Changing the Drug Delivery System: Does it Add to Non-Compliance Ramifications Control? A Simulation Study on the Pharmacokinetics and Pharmacodynamics of Atypical Antipsychotic Drug


Abstract
:
1. Introduction
2. Methods
2.1. Population Pharmacokinetic (PK) Model
2.2. Population Pharmacodynamics (PD) Model
2.3. Simulation Scenarios
- A.
- Dose adherence: The patient administers 200 mg of the IR formulation every 12 h (200 mg BID) and 400 mg of the XR formulation every 24 h (400 mg QD) on a regular basis without mistakes or cessation;
- B.
- Dose delay: One dose is administered at 25%, 50%, 75%, and 100% of the dosing interval (equivalent to 3, 6, 9, and 12 h for the BID and 6, 12, 18, and 24 h for the QD), with the next dose to be administered as scheduled;
- C.
- Dose omission: One dose is missed without replacement;
- D.
- Dose doubling: One dose is replicated twice by mistake;
- E.
- Dose discontinuation: The patient stops drug administration.
2.4. Monte-Carlo Simulations
2.5. Simulation Outcomes
- (1)
- Cmax: Peak plasma concentration at steady-state;
- (2)
- Cmin: Trough plasma concentration at steady-state;
- (3)
- BPRS)max: Peak %improvement (reduction) in BPRS scores from baseline at steady-state;
- (4)
- BPRS)min: Trough %improvement (reduction) in BPRS scores from baseline at steady-state;
- (5)
- ΔCmax, ΔCmin, ΔBPRS)max, ΔBPRS)min: Δ refers to %change in corresponding parameter value due to irregular administration relative to its value under regular administration conditions. In the case of non-adherence scenarios, the parameters were calculated after the administration of the next scheduled dose;
- (6)
- Trecovery: Time period required to restore regular steady-state plasma levels after the omission of a dose;
- (7)
- Therapeutic duration: Time period during which %reduction in BPRS scores from baseline exceeds 14%;
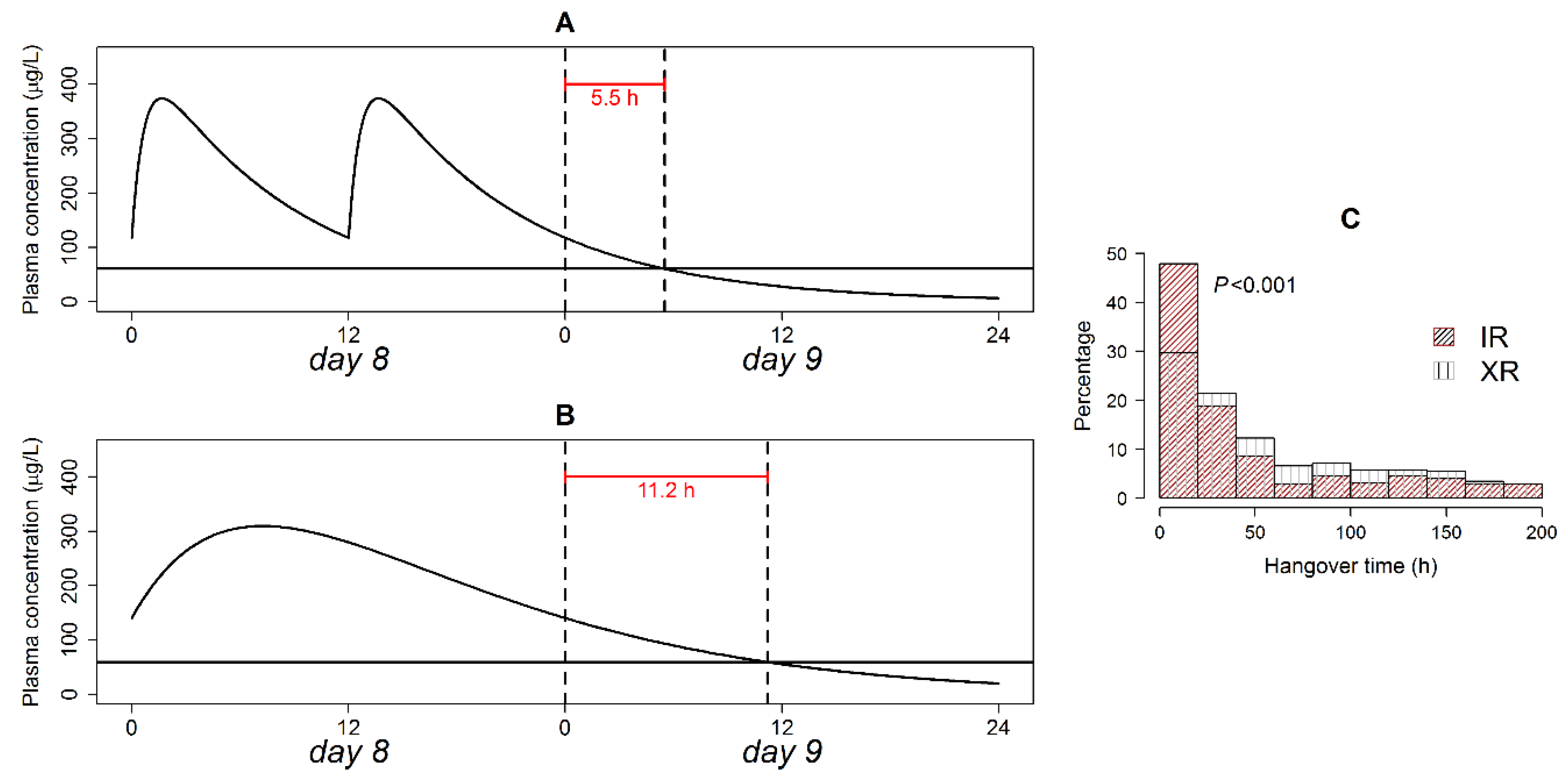
- (8)
- Thangover: The difference between Therapeutic duration and dosing interval when dose administration is discontinued;
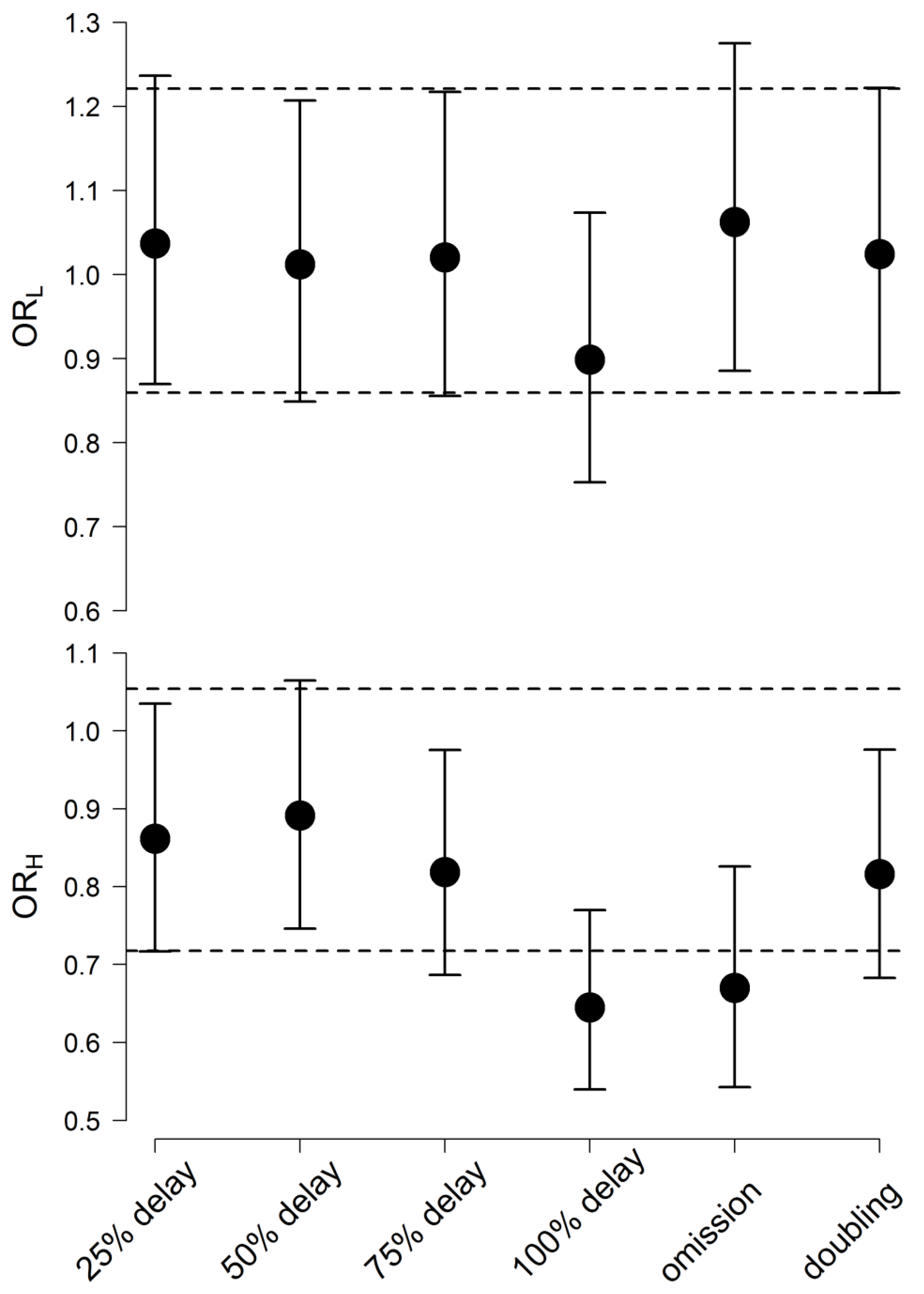
- (9)
- ORL: Odds ratio of obtaining %reduction in BPRS scores from a baseline of less than 14% in a patient on the XR versus a patient on the IR formulation;
- (10)
- ORH: Odds ratio of obtaining Quetiapine plasma concentrations of more than 500 µg/L in a patient on the XR versus a patient on the IR formulation.
3. Results
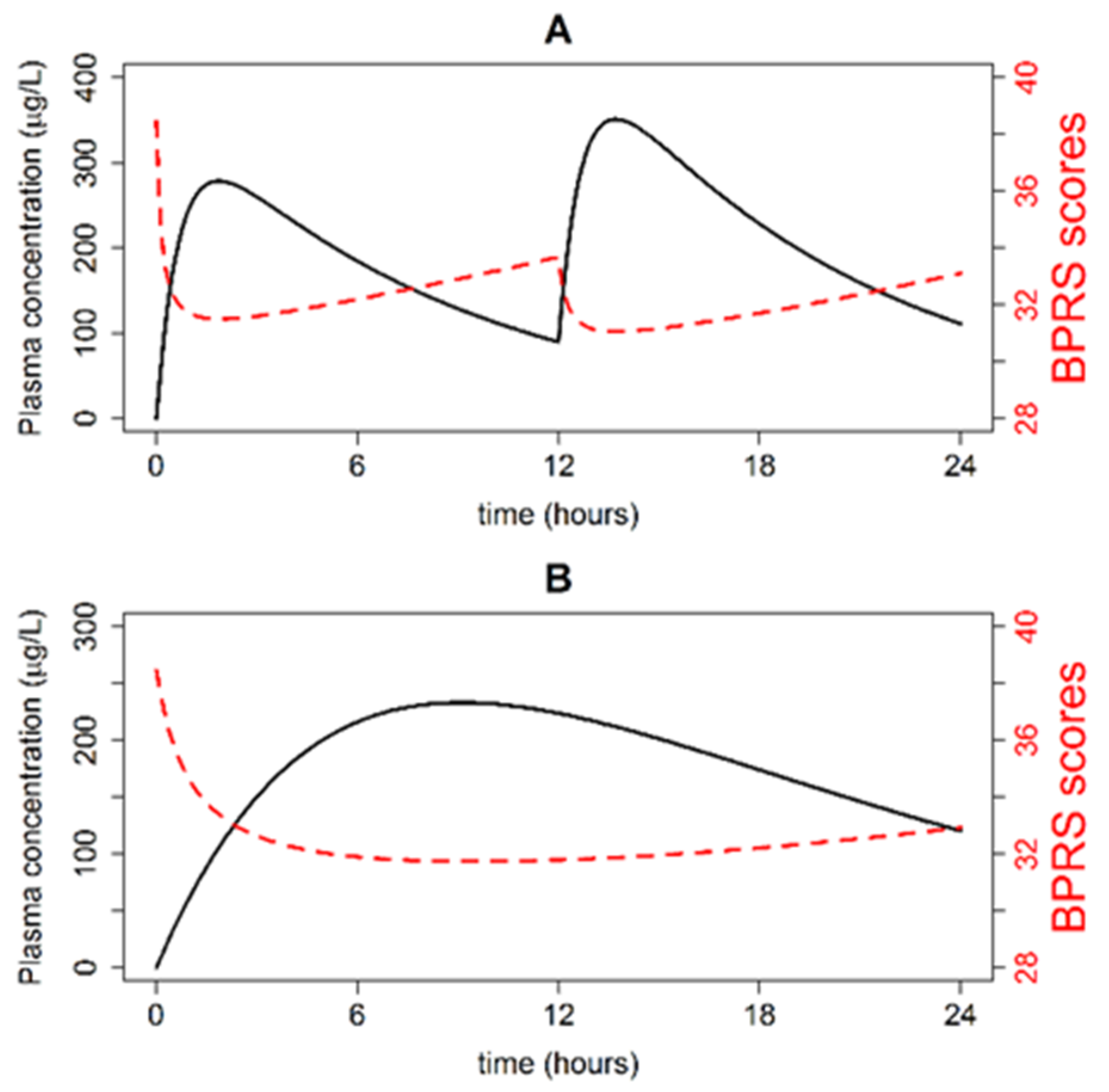
3.1. Simulations of Quetiapine Pre-Steady State PK/PD Profiles and Parameters
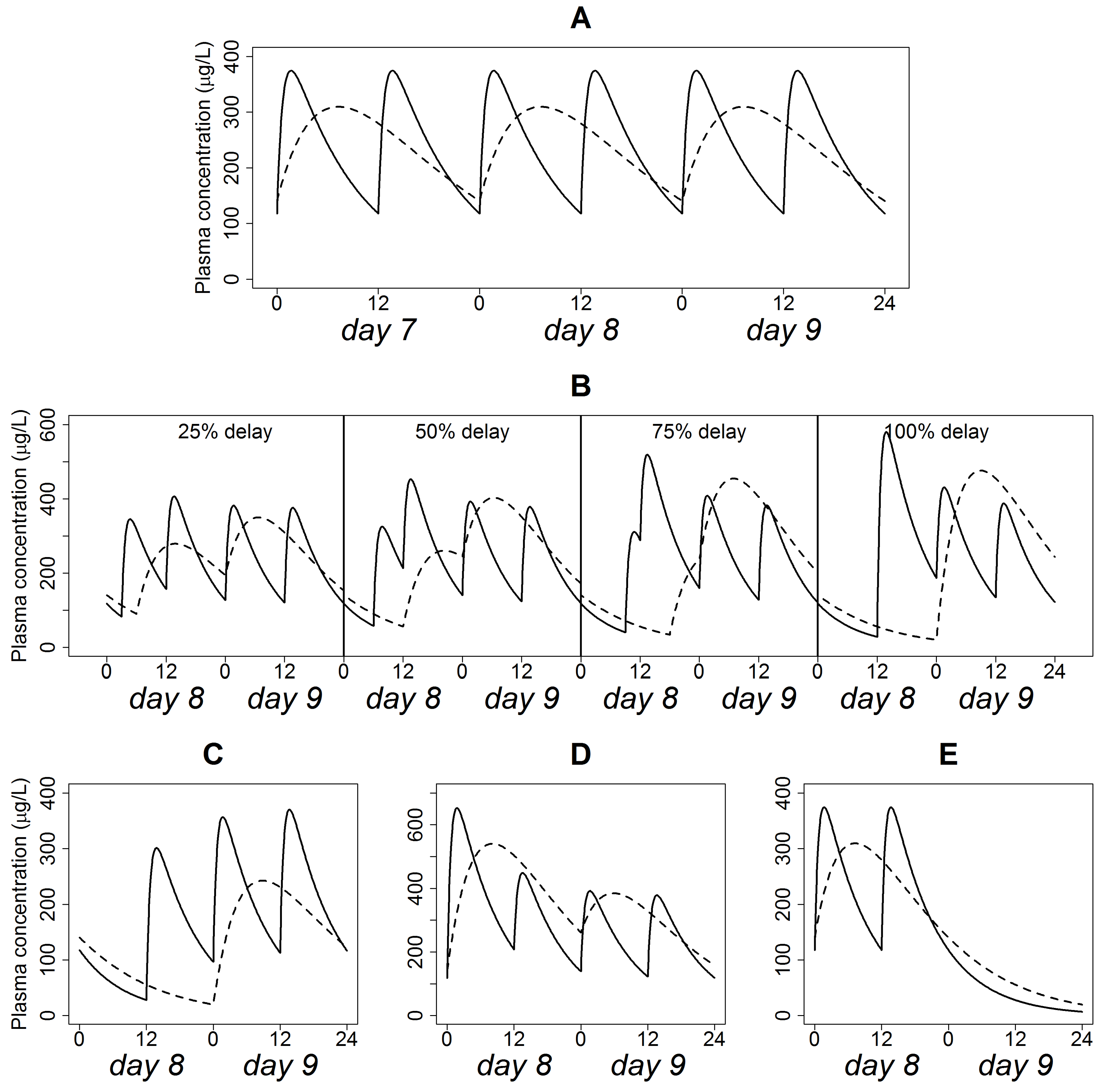
3.2. Simulations of Typical Steady-State PK Profiles
3.3. Simulations of Delayed Doses
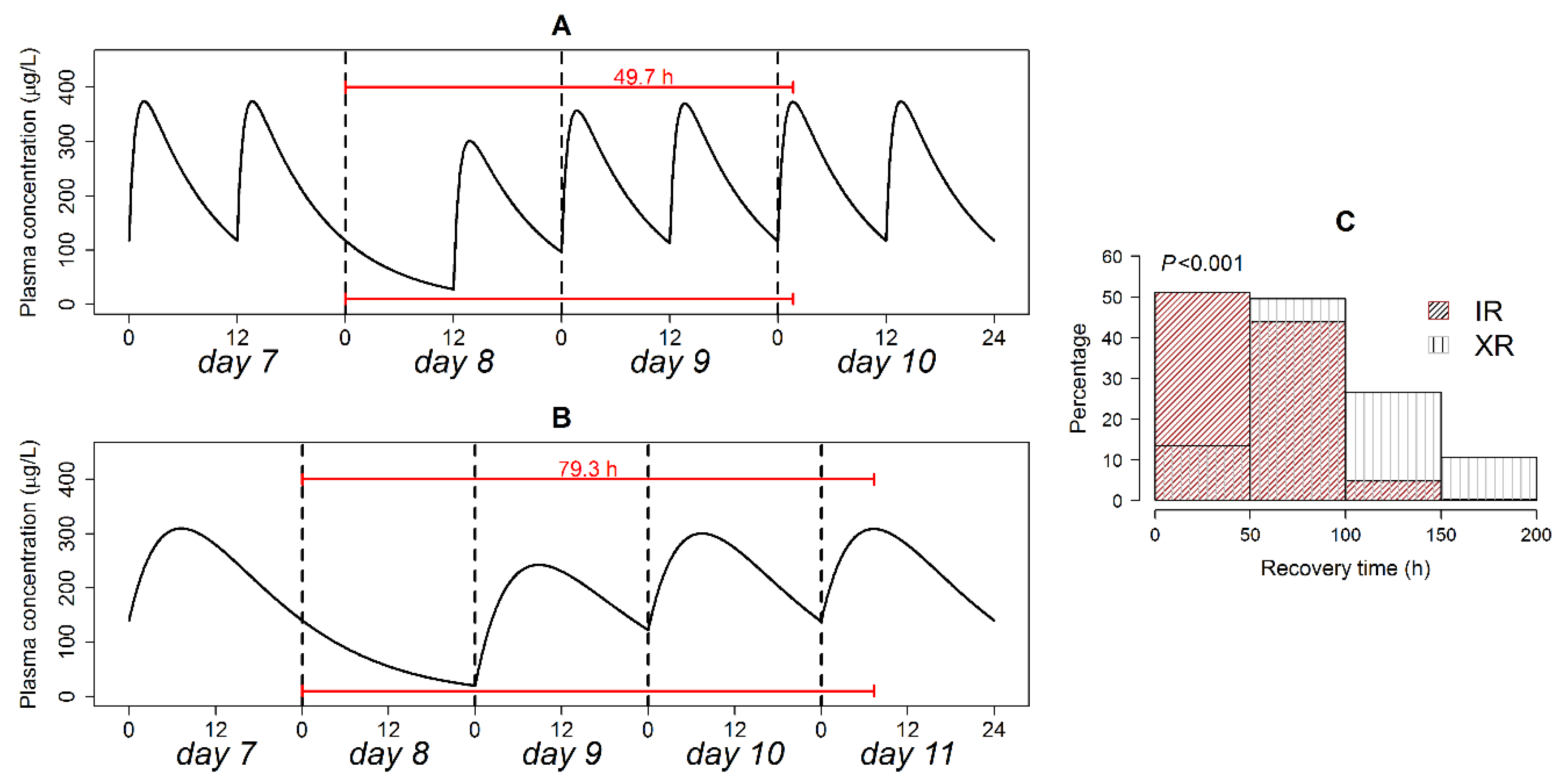
3.4. Simulation of Omitted Dose
3.5. Simulation of Doubled Dose
3.6. Simulations of Discontinued Administration
3.7. Risk Analysis
3.8. Therapeutic Plasma Concentration Hangover
3.9. Post-Irregularity Steady-State Recovery
4. Discussion
5. Conclusions
Supplementary Materials
Funding
Conflicts of Interest
References
- Haddad, P.M.; Brain, C.; Scott, J. Nonadherence with antipsychotic medication in schizophrenia: Challenges and management strategies. Patient Relat. Outcome Meas. 2014, 5, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barkhof, E.; Meijer, C.J.; de Sonneville, L.M.J.; Linszen, D.H.; de Haan, L. Interventions to improve adherence to antipsychotic medication in patients with schizophrenia—A review of the past decade. Eur. Psychiatry 2012, 27, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byerly, M.J.; Nakonezny, P.A.; Lescouflair, E. Antipsychotic medication adherence in schizophrenia. Psychiatr. Clin. N. Am. 2007, 30, 437–452. [Google Scholar] [CrossRef] [PubMed]
- Higashi, K.; Medic, G.; Littlewood, K.J.; Diez, T.; Granström, O.; De Hert, M. Medication adherence in schizophrenia: Factors influencing adherence and consequences of nonadherence, a systematic literature review. Ther. Adv. Psychopharmacol. 2013, 3, 200–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shuler, K.M. Approaches to improve adherence to pharmacotherapy in patients with schizophrenia. Patient Prefer. Adherence 2014, 8, 701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieberman, J.A.; Stroup, T.S.; McEvoy, J.P.; Swartz, M.S.; Rosenheck, R.A.; Perkins, D.O.; Keefe, R.; Thomas, S.M.; Davis, C.E.; Lebowit, B.D.; et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N. Engl. J. Med. 2005, 353, 1209–1223. [Google Scholar] [CrossRef] [Green Version]
- Alene, M.; Wiese, M.D.; Angamo, M.T.; Bajorek, B.V.; Yesuf, E.A.; Wabe, N.T. Adherence to medication for the treatment of psychosis: Rates and risk factors in an Ethiopian population. BMC Clin. Pharmacol. 2012, 12, 10. [Google Scholar] [CrossRef] [Green Version]
- Sajatovic, M.; Levin, J.; Fuentes-Casiano, E.; Cassidy, K.A.; Tatsuoka, C.; Jenkins, J.H. Illness experience and reasons for nonadherence among individuals with bipolar disorder who are poorly adherent with medication. Compr. Psychiatry. 2011, 52, 280–287. [Google Scholar] [CrossRef] [Green Version]
- Bialer, M. Extended-release formulations for the treatment of epilepsy. CNS Drugs 2007, 21, 765–774. [Google Scholar] [CrossRef]
- Brittain, S.T.; Wheless, J.W. Pharmacokinetic simulations of topiramate plasma concentrations following dosing irregularities with extended-release vs. immediate-release formulations. Epilepsy Behav. 2015, 52, 31–36. [Google Scholar] [CrossRef] [Green Version]
- DeVane, C.L.; Nemeroff, C.B. Clinical pharmacokinetics of quetiapine. Clin. Pharmacokinet. 2001, 40, 509–522. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Bui, K.H.; Li, J.; Al-Huniti, N. Population pharmacokinetic modeling of quetiapine after administration of seroquel and seroquel XR formulations to Western and Chinese patients with schizophrenia, schizoaffective disorder, or bipolar disorder. J. Clin. Pharmacol. 2015, 55, 1248–1255. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, C.; Brecher, M.; Hamer-Maansson, J.E.; Winter, H. Pharmacokinetic profiles of extended release quetiapine fumarate compared with quetiapine immediate release. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2009, 33, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Zhang, Y.; Jiao, Z.; Wang, Y. The effect of poor compliance on the pharmacokinetics of carbamazepine and its epoxide metabolite using Monte Carlo simulation. Acta Pharmacol Sin. 2012, 33, 1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkomy, M.H.; Drover, D.R.; Hammer, G.B.; Galinkin, J.L.; Ramamoorthy, C. Population pharmacokinetics of ketamine in children with heart disease. Int. J. Pharm. 2015, 478, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Elkomy, M.H.; Drover, D.R.; Galinkin, J.L.; Hammer, G.B.; Glotzbach, K.L. Pharmacodynamic analysis of morphine time-to-remedication events in infants and young children after congenital heart surgery. Clin Pharmacokinet. 2016, 55, 1217–1226. [Google Scholar] [CrossRef]
- Brookfield, K.F.; Elkomy, M.; Su, F.; Drover, D.R.; Carvalho, B. Optimization of Maternal Magnesium Sulfate Administration for Fetal Neuroprotection: Application of a Prospectively Constructed Pharmacokinetic Model to the BEAM Cohort. J. Clin. Pharmacol. 2017, 57, 1419–1424. [Google Scholar] [CrossRef]
- Elkomy, M.H.; Sultan, P.; Carvalho, B.; Peltz, G.; Wu, M.; Clavijo, C.; Galinkin, J.L.; Drover, D.R. Ondansetron pharmacokinetics in pregnant women and neonates: Towards a new treatment for neonatal abstinence syndrome. Clin. Pharmacol. Ther. 2015, 97, 167–176. [Google Scholar] [CrossRef] [Green Version]
- Elkomy, M.H.; Alruwaili, N.; Elmowafy, M.; Shalaby, K.; Drover, D.R.; Ramamoorthy, C. Assessment of Ketamine Adult Anesthetic Doses in Pediatrics using Pharmacokinetic Modeling and Simulations. Pharm. J. Hum. Pharmacol. Drug. Ther. 2019, 39, 454–462. [Google Scholar] [CrossRef]
- Damian, M.A.; Hammer, G.B.; Elkomy, M.H.; Frymoyer, A.; Drover, D.R.; Su, F. Pharmacokinetics of dexmedetomidine in infants and children after orthotopic liver transplantation. Anesth. Analg. 2020, 130, 209–216. [Google Scholar] [CrossRef]
- Carral, N.; Lukas, J.C.; Oteo, I.; Suarez, E. Impact of poor compliance with levofloxacin and moxifloxacin on respiratory tract infection antimicrobial efficacy: A pharmacokinetic/pharmacodynamic simulation study. Int. J. Antimicrob. Agents. 2015, 45, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Pellock, J.M.; Brittain, S.T. Use of computer simulations to test the concept of dose forgiveness in the era of extended-release (XR) drugs. Epilepsy Behav. 2016, 55, 21–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Wright, J.; Gidal, B.; Messenheimer, J. Assessing impact of real-world dosing irregularities with lamotrigine extended-release and immediate-release formulations by pharmacokinetic simulation. Ther Drug Monit. 2013, 35, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Kimko, H.C.; Reele, S.S.B.; Holford, N.H.G.; Peck, C.C. Prediction of the outcome of a phase 3 clinical trial of an antischizophrenic agent (quetiapine fumarate) by simulation with a population pharmacokinetic and pharmacodynamic model. Clin. Pharmacol. Ther. 2000, 68, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Gidal, B.E.; Majid, O.; Ferry, J.; Hussein, Z.; Yang, H.; Zhu, J.; Fain, R.; Laurenza, A. The practical impact of altered dosing on perampanel plasma concentrations: Pharmacokinetic modeling from clinical studies. Epilepsy Behav. 2014, 35, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Reed, R.C.; Dutta, S. Predicted serum valproic acid concentrations in patients missing and replacing a dose of extended-release divalproex sodium. Am. J. Heal. Pharm. 2004, 61, 2284–2289. [Google Scholar] [CrossRef]
- Garnett, W.R.; McLean, A.M.; Zhang, Y.; Clausen, S.; Tulloch, S.J. Simulation of the effect of patient nonadherence on plasma concentrations of carbamazepine from twice-daily extended-release capsules. Curr. Med. Res. Opin. 2003, 19, 519–525. [Google Scholar] [CrossRef]
- Dutta, S.; Reed, R.C. Effect of delayed and/or missed enteric-coated divalproex doses on valproic acid concentrations: Simulation and dose replacement recommendations for the clinician 1. J. Clin. Pharm. Ther. 2006, 31, 321–329. [Google Scholar] [CrossRef]
- Nemeroff, C.B.; Kinkead, B.; Goldstein, J. Quetiapine: Preclinical studies, pharmacokinetics, drug interactions, and dosing. J. Clin. Psychiatry. 2002, 63, 5–11. [Google Scholar]
- Storosum, J.G.; Elferink, A.J.A.; Van Zwieten, B.J. Schizophrenia: Do we really need placebo-controlled studies? Eur. Neuropsychopharmacol. 1998, 8, 279–286. [Google Scholar] [CrossRef]
- Handley, S.A.; Bowskill, S.V.J.; Patel, M.X.; Flanagan, R.J. Plasma quetiapine in relation to prescribed dose and other factors: Data from a therapeutic drug monitoring service, 2000–2011. Ther. Adv. Psychopharmacol. 2013, 3, 129–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | 25% Delay | 50% Delay | 75% Delay | 100% Delay | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IR | XR | p * | IR | XR | p * | IR | XR | p * | IR | XR | p * | |
| ΔCmax | 7.9(0.2) | 7.5(0.5) | <0.001 | 19.6(0.2) | 18.6(0.6) | <0.001 | 36.7(0.3) | 33.8(0.6) | <0.001 | 53.4(0.4) | 45.9(0.7) | <0.001 |
| ΔCmin | 1.7(0.4) | 5.2(0.6) | <0.001 | 4.1(0.4) | 12.7(0.7) | <0.001 | 7.2(0.4) | 21(1.1) | <0.001 | 12.3(0.2) | 56.5(0.5) | <0.001 |
| ΔBPRS)max | 1.2(4.6) | 1.7(5.1) | <0.001 | 3.2(4) | 4.2(6.2) | <0.001 | 5.9(3.6) | 7.4(7.7) | <0.01 | 8.2(3.4) | 9.5(8.4) | <0.05 |
| ΔBPRS)min | 2.6(1.9) | 3.3(2.4) | <0.001 | 3.9(2.9) | 5.3(4) | <0.001 | 5(3.9) | 6.9(7.5) | <0.001 | 6.5(4.8) | 15(8.4) | <0.001 |
| Dose Omission | Dose Doubling | |||||||
|---|---|---|---|---|---|---|---|---|
| ΔCmax | ΔCmin | BPRS)max | BPRS)min | ΔCmax | ΔCmin | BPRS)max | BPRS)max | |
| IR | −5.6(0.7) | −74.6(0.2) | −1.6(5.8) | −34.5(5.8) | 19.5(0.3) | 5.1(0.7) | 3(4.2) | 4.3(3.3) |
| XR | −19.3(0.5) | −71(0.4) | −4.6(2.7) | −33.3(5) | 20.5(0.5) | 15.3(0.6) | 3.7(3.2) | 5.3(6.3) |
| p * | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elkomy, M.H. Changing the Drug Delivery System: Does it Add to Non-Compliance Ramifications Control? A Simulation Study on the Pharmacokinetics and Pharmacodynamics of Atypical Antipsychotic Drug. Pharmaceutics 2020, 12, 297. https://doi.org/10.3390/pharmaceutics12040297
Elkomy MH. Changing the Drug Delivery System: Does it Add to Non-Compliance Ramifications Control? A Simulation Study on the Pharmacokinetics and Pharmacodynamics of Atypical Antipsychotic Drug. Pharmaceutics. 2020; 12(4):297. https://doi.org/10.3390/pharmaceutics12040297
Chicago/Turabian StyleElkomy, Mohammed H. 2020. "Changing the Drug Delivery System: Does it Add to Non-Compliance Ramifications Control? A Simulation Study on the Pharmacokinetics and Pharmacodynamics of Atypical Antipsychotic Drug" Pharmaceutics 12, no. 4: 297. https://doi.org/10.3390/pharmaceutics12040297
APA StyleElkomy, M. H. (2020). Changing the Drug Delivery System: Does it Add to Non-Compliance Ramifications Control? A Simulation Study on the Pharmacokinetics and Pharmacodynamics of Atypical Antipsychotic Drug. Pharmaceutics, 12(4), 297. https://doi.org/10.3390/pharmaceutics12040297