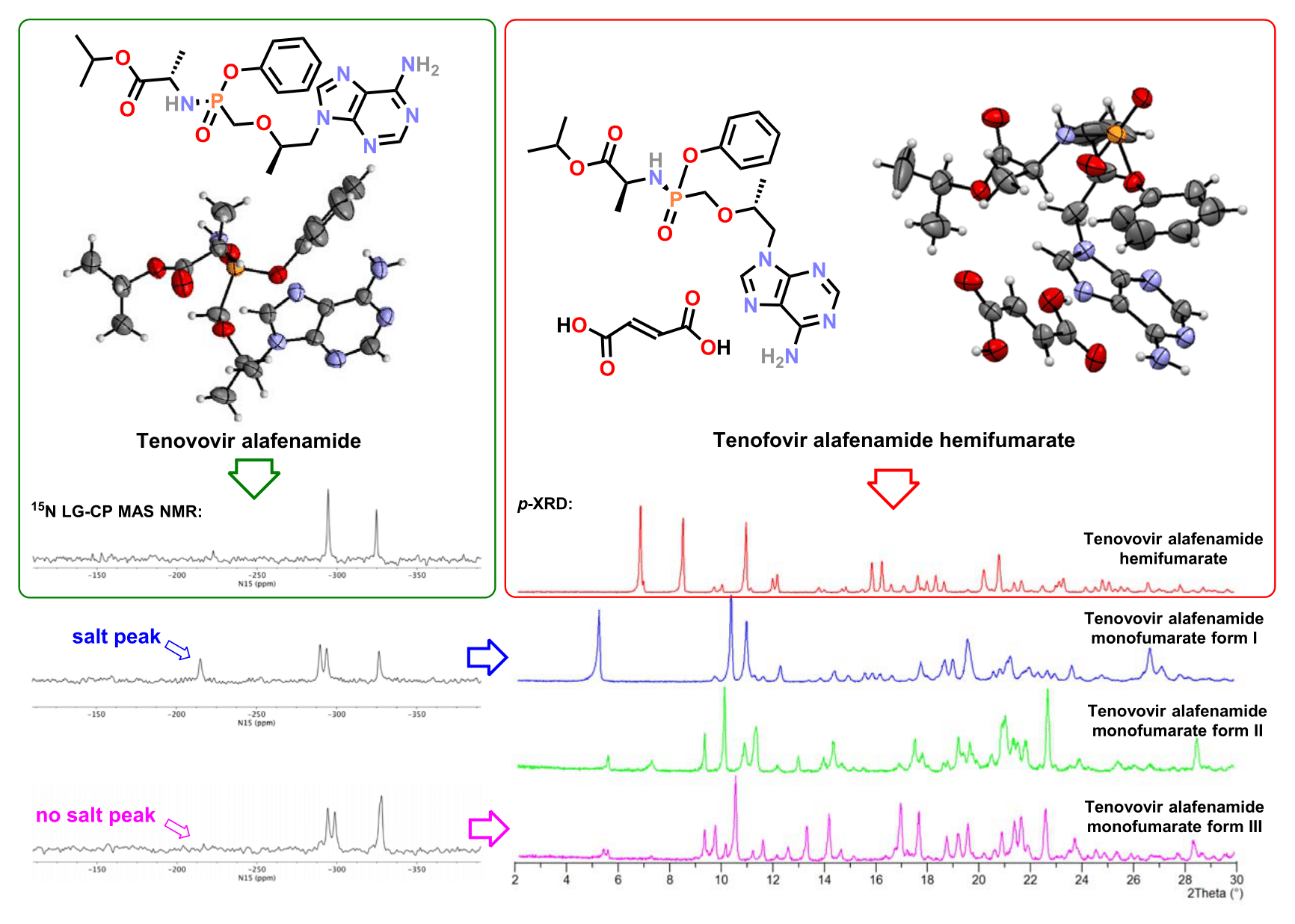
3.1. Synthesis of Tenofovir Alafenamide Derivatives
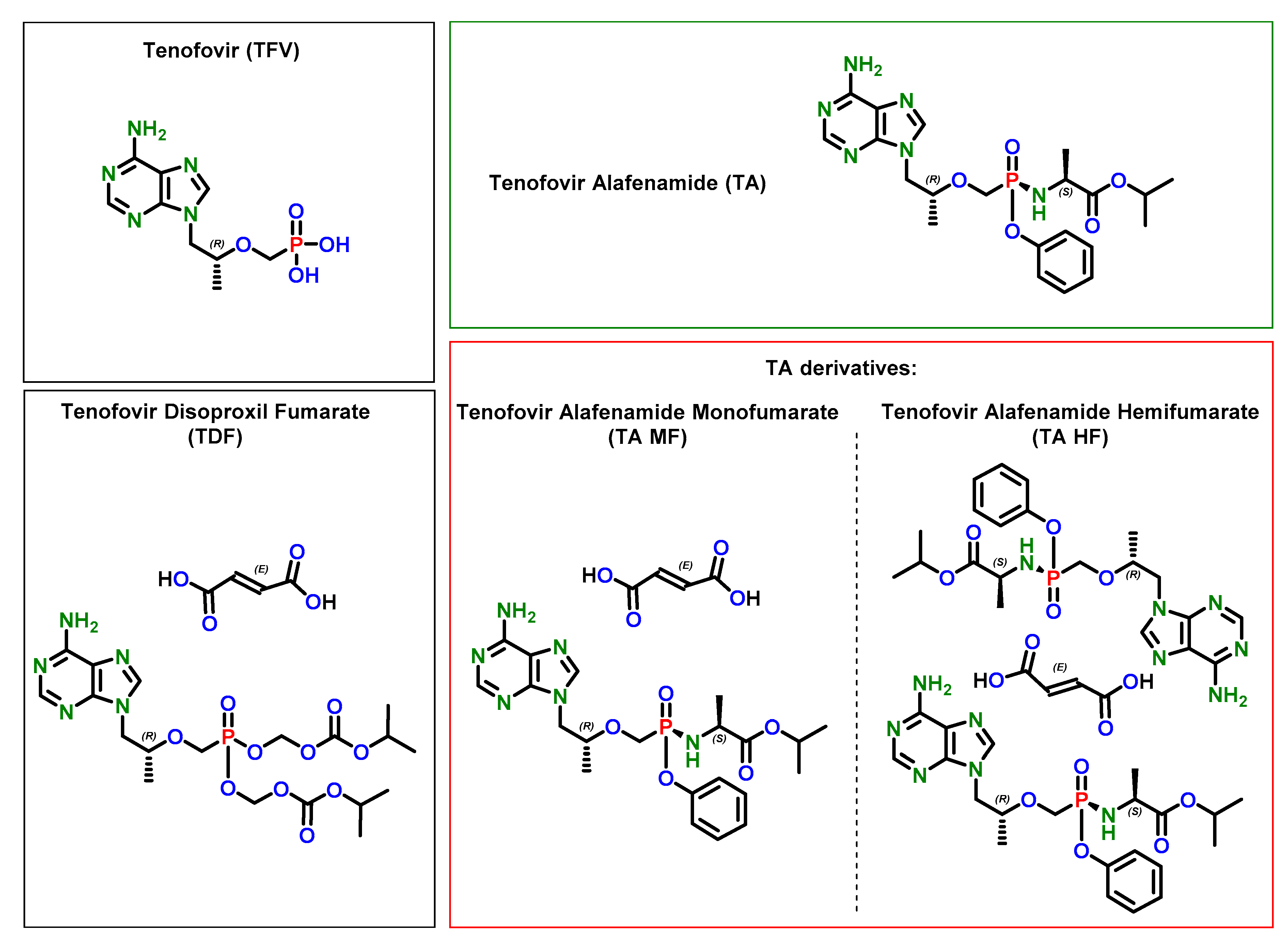
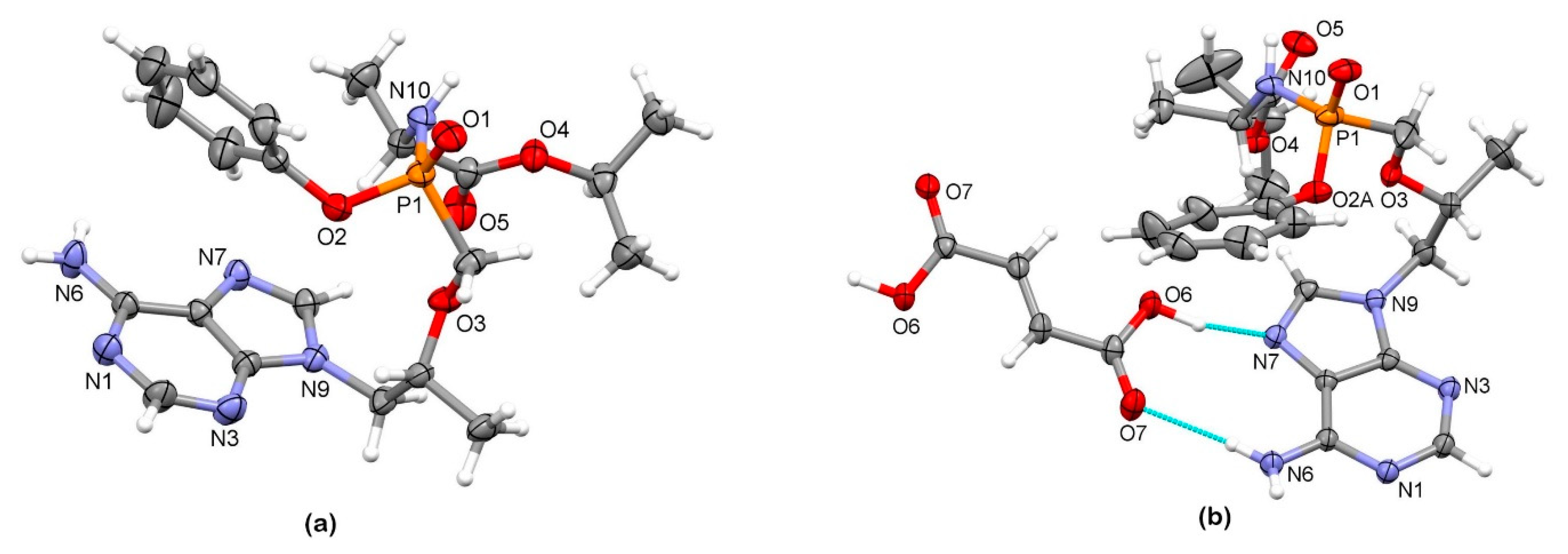
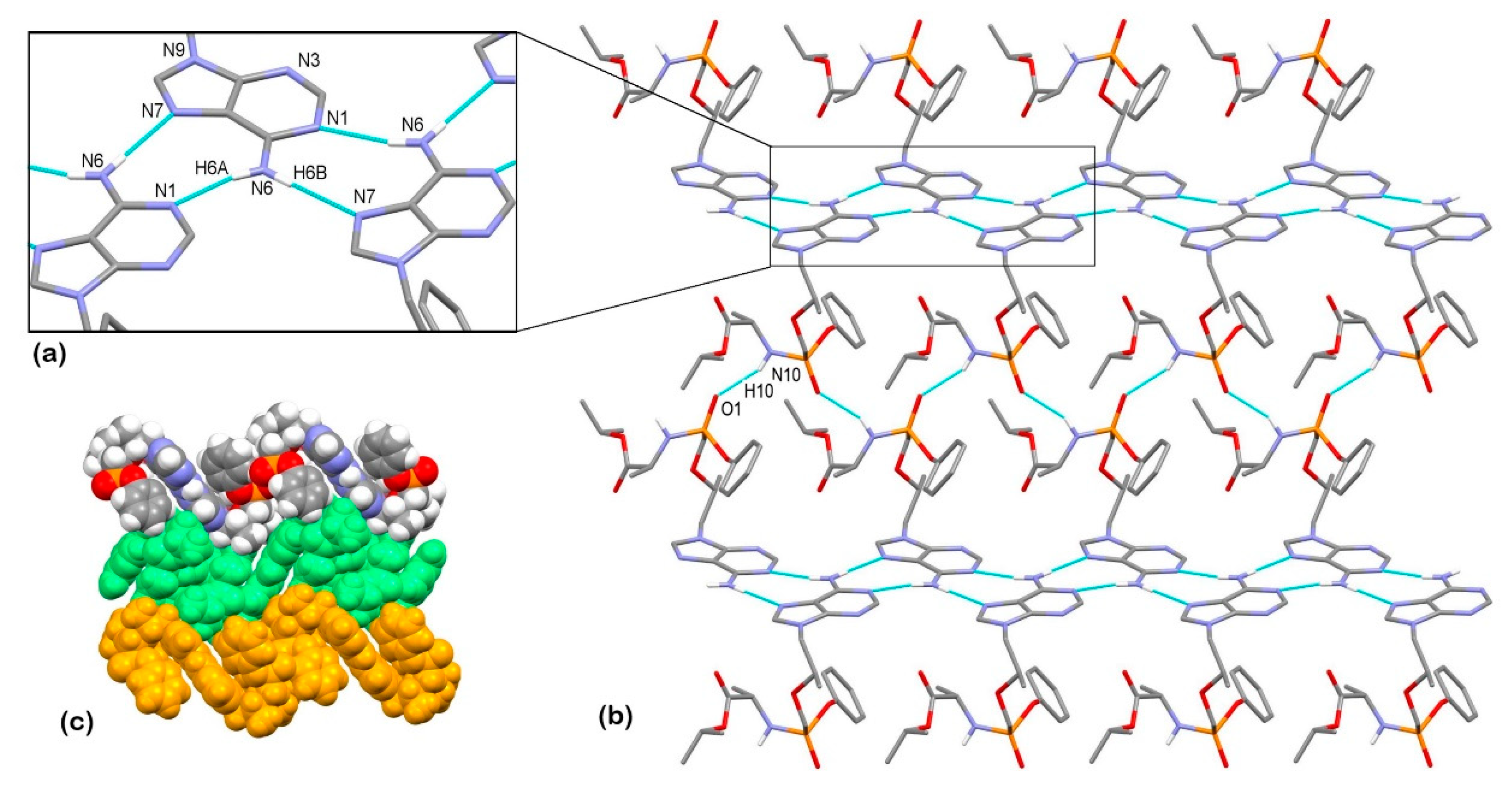
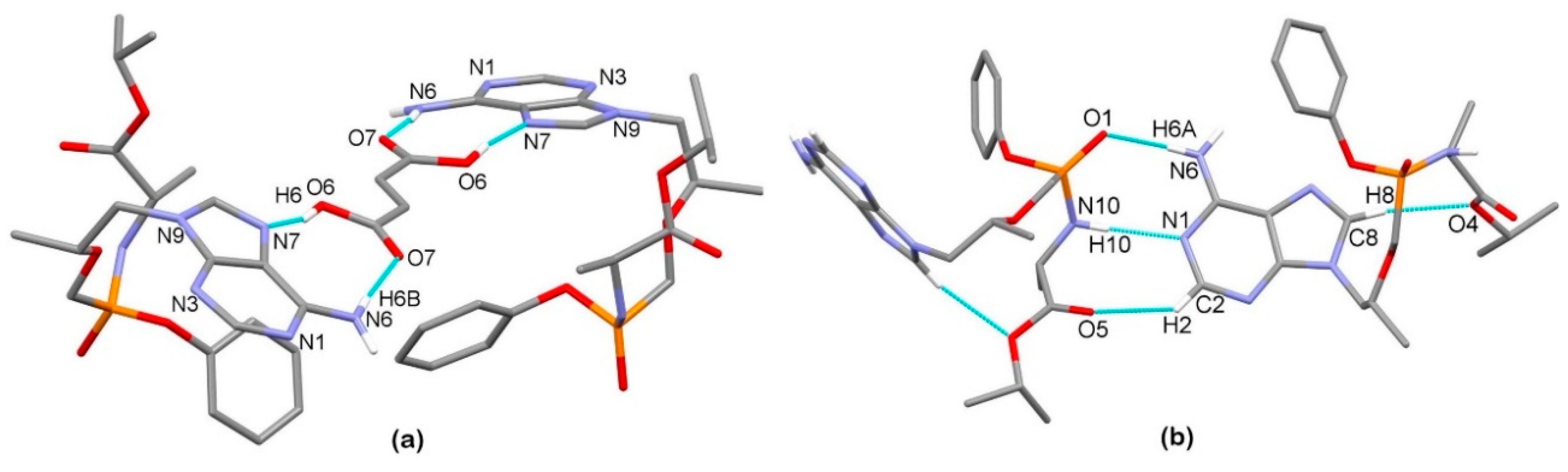
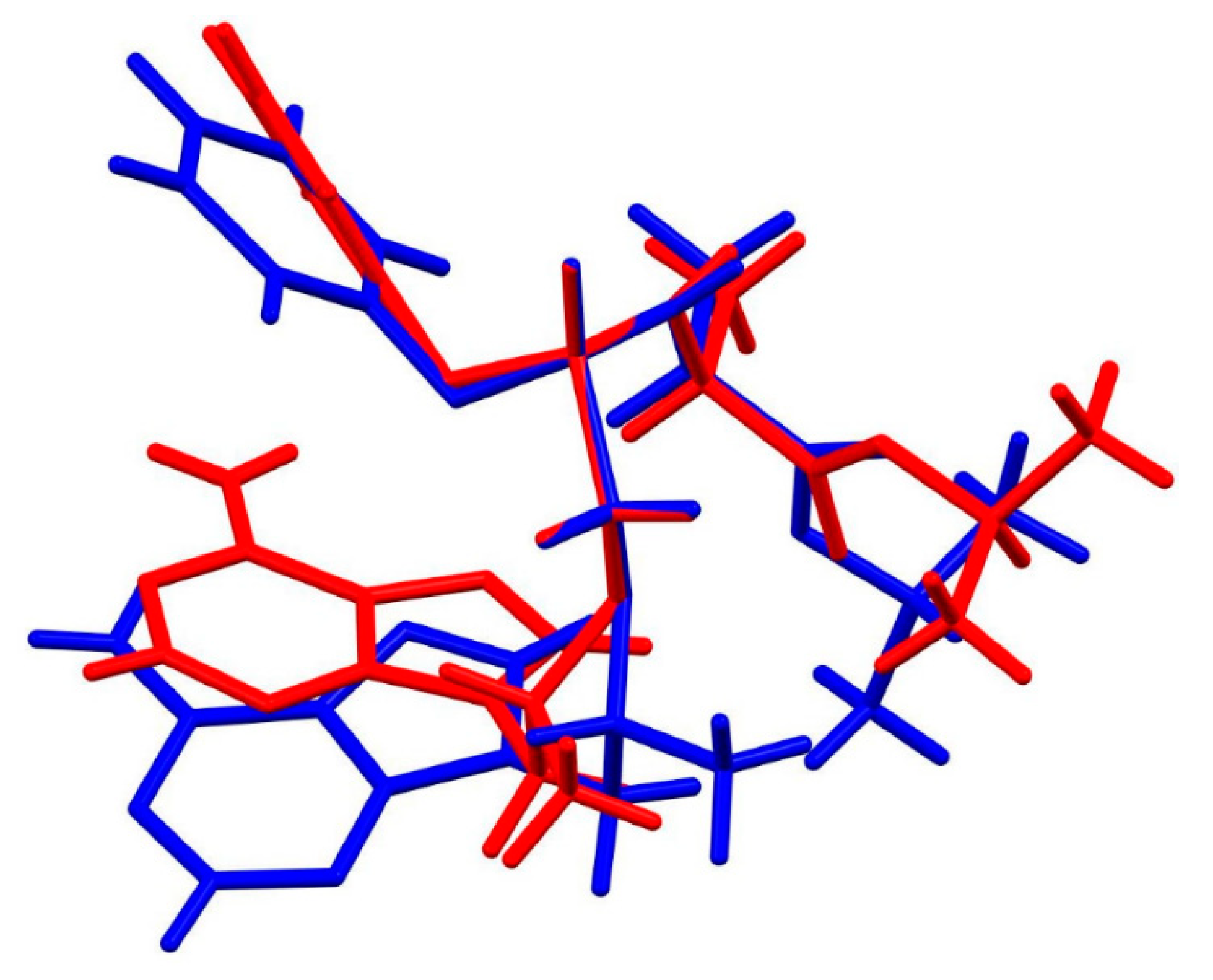
3.2. X-Ray Single Crystal Determination
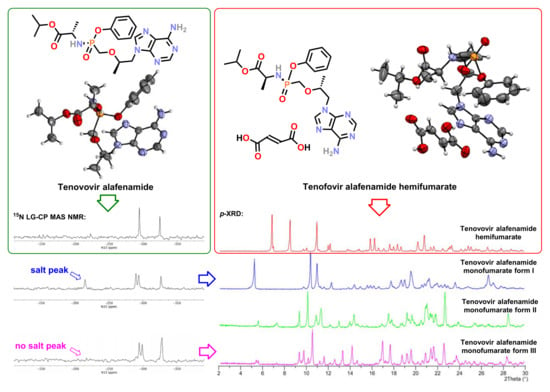
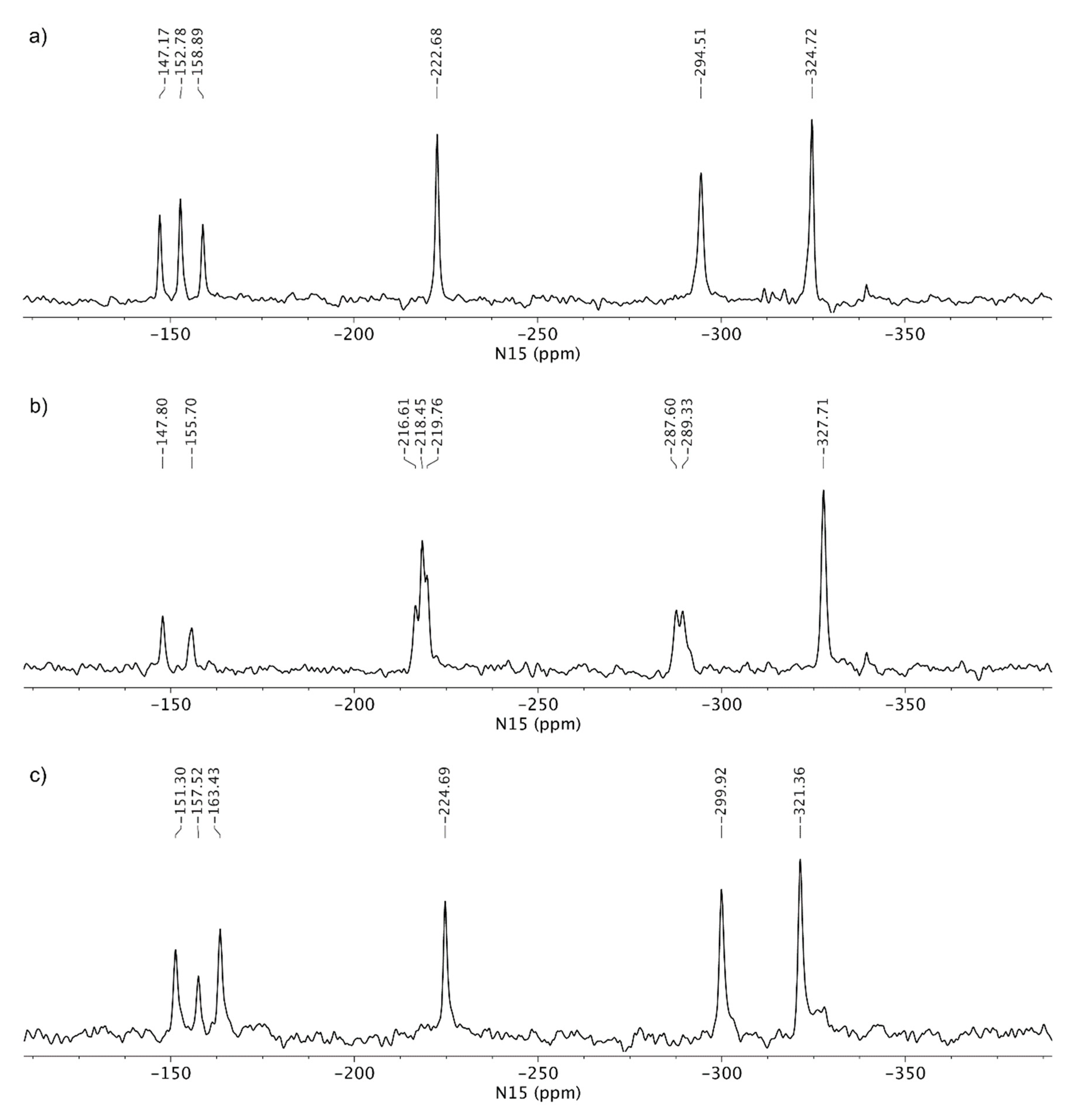
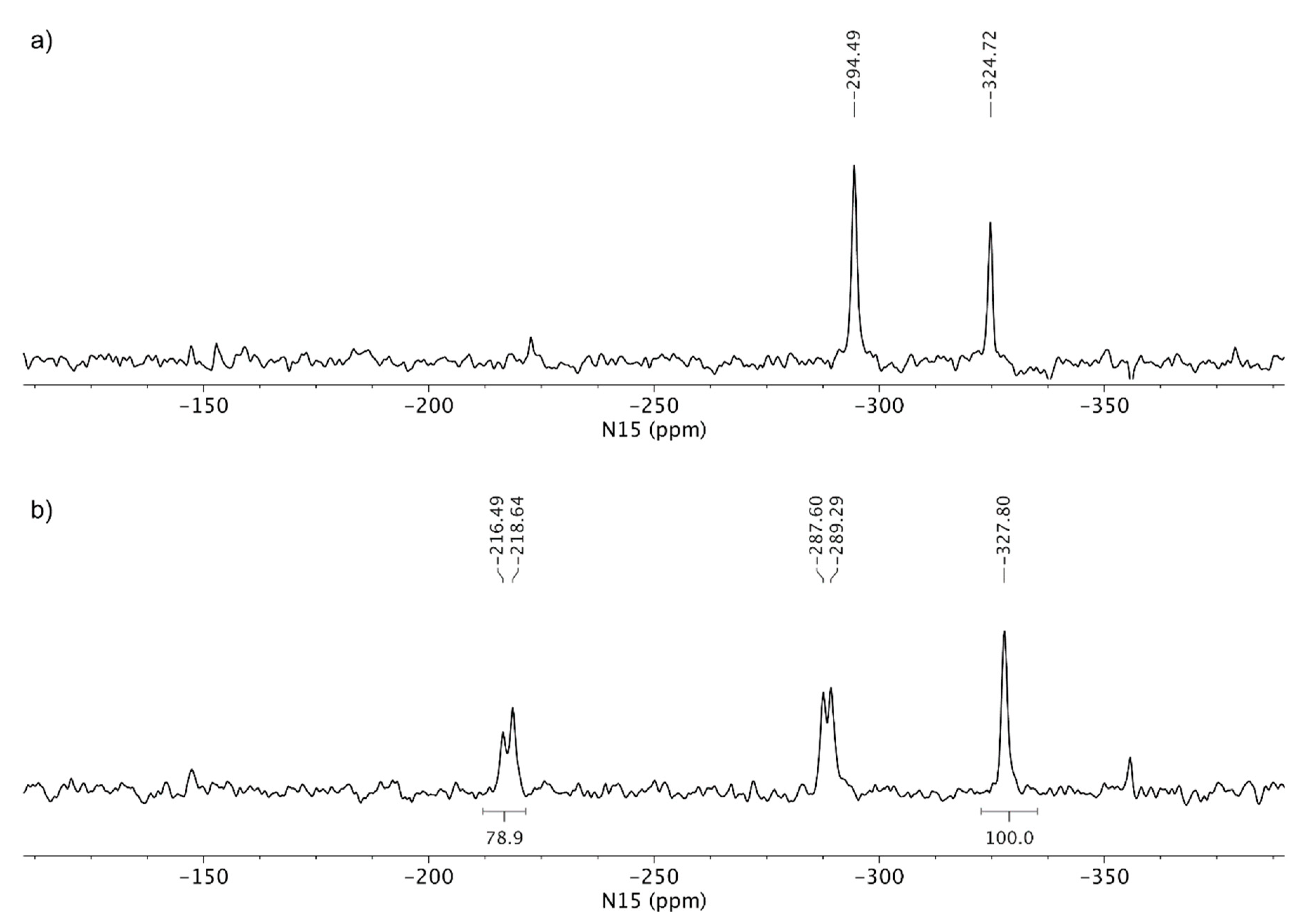
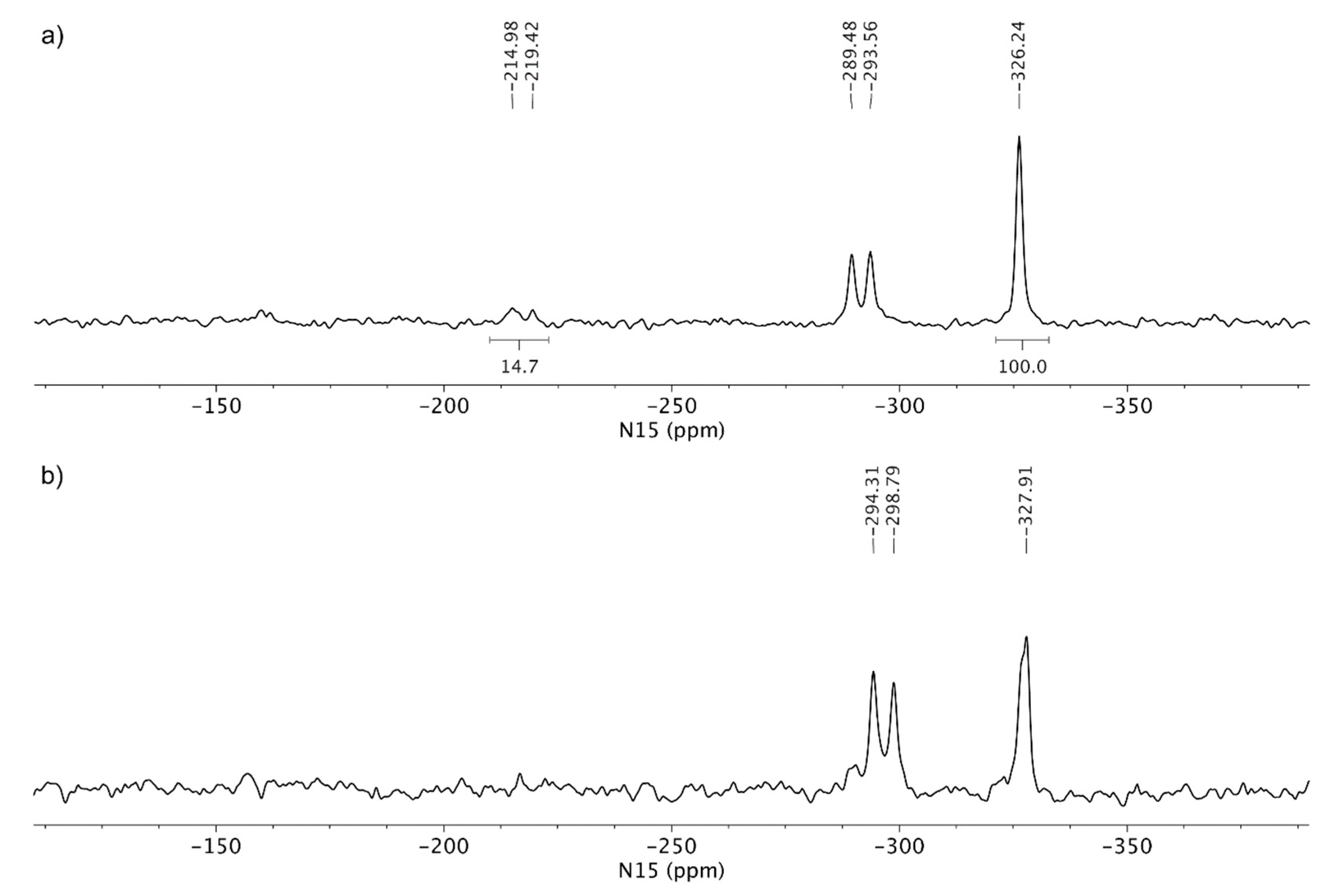
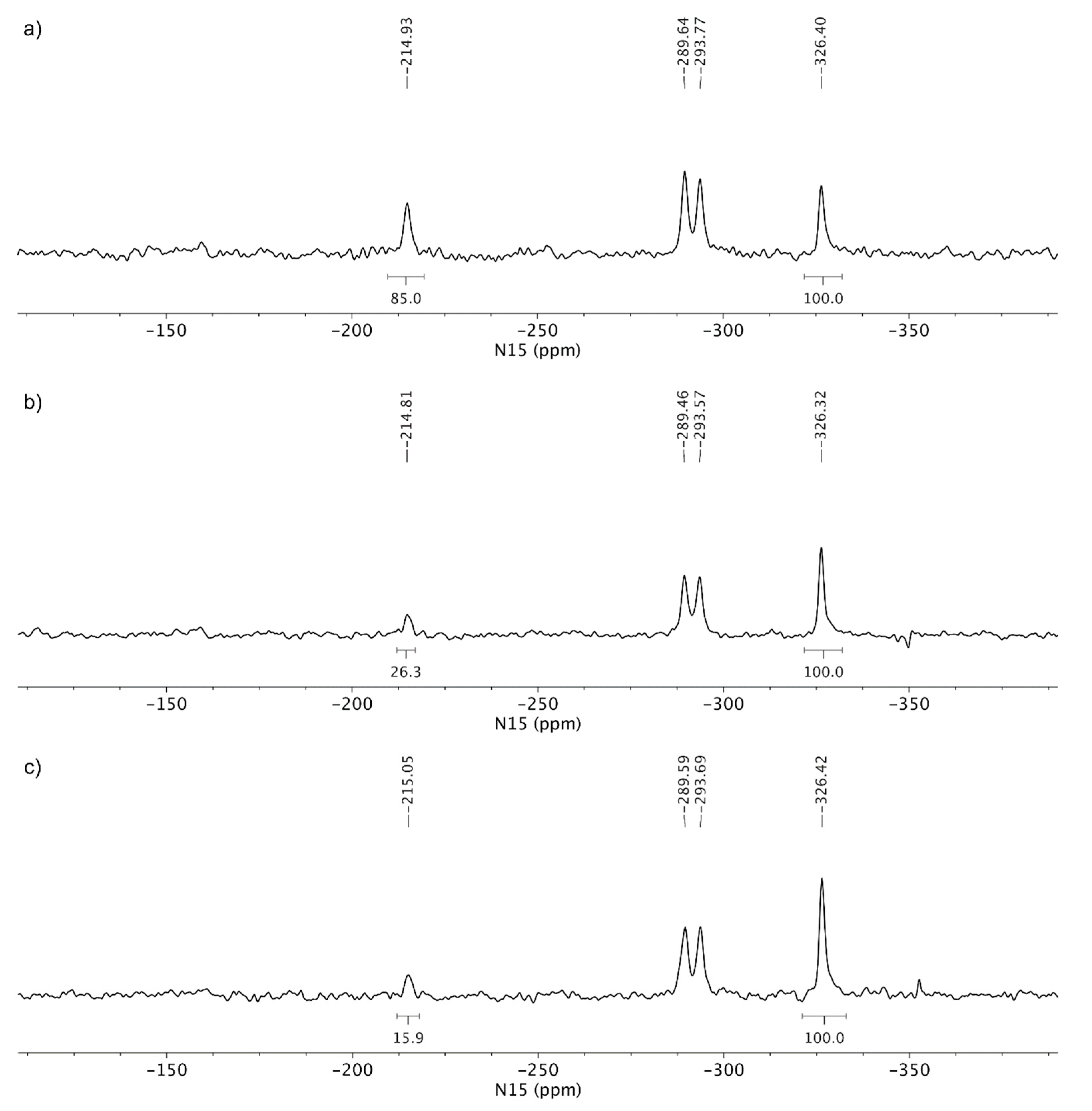
3.3. Solid-State Nuclear Magnetic Resonance Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- The Top 10 Causes of death. Available online: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 12 March 2020).
- Global Factsheets 2018. HIV and AIDS Estimates. Available online: http://aidsinfo.unaids.org/ (accessed on 12 March 2020).
- Kinch, M.S.; Patridge, E. An analysis of FDA-approved drugs for infectious disease: HIV/AIDS drugs. Drug Discov. Today 2014, 19, 1510–1513. [Google Scholar] [CrossRef] [PubMed]
- Blanco, J.L.; Whitlock, G.; Milinkovic, A.; Moyle, G. HIV integrase inhibitors: A new era in the treatment of HIV. Expert Opin. Pharmacother. 2015, 16, 1313–1324. [Google Scholar] [CrossRef] [PubMed]
- Caplan, M.R.; Daar, E.S.; Corado, K.C. Next generation fixed dose combination pharmacotherapies for treating HIV. Expert Opin. Pharmacother. 2018, 19, 589–596. [Google Scholar] [CrossRef]
- De Clercq, E. Tenofovir alafenamide (TAF) as the successor of tenofovir disoproxil fumarate (TDF). Biochem. Pharmacol. 2016, 119, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.S.; Fordyce, M.W.; Hitchcock, M.J.M. Tenofovir alafenamide: A novel prodrug of tenofovir for the treatment of human immunodeficiency virus. Antivir. Res. 2016, 125, 63–70. [Google Scholar] [CrossRef]
- Sampath, R.; Zeuli, J.; Rizza, S.; Temesgen, Z. Tenofovir alafenamide fumarate for the treatment of HIV infection. Drugs Today 2016, 52, 617–625. [Google Scholar] [CrossRef]
- Holy, A.; Rosenberg, I. 9-(Phosponylmethoxyalkyl) adenines, the method of preparation and utilization thereof. European Patent Application EP0206459 A2, 30 December 1986. [Google Scholar]
- Deeks, S.G.; Barditch-Crovo, P.; Lietman, P.S.; Hwang, F.; Cundy, K.C.; Rooney, J.F.; Hellmann, N.S.; Safrin, S.; Kahn, J.O. Safety, pharmacokinetics, and antiretroviral activity of intravenous 9-[2-(R)-(phosphonomethoxy)propyl]adenine, a novel anti-human immunodeficiency virus (HIV) therapy, in HIV-infected adults. Antimicrob. Agents Chemother. 1998, 42, 2380–2384. [Google Scholar] [CrossRef]
- Murphy, R.A.; Valentovic, M.A. Factors contributing to the antiviral effectiveness of tenofovir. J. Pharmacol. Exp. Ther. 2017, 363, 156–163. [Google Scholar] [CrossRef]
- Arimilli, M.N.; Cundy, K.C.; Dougherty, J.P.; Kim, C.U.; Oliyai, R.; Stella, V.J. Antiviral phosphonomethyoxy nucleotide analogs having increased oral bioavailability. International Patent Application WO9804569 A1, 5 February 1998. [Google Scholar]
- Arimilli, M.N.; Kim, C.U.; Dougherty, J.; Mulato, A.; Oliyai, R.; Shaw, J.P.; Crundy, K.U.; Bischofberger, N. Synthesis, in vitro biological evaluation and oral bioavailability of 9-[2-(phosphonomethoxy)propyl]adenine (PMPA) prodrugs. Antivir. Chem. Chemother. 1997, 8, 557–564. [Google Scholar] [CrossRef]
- Robbins, B.L.; Srinivas, R.V.; Kim, C.; Bischofberger, N.; Fridland, A. Anti-human immunodeficiency virus activity and cellular metabolism of a potential prodrug of the acyclic nucleoside phosphonate 9-R-(2-phosphonomethoxypropyl)adenine (PMPA), bis(isopropyloxymethylcarbonyl)PMPA. Antimicrob. Agents Chemother. 1998, 42, 612–617. [Google Scholar] [CrossRef]
- Grim, S.A.; Romanelli, F. Tenofovir disoproxil fumarate. Ann. Pharmacother. 2003, 37, 849–859. [Google Scholar] [CrossRef] [PubMed]
- Basit, S.A.; Dawood, A.; Ryan, J.; Gish, R. Tenofovir alafenamide for the treatment of chronic hepatitis B virus infection. Expert Rev. Clin. Pharmacol. 2017, 10, 707–716. [Google Scholar] [CrossRef]
- Hsu, Y.-C.; Wei, M.T.; Nguyen, M.H. Tenofovir alafenamide as compared to tenofovir disoproxil fumarate in the management of chronic hepatitis B with recent trends in patient demographics. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.J.; Chan, H.L.Y. Tenofovir alafenamide: A review in chronic hepatitis B. Drugs 2017, 77, 1017–1028. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, E.; Furusyo, N.; Nguyen, M.H. Tenofovir alafenamide in the treatment of chronic hepatitis B: Design, development, and place in therapy. Drug Des. Dev. Ther. 2017, 11, 3197–3204. [Google Scholar] [CrossRef]
- Childs-Kean, L.M.; Egelund, E.F.; Jourjy, J. Tenofovir alafenamide for the treatment of chronic hepatitis B monoinfection. Pharmacotherapy 2018, 38, 1051–1057. [Google Scholar] [CrossRef]
- Center for Drug Evaluation and Research Application Number: 208464Orig1s000, Labeling. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/208464Orig1s000Lbl.pdf (accessed on 15 January 2020).
- Greig, S.L.; Deeks, E.D. Elvitegravir/cobicistat/emtricitabine/tenofovir alafenamide: A review in HIV-1 infection. Drugs 2016, 76, 957–968. [Google Scholar] [CrossRef]
- Imaz, A.; Podzamczer, D. Tenofovir alafenamide, emtricitabine, elvitegravir, and cobicistat combination therapy for the treatment of HIV. Expert Rev. Anti-Infect. Ther. 2017, 15, 195–209. [Google Scholar] [CrossRef]
- Angione, S.A.; Cherian, S.M.; Özdener, A.E. A Review of the efficacy and safety of Genvoya® (elvitegravir, cobicistat, emtricitabine, and tenofovir alafenamide) in the management of HIV-1 infection. J. Pharm. Pract. 2018, 31, 216–221. [Google Scholar] [CrossRef]
- Ogbuagu, O. Rilpivirine, emtricitabine and tenofovir alafenamide: Single-tablet combination for the treatment of HIV-1 infection in selected patients. Expert Rev. Anti-Infect. Ther. 2016, 14, 1113–1126. [Google Scholar] [CrossRef]
- Corado, K.C.; Daar, E.S. Emtricitabine + tenofovir alafenamide for the treatment of HIV. Expert Opin. Pharmacother. 2017, 18, 427–432. [Google Scholar] [CrossRef]
- Negredo, E.; Clotet, B. Efficacy of single-tablet darunavir, cobicistat, emtricitabine, and tenofovir alafenamide in the treatment of HIV-1. Expert Opin. Pharmacother. 2018, 19, 929–934. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D. Darunavir/cobicistat/emtricitabine/tenofovir alafenamide: A review in HIV-1 infection. Drugs 2018, 78, 1013–1024. [Google Scholar] [CrossRef] [PubMed]
- Squillace, N.; Bozzi, G.; Colella, E.; Gori, A.; Bandera, A. Darunavir–cobicistat–emtricitabine–tenofovir alafenamide: Safety and efficacy of a protease inhibitor in the modern era. Drug Des. Dev. Ther. 2018, 12, 3635–3643. [Google Scholar] [CrossRef]
- Callebaut, C.; Stepan, G.; Tian, Y.; Miller, M.D. In vitro virology profile of tenofovir alafenamide, a novel oral prodrug of tenofovir with improved antiviral activity compared to that of tenofovir disoproxil fumarate. Antimicrob. Agents Chemother. 2015, 59, 5909–5916. [Google Scholar] [CrossRef] [PubMed]
- Gibson, A.K.; Shah, B.M.; Nambiar, P.H.; Schafer, J.J. Tenofovir Alafenamide: A review of its use in the treatment of HIV-1 infection. Ann. Pharmacother. 2016, 50, 942–952. [Google Scholar] [CrossRef]
- Shafran, S.; Di Perri, G.; Esser, S.; Lelièvre, J.-D.; Parczewski, M. Planning HIV therapy to prevent future comorbidities: Patient years for tenofovir alafenamide. HIV Med. 2019, 20, 1–16. [Google Scholar] [CrossRef]
- Cazorla-Luna, R.; Notario-Pérez, F.; Martín-Illana, A.; Ruiz-Caro, R.; Tamayo, A.; Rubio, J.; Veiga, M.D. Chitosan-based mucoadhesive vaginal tablets for controlled release of the anti-HIV drug tenofovir. Pharmaceutics 2019, 11, 20. [Google Scholar] [CrossRef]
- Notario-Pérez, F.; Cazorla-Luna, R.; Martín-Illana, A.; Ruiz-Caro, R.; Peña, J.; Veiga, M.-D. Tenofovir hot-melt granulation using Gelucire® to develop sustained-release vaginal systems for weekly protection against sexual transmission of HIV. Pharmaceutics 2019, 11, 137. [Google Scholar] [CrossRef]
- Puri, A.; Bhattaccharjee, S.A.; Zhang, W.; Clark, M.; Singh, O.N.; Doncel, G.F.; Banga, A.K. Development of a transdermal delivery system for tenofovir alafenamide, a prodrug of tenofovir with potent antiviral activity against HIV and HBV. Pharmaceutics 2019, 11, 173. [Google Scholar] [CrossRef]
- Grande, F.; Ioele, G.; Occhiuzzi, M.A.; De Luca, M.; Mazzotta, E.; Ragno, G.; Garofalo, A.; Muzzalupo, R. Reverse transcriptase inhibitors nanosystems designed for drug stability and controlled delivery. Pharmaceutics 2019, 11, 197. [Google Scholar] [CrossRef] [PubMed]
- Martín-Illana, A.; Notario-Pérez, F.; Cazorla-Luna, R.; Ruiz-Caro, R.; Veiga, M.D. Smart freeze-dried bigels for the prevention of the sexual transmission of HIV by accelerating the vaginal release of tenofovir during intercourse. Pharmaceutics 2019, 11, 232. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.M.; Krovi, S.A.; Li, L.; Girouard, N.; Demkovich, Z.R.; Myers, D.; Creelman, B.; van der Straten, A. Characterization of a reservoir-style implant for sustained release of tenofovir alafenamide (TAF) for HIV pre-exposure prophylaxis (PrEP). Pharmaceutics 2019, 11, 315. [Google Scholar] [CrossRef] [PubMed]
- Peet, M.M.; Agrahari, V.; Anderson, S.M.; Hanif, H.; Singh, O.N.; Thurman, A.R.; Doncel, G.F.; Clark, M.R. Topical Inserts: A versatile delivery form for HIV prevention. Pharmaceutics 2019, 11, 374. [Google Scholar] [CrossRef]
- Faria, M.J.; Machado, R.; Ribeiro, A.; Gonçalves, H.; Real Oliveira, M.E.C.D.; Viseu, T.; das Neves, J.; Lúcio, M. Rational development of liposomal hydrogels: A strategy for topical vaginal antiretroviral drug delivery in the context of HIV prevention. Pharmaceutics 2019, 11, 485. [Google Scholar] [CrossRef]
- Polymorphism in Pharmaceutical Solids, 2nd ed.; Brittain, H.G., Ed.; Informa Healthcare Inc.: New York, NY, USA, 1999. [Google Scholar]
- Polymorphism in the Pharmaceutical Industry: Solid Form and Drug Development; Hilfiker, R., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006. [Google Scholar]
- Pharmaceutical Salts and Co-crystals; Wouters, J., Quéré, L., Eds.; Royal Society of Chemistry: Cambridge, UK, 2011. [Google Scholar]
- Polymorphism in the Pharmaceutical Industry: Solid Form and Drug Development, 2nd ed.; Hilfiker, R., von Raumer, M., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2019. [Google Scholar]
- Yu, L.X.; Furness, M.S.; Raw, A.; Woodland Outlaw, K.P.; Nashed, N.E.; Ramos, E.; Miller, S.P.F.; Adams, R.C.; Fang, F.; Patel, R.M.; et al. Scientific considerations of pharmaceutical solid polymorphism in abbreviated new drug applications. Pharm. Res. 2003, 20, 531–536. [Google Scholar] [CrossRef]
- Herbstein, F.H. Diversity amidst similarity: a multidisciplinary approach to phase relationships, solvates, and polymorphs. Cryst. Growth Des. 2004, 4, 1419–1429. [Google Scholar] [CrossRef]
- Nangia, A. Pseudopolymorph: retain this widely accepted term. Cryst. Growth Des. 2006, 6, 2–4. [Google Scholar] [CrossRef]
- Stahly, G.P. Diversity in single- and multiple-component crystals. The search for and prevalence of polymorphs and cocrystals. Cryst. Growth Des. 2007, 7, 1007–1026. [Google Scholar] [CrossRef]
- Desiraju, G.R. Crystal engineering: A holistic view. Angew. Chem. Int. Ed. 2007, 46, 8342–8356. [Google Scholar] [CrossRef]
- Lu, J.; Li, Z.; Jiang, X. Polymorphism of pharmaceutical molecules: Perspectives on nucleation. Front. Chem. Eng. China 2010, 4, 37–44. [Google Scholar] [CrossRef]
- Palucki, M.; Higgins, J.D.; Kwong, E.; Templeton, A.C. Strategies at the interface of drug discovery and development: Early optimization of the solid state phase and preclinical toxicology formulation for potential drug candidates. J. Med. Chem. 2010, 53, 5897–5905. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, J. Polymorphism—A perspective. Cryst. Growth Des. 2011, 11, 632–650. [Google Scholar] [CrossRef]
- Aitipamula, S.; Banerjee, R.; Bansal, A.K.; Biradha, K.; Cheney, M.L.; Choudhury, A.R.; Desiraju, G.R.; Dikundwar, A.G.; Dubey, R.; Duggirala, N.; et al. Polymorphs, Salts, and Cocrystals: What’s in a Name? Cryst. Growth Des. 2012, 12, 2147–2152. [Google Scholar] [CrossRef]
- Desiraju, G.R. Crystal engineering: From molecule to crystal. J. Am. Chem. Soc. 2013, 135, 9952–9967. [Google Scholar] [CrossRef]
- Cruz-Cabeza, A.J.; Bernstein, J. Conformational polymorphism. Chem. Rev. 2014, 114, 2170–2191. [Google Scholar] [CrossRef]
- Cruz-Cabeza, A.J.; Reutzel-Edens, S.M.; Bernstein, J. Facts and fictions about polymorphism. Chem. Soc. Rev. 2015, 44, 8619–8635. [Google Scholar] [CrossRef]
- Censi, R.; Di Martino, P. Polymorph impact on the bioavailability and stability of poorly soluble drugs. Molecules 2015, 20, 18759–18776. [Google Scholar] [CrossRef]
- Higashi, K.; Ueda, K.; Moribe, K. Recent progress of structural study of polymorphic pharmaceutical drugs. Adv. Drug Deliv. Rev. 2017, 117, 71–85. [Google Scholar] [CrossRef]
- Qiao, Y.; Qiao, R.; He, Y.; Shi, C.; Liu, Y.; Hao, H.; Su, J.; Zhong, J. Instrumental analytical techniques for the characterization of crystals in pharmaceutics and foods. Cryst. Growth Des. 2017, 17, 6138–6148. [Google Scholar] [CrossRef]
- Newman, A.; Chen, C.; Sanrame, C. Salt and cocrystal screening. In Early Drug Development: Bringing a Preclinical Candidate to the Clinic, 1st ed.; Giordanetto, F., Ed.; Wiley-VCH Verlag GmbH & Co.KGaA: Weinheim, Germany, 2018; Volume 1, pp. 229–270. [Google Scholar] [CrossRef]
- Jones, W.; Motherwell, W.; Trask, A. Pharmaceutical cocrystals: An emerging approach to physical property enhancement. MRS Bull. 2006, 31, 875–879. [Google Scholar] [CrossRef]
- Trask, A.V. An overview of pharmaceutical cocrystals as intellectual property. Mol. Pharm. 2007, 4, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Aakeröy, C.B.; Fasulo, M.E.; Desper, J. Cocrystal or salt: Does it really matter? Mol. Pharm. 2007, 4, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Childs, S.L.; Stahly, G.P.; Park, A. The salt-cocrystal continuum: The influence of crystal structure on ionization state. Mol. Pharm. 2007, 4, 323–338. [Google Scholar] [CrossRef]
- Nangia, A. Crystal engineering—inclusion compounds, molecular networks, cocrystals, and polymorphs. Cryst. Growth Des. 2008, 8, 1079–1081. [Google Scholar] [CrossRef]
- Miroshnyk, I.; Mirza, S.; Sandler, N. Pharmaceutical co-crystals–an opportunity for drug product enhancement. Expert Opin. Drug Deliv. 2009, 6, 333–341. [Google Scholar] [CrossRef]
- Schultheiss, N.; Newman, A. Pharmaceutical cocrystals and their physicochemical properties. Cryst. Growth Des. 2009, 9, 2950–2967. [Google Scholar] [CrossRef]
- Babu, N.J.; Nangia, A. Solubility Advantage of Amorphous Drugs and Pharmaceutical Cocrystals. Cryst. Growth Des. 2011, 11, 2662–2679. [Google Scholar] [CrossRef]
- Elder, D.P.; Holm, R.; Lopez de Diego, H. Use of pharmaceutical salts and cocrystals to address the issue of poor solubility. Int. J. Pharm. 2013, 453, 88–100. [Google Scholar] [CrossRef]
- Sun, C.C. Cocrystallization for successful drug delivery. Expert Opin. Drug Deliv. 2013, 10, 201–213. [Google Scholar] [CrossRef]
- Brittain, H.G. Pharmaceutical cocrystals: The coming wave of new drug substances. J. Pharm. Sci. 2013, 102, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Kelley, S.P.; Narita, A.; Holbrey, J.D.; Green, K.D.; Reichert, W.M.; Rogers, R.D. Understanding the effects of ionicity in salts, solvates, co-crystals, ionic co-crystals, and ionic liquids, rather than nomenclature, is critical to understanding their behavior. Cryst. Growth Des. 2013, 13, 965–975. [Google Scholar] [CrossRef]
- Korotkova, E.I.; Kratochvíl, B. Pharmaceutical cocrystals. Procedia Chem. 2014, 10, 473–476. [Google Scholar] [CrossRef]
- Stoler, E.; Warner, J.C. Non-covalent derivatives: Cocrystals and eutectics. Molecules 2015, 20, 14833–14848. [Google Scholar] [CrossRef] [PubMed]
- Kuminek, G.; Cao, F.; de Oliveira da Rocha, A.B.; Cardoso, S.G.; Rodríguez-Hornedo, N. Cocrystals to facilitate delivery of poorly soluble compounds beyond-rule-of-5. Adv. Drug Deliv. Rev. 2016, 101, 143–166. [Google Scholar] [CrossRef]
- Pratik, S.M.; Datta, A. Nonequimolar mixture of organic acids and bases: An exception to the rule of thumb for salt or cocrystal. J. Phys. Chem. B 2016, 120, 7606–7613. [Google Scholar] [CrossRef]
- Berry, D.J.; Steed, J.W. Pharmaceutical cocrystals, salts and multicomponent systems; intermolecular interactions and property based design. Adv. Drug Deliv. Rev. 2017, 117, 3–24. [Google Scholar] [CrossRef]
- Healy, A.M.; Worku, Z.A.; Kumar, D.; Madi, A.M. Pharmaceutical solvates, hydrates and amorphous forms: A special emphasis on cocrystals. Adv. Drug Deliv. Rev. 2017, 117, 25–46. [Google Scholar] [CrossRef]
- Cerreia Vioglio, P.; Chierotti, M.R.; Gobetto, R. Pharmaceutical aspects of salt and cocrystal forms of APIs and characterization challenges. Adv. Drug Deliv. Rev. 2017, 117, 86–110. [Google Scholar] [CrossRef]
- Pindelska, E.; Sokal, A.; Kolodziejski, W. Pharmaceutical cocrystals, salts and polymorphs: Advanced characterization techniques. Adv. Drug Deliv. Rev. 2017, 117, 111–146. [Google Scholar] [CrossRef]
- Kale, D.P.; Zode, S.S.; Bansal, A.K. Challenges in translational development of pharmaceutical cocrystals. J. Pharm. Sci. 2017, 106, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Rajput, L.; Banik, M.; Yarava, J.R.; Joseph, S.; Pandey, M.K.; Nishiyama, Y.; Desiraju, G.R. Exploring the salt–cocrystal continuum with solidstate NMR using natural-abundance samples: Implications for crystal engineering. IUCrJ 2017, 4, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Nanda, A. Approaches to design of pharmaceutical cocrystals: A review. Mol. Cryst. Liq. Cryst. 2018, 667, 54–77. [Google Scholar] [CrossRef]
- Cavanagh, K.L.; Maheshwari, C.; Rodríguez-Hornedo, N. Understanding the differences between cocrystal and salt aqueous solubilities. J. Pharm. Sci. 2018, 107, 113–120. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Orton, E.; Yang, D. The selection of a pharmaceutical salt—the effect of the acidity of the counterion on its solubility and potential biopharmaceutical performance. J. Pharm. Sci. 2018, 107, 419–425. [Google Scholar] [CrossRef]
- Thakuria, R.; Sarma, B. Drug-drug and drug-nutraceutical cocrystal/salt as alternative medicine for combination therapy: A crystal engineering approach. Crystals 2018, 8, 101. [Google Scholar] [CrossRef]
- Shaikh, R.; Singh, R.; Walker, G.M.; Croker, D.M. Pharmaceutical cocrystal drug products: An outlook on product development. Trends Pharmacol. Sci. 2018, 39, 1033–1048. [Google Scholar] [CrossRef]
- Zhao, L.; Hanrahan, M.P.; Chakravarty, P.; DiPasquale, A.G.; Sirois, L.E.; Nagapudi, K.; Lubach, J.W.; Rossini, A.J. Characterization of pharmaceutical cocrystals and salts by dynamic nuclear polarization-enhanced solid-state NMR spectroscopy. Cryst. Growth Des. 2018, 18, 2588–2601. [Google Scholar] [CrossRef]
- Karimi-Jafari, M.; Padrela, L.; Walker, G.M.; Croker, D.M. Creating cocrystals: A review of pharmaceutical cocrystal preparation routes and applications. Cryst. Growth Des. 2018, 18, 6370–6387. [Google Scholar] [CrossRef]
- Cao, H.-L.; Zhou, J.-R.; Cai, F.-Y.; Lü, J.; Cao, R. Two-component pharmaceutical cocrystals regulated by supramolecular synthons comprising primary N···H···O interactions. Cryst. Growth Des. 2019, 19, 3–16. [Google Scholar] [CrossRef]
- Zhang, C.; Xiong, Y.; Jiao, F.; Wang, M.; Li, H. Redefining the term of “cocrystal” and broadening its intention. Cryst. Growth Des. 2019, 19, 1471–1478. [Google Scholar] [CrossRef]
- Yousef, M.A.E.; Vangala, V.R. Pharmaceutical cocrystals: Molecules, crystals, formulations, medicines. Cryst. Growth Des. 2019, 19, 7420–7438. [Google Scholar] [CrossRef]
- 3 Cocrystal API’s Now on the Market. Available online: https://www.scientificupdate.com/process-chemistry-articles/3-cocrystal-apis-now-on-the-market/ (accessed on 18 January 2020).
- Lee, E.H.; Smith, D.T.; Fanwick, P.E.; Byrn, S.R. Characterization and anisotropic lattice expansion/contraction of polymorphs of tenofovir disoproxil fumarate. Cryst. Growth Des. 2010, 10, 2314–2322. [Google Scholar] [CrossRef]
- Lee, J.; Boerrigter, S.X.M.; Jung, Y.W.; Byun, Y.; Yuk, S.H.; Byrn, S.R.; Lee, E.H. Organic vapor sorption method of isostructural solvates and polymorph of tenofovir disoproxil fumarate. Eur. J. Pharm. Sci. 2013, 50, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Sládková, V.; Dammer, O.; Kratochvíl, B. Solid forms of tenofovir disoproxil fumarate, their mutual conversion, and stabilization of form I in formulation. J. Pharm. Sci. 2016, 105, 3136–3142. [Google Scholar] [CrossRef] [PubMed]
- Gomes, E.C.D.; Mussel, W.N.; Resende, J.M.; Fialho, S.L.; Barbosa, J.; Carignani, E.; Geppi, M.; Yoshida, M.I. Characterization of tenofovir disoproxil fumarate and its behavior under heating. Cryst. Growth Des. 2015, 15, 1915–1922. [Google Scholar] [CrossRef]
- An, J.-H.; Kiyonga, A.N.; Yoon, W.; Ryu, H.C.; Kim, J.-S.; Kang, C.; Park, M.; Yun, H.; Jung, K. Crystal structure analysis of the first discovered stability-enhanced solid state of tenofovir disoproxil free base using single crystal X-ray diffraction. Molecules 2017, 22, 1182. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Choi, H.-G. Development of novel tenofovir disoproxil phosphate salt with stability enhancement and bioequivalence to the commercial tenofovir disoproxil fumarate salt in rats and beagle dogs. Int. J. Pharm. 2019. [Google Scholar] [CrossRef]
- Becker, M.W.; Chapman, H.H.; Cihlar, T.; Eisenberg, E.J.; He, G.-X.; Kernan, M.R.; Lee, W.A.; Prisbe, E.J.; Rohloff, J.C.; Sparacino, M.L. Prodrugs of phosphonate nucleotide analogues and methods for selecting and making same. International Patent Application WO02008241 A2, 31 January 2002. [Google Scholar]
- Liu, D.; Shi, B.; Wang, F.; Yu, R.H.C. Tenofovir alafenamide hemifumarate. International Patent Application WO2013025788 A1, 21 February 2013. [Google Scholar]
- Hotter, A.; Lengauer, H. Crystalline forms of tenofovir alafenamide monofumarate. International Patent Application WO2017134089 A1, 10 August 2017. [Google Scholar]
- Camargo, A.P.M.; Baumgärtel, H.; Donner, C. Coadsorption of the DNA bases thymine and adenine at the Au(111) electrode. Phys. Chem. Chem. Phys. 2003, 5, 1657–1664. [Google Scholar] [CrossRef]
- Gowri, S.; Devi, T.U.; Sajan, D.; Lawrence, N. Spectral, thermal and optical properties of adenosinium picrate: A nonlinear optical single crystal. Spectrochim. Acta Part A 2012, 89, 119–122. [Google Scholar] [CrossRef]
- Prieto, F.; Alvarez-Malmagro, J.; Rueda, M. Electrochemical Impedance Spectroscopy study of the adsorption of adenine on Au(111) electrodes as a function of the pH. J. Electroanal. Chem. 2017, 793, 209–217. [Google Scholar] [CrossRef]
- Sedghiniya, S.; Soleimannejad, J.; Janczak, J. The salt–cocrystal spectrum in salicylic acid–adenine: The influence of crystal structure on proton-transfer balance. Acta Cryst. C 2019, 75, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Popović-Nikolić, M.R.; Popović, G.V.; Stojilković, K.; Dobrosavljević, M.; Agbaba, D.D. Acid–base equilibria of rupatadine fumarate in aqueous media. J. Chem. Eng. Data 2018, 63, 3150–3156. [Google Scholar] [CrossRef]
- Regulatory Classification of Pharmaceutical Co-crystals. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/regulatory-classification-pharmaceutical-co-crystals (accessed on 19 January 2020).
- Lee, M.; Goldburg, W. Nuclear magnetic resonance line narrowing by a rotating RF field. Phys. Rev. 1965, 140, 1261–1271. [Google Scholar] [CrossRef]
- Ladizhansky, V.; Vega, S. Polarization transfer dynamics in Lee– Goldburg cross polarization under magnetic resonance experiments on rotating solids. J. Chem. Phys. 2000, 112, 7158–7168. [Google Scholar] [CrossRef]
- van Rossum, B.-J.; de Groot, C.P.; Ladizhansky, V.; Vega, S.; de Groot, H.J.M. A method for measuring heteronuclear (1H– 13C) distances in high speed MAS NMR. J. Am. Chem. Soc. 2000, 122, 3465–3472. [Google Scholar] [CrossRef]
- Agilent Technologies Ltd. CrysAlisPro, Version 1.171.36.28; Agilent Technologies: Yarnton, UK, 2013; Available online: https://www.agilent.com/cs/library/usermanuals/Public/CrysAlis_Pro_User_Manual.pdf (accessed on 12 March 2020).
- Palatinus, L.; Chapuis, G. Superflip—A computer program for the solution of crystal structures by charge flipping in arbitrary dimensions. J. Appl. Crystallogr. 2007, 40, 786–790. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Chapman, H.; Kernan, M.; Prisbe, E.; Rohloff, J.; Sparacino, M.; Terhorst, T.; Yu, R. Practical synthesis, separation, and stereochemical assignment of the PMPA pro-drug GS-7340. Nucleosides Nucleotides Nucleic Acids 2001, 20, 621–628. [Google Scholar] [CrossRef]
- Dammer, O.; Skalicka, T.; Krejcik, L.; Pekarek, T.; Ridvan, L. Solid forms of Tenofovir alafenamide. International Patent Application WO2016192692 A1, 8 December 2016. [Google Scholar]
- Cardew, P.T.; Davey, R.J. The kinetics of solvent-mediated phase transformations. Proc. R. Soc. Lond. A 1985, 398, 415–428. [Google Scholar]
- Ferrari, E.S.; Davey, R.J.; Cross, W.I.; Gillon, A.L.; Towler, C.S. Crystallization in polymorphic systems: the solution-mediated transformation of β to α glycine. Cryst. Growth Des. 2003, 3, 53–60. [Google Scholar] [CrossRef]
- Saranteas, K.; Bakale, R.; Hong, Y.; Luong, H.; Foroughi, R.; Wald, S. Process design and scale-up elements for solvent mediated polymorphic controlled tecastemizole crystallization. Org. Process Res. Dev. 2005, 9, 911–922. [Google Scholar] [CrossRef]
- Bernstein, J.; Davis, R.E.; Shimoni, L.; Chang, N.L. Patterns in hydrogen bonding: Functionality and graph set analysis in crystals. Angew. Chem. Int. Ed. 1995, 34, 1555–1573. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TA | TA HF | |
|---|---|---|
| CCDC number | 1990630 | 1990631 |
| Formula | C21H29N6O5P | C46H62N12O14P2 |
| Mr | 476.47 | 1069.01 |
| T (K) | 293(2) K | 150(2) K |
| Crystal system | Orthorhombic | Tetragonal |
| Space group | P212121 | P42212 |
| a (Å) | 8.4062(2) Å | 18.0407(2) |
| b (Å) | 15.7401(3) | 18.0407(2) |
| c (Å) | 18.2196(4) | 17.5003(2) |
| Volume (Å3) | 2410.72(9) | 5695.77(14) |
| Z | 4 | 4 |
| Dc (g/cm3) | 1.313 | 1.247 |
| μ (mm–1) | 1.385 | 1.283 |
| F(000) | 1008 | 2256 |
| Reflections collected | 9201 | 23148 |
| Independent reflections (Rint) | 4403 (0.0336) | 5834 (0.0316) |
| Data/restraints/parameters | 4403 / 3 / 312 | 5834 / 3 / 432 |
| R, wR2 [I > 2σ(I)] a | 0.0380, 0.0966 | 0.0383, 0.0976 |
| R, wR2 (all data) a | 0.0452, 0.1013 | 0.0430, 0.1029 |
| GOF, S b | 1.024 | 1.016 |
| Largest diff. peak/hole / e Å−3 | 0.299/−0.200 | 0.177/−0.369 |
| Flack parameter | 0.023(17) | −0.020(10) |
| Sample | Isolated | Result according to powder X-ray diffraction |
|---|---|---|
| A | 6 days | TA MF2 + traces TA MF1 |
| B | 10 days | TA MF2 |
| C | 11 days | TA MF2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lengauer, H.; Makuc, D.; Šterk, D.; Perdih, F.; Pichler, A.; Trdan Lušin, T.; Plavec, J.; Časar, Z. Co-crystals, Salts or Mixtures of Both? The Case of Tenofovir Alafenamide Fumarates. Pharmaceutics 2020, 12, 342. https://doi.org/10.3390/pharmaceutics12040342
Lengauer H, Makuc D, Šterk D, Perdih F, Pichler A, Trdan Lušin T, Plavec J, Časar Z. Co-crystals, Salts or Mixtures of Both? The Case of Tenofovir Alafenamide Fumarates. Pharmaceutics. 2020; 12(4):342. https://doi.org/10.3390/pharmaceutics12040342
Chicago/Turabian StyleLengauer, Hannes, Damjan Makuc, Damjan Šterk, Franc Perdih, Arthur Pichler, Tina Trdan Lušin, Janez Plavec, and Zdenko Časar. 2020. "Co-crystals, Salts or Mixtures of Both? The Case of Tenofovir Alafenamide Fumarates" Pharmaceutics 12, no. 4: 342. https://doi.org/10.3390/pharmaceutics12040342
APA StyleLengauer, H., Makuc, D., Šterk, D., Perdih, F., Pichler, A., Trdan Lušin, T., Plavec, J., & Časar, Z. (2020). Co-crystals, Salts or Mixtures of Both? The Case of Tenofovir Alafenamide Fumarates. Pharmaceutics, 12(4), 342. https://doi.org/10.3390/pharmaceutics12040342