1. Introduction
Achieving appropriate glycemic control for type 2 diabetes patients is a prerequisite for preventing cardiovascular and microvascular complications, and this can be guided by a combination of antidiabetic drugs with different modes of action [
1].
Sodium-glucose cotransporter 2 (SGLT2) inhibitors are the latest class of antidiabetic drugs that act through the inhibition of renal tubular glucose reabsorption and a reduction of blood glucose levels without stimulating insulin release [
2]. SGLT2 is expressed in the S1 segment of proximal kidney tubules and is responsible for roughly 90% of the reabsorption of filtered glucose [
3]. Additionally, SGLT2 inhibitors exhibit low hypoglycemia risk and have been associated with a significant reduction in major adverse cardiovascular events in clinical trials [
4], garnering SGLT2 inhibitors increased attention.
Several SGLT2 inhibitors have been approved for the treatment of type 2 diabetes, including canagliflozin (Invokana
®), dapagliflozin (Farxiga
®), empagliflozin (Jardiance
®), ipragliflozin (Suglat
®), and tofogliflozin (Apleway
®) [
3,
5].
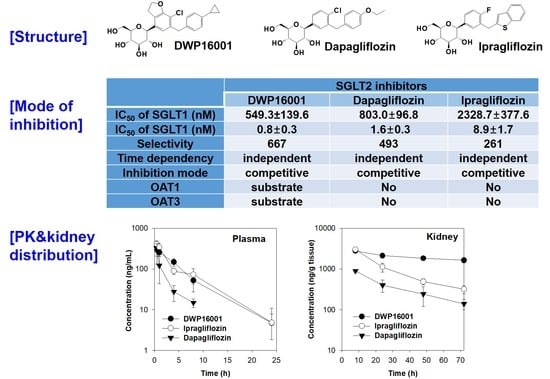
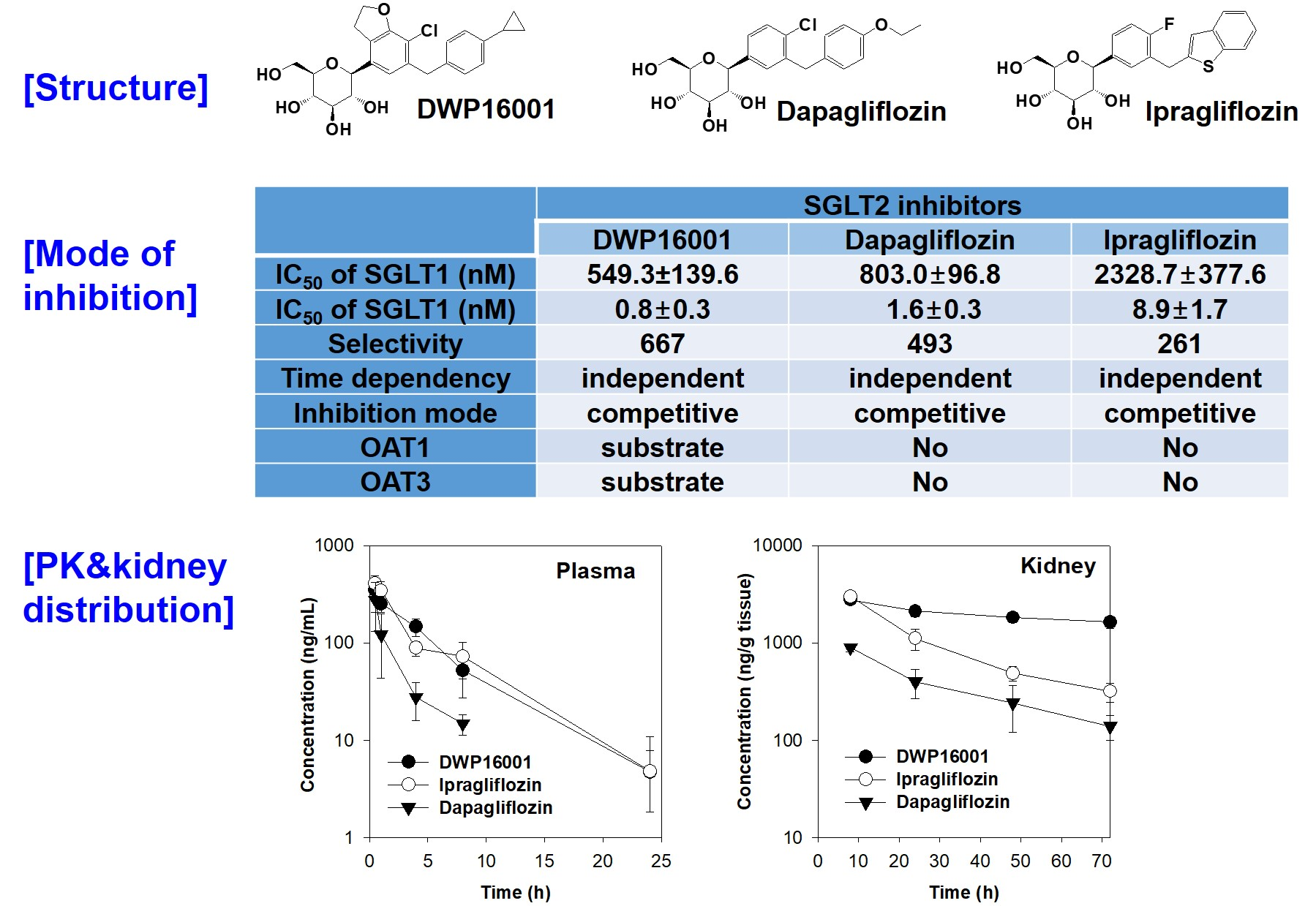
Additionally, there are several other similar compounds in the pipeline that may be approved in the near future. DWP16001 (
Figure 1), a selective SGLT2 inhibitor, is under development by Daewoong Pharmaceutical Co. Ltd. (Yongin, Korea) and is currently undergoing phase 2 clinical trials (Registration No. NCT04014023).
Recently, Tahara et al. [
5] compared the pharmacokinetics, pharmacodynamics, and pharmacological characteristics of six SGLT2 inhibitors, such as ipragliflozin, dapagliflozin, tofogliflozin, canagliflozin, empagliflozin, and luseogliflozin. The study showed that all the SGLT2 inhibitors induced urinary glucose excretion in a dose-dependent manner but the duration of action differed among the six drugs. Ipragliflozin and dapagliflozin showed persistent duration of action; these two drugs exhibited increased urinary glucose excretion even 18 h post dose but the others showed about 12 h of duration. In addition, ipragliflozin and dapagliflozind showed the lowest blood glucose and insulin level following the same daily dose (3 mg/kg) in diabetic mice. The long duration of action and glucose lowering efficacy of these two drugs closely correlated with the drug distribution and retention in the kidney and elimination half-life [
5], suggesting the importance of kidney distribution and elimination profile is prerequisite in the efficacy of SGLT2 inhibitors, as well as the potent SGLT2 inhibition. Therefore, this study aimed to compare the pharmacokinetic properties and kidney distribution of DWP16001 with those of dapagliflozin and ipragliflozin, representative SGLT2 inhibitors that showed potent and long duration efficacy [
5] and to compare the selectivity and mode of inhibition of DWP16001 on SGLT2 with dapagliflozin and ipragliflozin to evaluate the potency of DWP16001 over other SGLT2 inhibitors.
2. Materials and Methods
2.1. Materials
DWP16001 and D4-DWP16001 (for internal standard (IS)), were obtained from Daewoong Pharmaceutical Co. Ltd. (Yongin, Korea). Dapagliflozin and ipragliflozin were obtained from Toronto Research Chemicals Inc. (North York, ON, Canada) (
Figure 1).
Para-aminohipuric acid (PAH), methyl a-D-glucopyranoside (AMG), sodium dodecyl sulfate (SDS), G418, non-essential amino acids, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), and Hank’s balances salts solution (HBSS, pH 7.4) were purchased from Sigma–Aldrich Chemical Co. (St. Louis, MO, USA). [14C]Methyl-α-D-glucopyranoside (AMG) (290 mCi/mmol) was purchased from Moravek (Brea, CA, USA). [3H]Estrone-3-sulfate (ES) (2.12 TBq/mmol) and [3H]para-aminohipuric acid (PAH) (0.13 TBq/mmol) were purchased from Perkin Elmer Inc. (Boston, MA, USA). Dulbecco’s modified Eagle’s medium (DMEM), RPMI1640 medium, fetal bovine serum (FBS), and poly-D-lysine-coated 24-well plates were purchased from Corning (Tewksbury, MA, USA). All other chemicals and solvents were reagent or analytical grade.
CHO cells overexpressing SGLT1 and SGLT2 (CHO-SGLT1 and -SGLT2, respectively) and CHO-mock cells were obtained from Daewoong Pharmaceutical Co. Ltd. (Yongin, Korea). HEK293 cells overexpressing organic anion transporter 1 (OAT1) and OAT3 (HEK293-OAT1 and -OAT3, respectively) and HEK293-mock cells were purchased from Corning (Tewksbury, MA, USA).
2.2. Animals and Ethical Approval
Male Institute of Cancer Research (ICR) mice (7–8-weeks-year-old, 30−35 g) were purchased from Samtako Co. (Osan, Korea). Animals were acclimatized for 1 week in an animal facility at Kyungpook National University. Food and water were available ad libitum. All animal procedures were approved by the Animal Care and Use Committee of Kyungpook National University (Approval No. 2016-0138) and carried out in accordance with the National Institutes of Health guidance for the care and use of laboratory animals.
2.3. Pharmacokinetic Study
ICR mice were randomly divided into three groups and were fasted for at least 12 h before the oral administration of DWP16001, dapagliflozin, and ipragliflozin but had free access to water. On the day of pharmacokinetic study, the mice received DWP16001, dapagliflozin, or ipragliflozin solution at a dose of 1 mg/kg (dissolved in a mixture of 10% DMSO and 90% saline) using oral gavage. Blood samples were collected at 0.5, 1, 2, 4, 8, 24, 48, and 72 h following the oral administration (1 mg/kg each) of DWP16001, dapagliflozin, or ipragliflozin and centrifuged at 12,000× g for 1 min to separate the plasma. An aliquot (30 µL) of each plasma sample was stored at −80 °C until the analysis. Kidney samples were also isolated at 8, 24, 48, and 72 h following the oral administration of DWP16001, dapagliflozin, or ipragliflozin, minced thoroughly, and homogenized with four volumes of saline using tissue grinder. An aliquot (50 µL) of each kidney homogenate sample was stored at −80 °C until the analysis.
For the analysis of SGLT2 inhibitors, aliquots of plasma (30 µL) and kidney homogenate (50 µL) were added to 100 μL of aqueous solution of D4-DWP16001 (IS, 20 ng/mL), and vigorously mixed with 500 μL methyl tert-butyl ether (MTBE) for 15 min. After centrifugation at 16,000 g for 5 min, samples were kept for 1 h at −80 °C to make an aqueous layer freeze. An organic upper layer was transferred to a clean tube and evaporated to dryness under a gentle stream of nitrogen. Then, the dried extract was reconstituted in 150 µL of mobile phase, and a 3 µL aliquot of the reconstituent was injected into a liquid chromatography–tandem mass spectrometry (LC–MS/MS) system.
Pharmacokinetic parameters, such as the area under plasma concentration-time curve from zero to infinity (AUC), were calculated from plasma concentration vs time curves using non-compartment analysis with WinNonlin (version 5.1; Pharsights, Cary, NC, USA). The AUC ratios (i.e., ratios of kidney AUC to plasma AUC) were calculated by dividing the AUC of the three SGLT2 inhibitors in the kidney by the plasma AUC values of the three SGLT2 inhibitors.
2.4. Protein Binding
The protein binding of DWP16001, dapagliflozin, and ipragliflozin (1000 ng/mL) in mouse plasma and 20% kidney homogenate was determined using a rapid equilibrium dialysis kit (ThermoFisher Scientific Korea, Seoul, Korea) according to the manufacturer’s instructions. Briefly, 100 μL of mouse plasma and 20% kidney homogenate samples containing 1000 ng/mL of DWP16001, dapagliflozin, or ipragliflozin were added to the sample chamber of a semipermeable membrane (molecular weight cut-off 8000 Da) and 300 μL of phosphate buffered saline (PBS) was added to the outer buffer chamber. Four hours after incubation at 37 °C on a shaking incubator at 300 rpm, aliquots (50 μL) were collected from both the sample and buffer chambers and treated with equal volumes of fresh PBS and blank plasma or blank kidney homogenate, respectively, to match the sample matrices. The matrix-matched sample (100 μL) was added 100 μL of aqueous solution of D4-DWP16001 (IS, 20 ng/mL), and vigorously mixed with 1000 μL MTBE for 15 min. After centrifugation at 16,000 g for 5 min, samples were kept for 1 h at −80 °C. An organic upper layer was transferred to a clean tube and evaporated to dryness under a gentle stream of nitrogen. Then, the dried extract was reconstituted in 300 µL of mobile phase and a 3 µL aliquot of the reconstituent was injected into the LC-MS/MS system.
Plasma protein binding was calculated using the following Equation (1) [
6,
7].
Kidney protein binding was calculated using the following equations, Equations (2) and (3), and a dilution factor (D as a value of 5) for tissue homogenates was used since we used 20% kidney homogenates [
6,
7].
2.5. Substrate Specificity of DWP16001, Dapagliflozin, and Ipragliflozin for OAT1 and OAT3
HEK293-mock cells and HEK293 cells overexpressing OAT1 and OAT3 transporters (HEK293-OAT1 and -OAT3, respectively) were seeded in poly-D-lysine-coated 24-well plates at a density of 4 × 105 cells/well and cultured for 24 h in DMEM supplemented with 10% FBS and 5 mM non-essential amino acids at 37 °C in 8% CO2 condition.
For each experiment, the growth medium was discarded after 24 h, and the attached cells were washed with pre-warmed HBSS and incubated with pre-warmed HBSS for 20 min at 37 °C. To confirm the functionality of OAT1 and OAT3, we measured the uptake of 0.1 µM [
3H]PAH and 0.1 µM [
3H]ES, representative substrates for OAT1 and OAT3, respectively, into in the HEK293-mock cells and HEK293-OAT1 and -OAT3 cells, respectively, for 5 min in the presence and absence of 20 µM probenecid, a typical inhibitor for both OAT1 and OAT3 [
8,
9]. The cells were then washed three times with 500 μL of ice-cold HBSS immediately after placing the plates on ice. Subsequently, cells were lysed with 10% sodium dodecyl sulfate and mixed with Optiphase cocktail solution overnight. The radioactivity of the cell lysate was measured using a liquid scintillation counter (Microbeta 2; Perkin Elmer Inc., Boston, MA, USA).
The uptake of DWP16001, dapagliflozin, and ipragliflozin (2 µM each) was measured for 5 min at 37 °C in the HEK293-mock cells and HEK293-OAT1 and -OAT3 cells, respectively, in the absence and presence of 20 µM probenecid. For the concentration dependency in the uptake of DWP16001, the uptake of DWP16001 in a concentration range of 0.5–50 μM dissolved in HBSS was measured for 5 min at 37 °C in the mock cells and HEK293-OAT1 and -OAT3 cells, respectively. After 5 min, the cells were washed three times with 500 μL of ice-cold HBSS immediately after placing the plates on ice. Subsequently, the cells were scraped using a cell scraper with 100 μL of PBS, and cell suspensions were transferred to a clean tube, combined with 100 μL of aqueous solution of D4-DWP16001 (IS, 20 ng/mL), and vigorously mixed with 1000 μL MTBE for 15 min. After centrifugation at 16,000 g for 5 min, samples were kept for 1 h at −80 °C. An organic upper layer was transferred to a clean tube and evaporated to dryness under a gentle stream of nitrogen. Then, the dried extract was reconstituted in 300 µL of mobile phase and a 3 µL aliquot of the reconstituent was injected into the LC-MS/MS system.
In the concentration-dependent uptake studies, the transporter-mediated uptake of DWP16001 was calculated by the subtraction of the transport rates of DWP16001 into the mock cells from those of the HEK293-OAT1 and -OAT3 cells. Kinetic parameters for the OAT1- and OAT3-mediated transport of DWP16001 were determined using the Michaelis-Menten equation [
V =
Vmax⋅S/(
Km + S)] [
10].
2.6. LC-MS/MS Analysis of DWP16001, Dapagliflozin, and Ipragliflozin
Concentrations of DWP16001, dapagliflozin, and ipragliflozin in plasma and kidney homogenate samples were analyzed using an Agilent 6470 triple quadrupole LC–MS/MS system (Agilent, Wilmington, DE, USA).
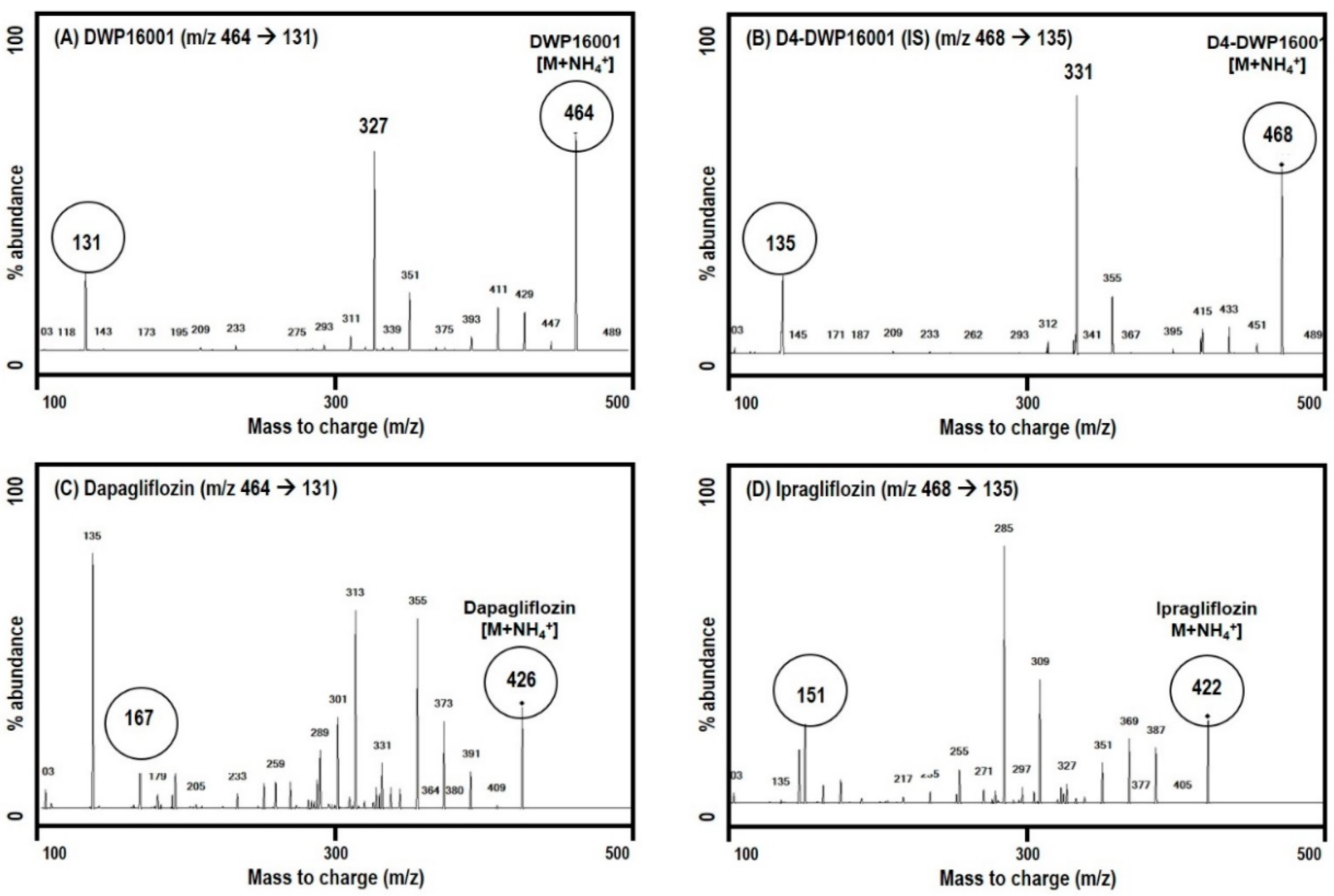
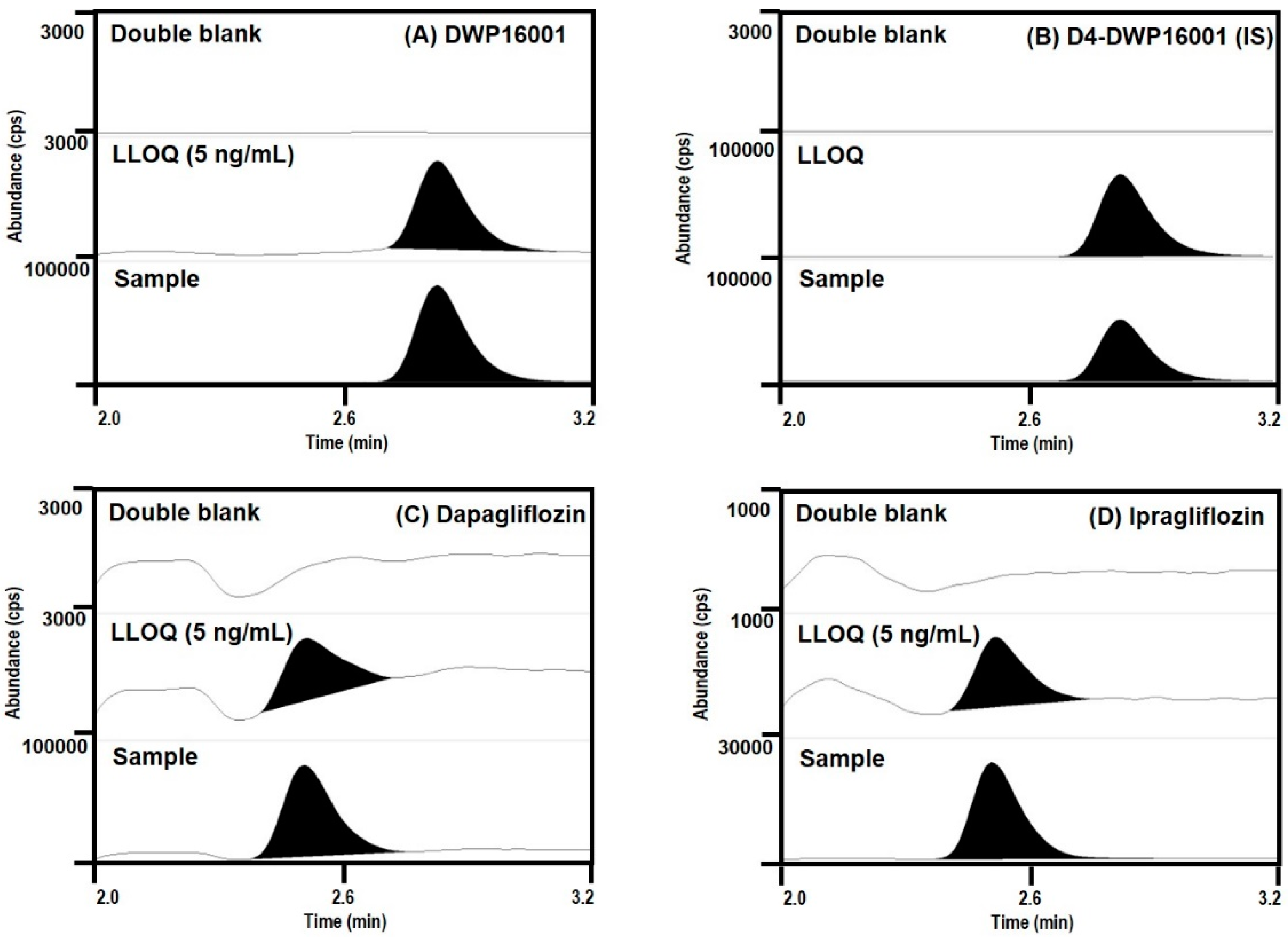
DWP16001, dapagliflozin, and ipragliflozin were separated on a Synergi Polar RP column (2.0 × 150 mm, 4 μm particle size; Phenomenex, Torrence, CA) using a mobile phase consisting of water (15%) and methanol (85%) containing 0.1% formic acid at a flow rate of 0.25 mL/min.
Quantification of a separated analyte peak was performed at m/z 464 → 131 for DWP16001 (TR (retention time) 2.8 min), m/z 422 → 151 for ipragliflozin (TR 2.5 min), m/z 426 → 167 for dapagliflozin (TR 2.5 min), m/z 468 → 135 for D4-DWP16001 (TR 2.8 min), in the positive ionization mode with a collision energy (CE) of 25 eV. The calibration standards of a mixture of DWP16001, dapagliflozin, and ipragliflozin in mouse plasma were 5–1000 ng/mL, and intraday and interday precision and accuracy were less than 14.7% in all samples. The calibration standards of a mixture of DWP16001, dapagliflozin, and ipragliflozin in mouse kidney homogenate were 5–1000 ng/mL, and intraday and interday precision and accuracy were less than 13.8% in all samples.
2.7. Inhibitory Effects of DWP16001, Dapagliflozin, and Ipragliflozin on the SGLT1 and SGLT2 Activities
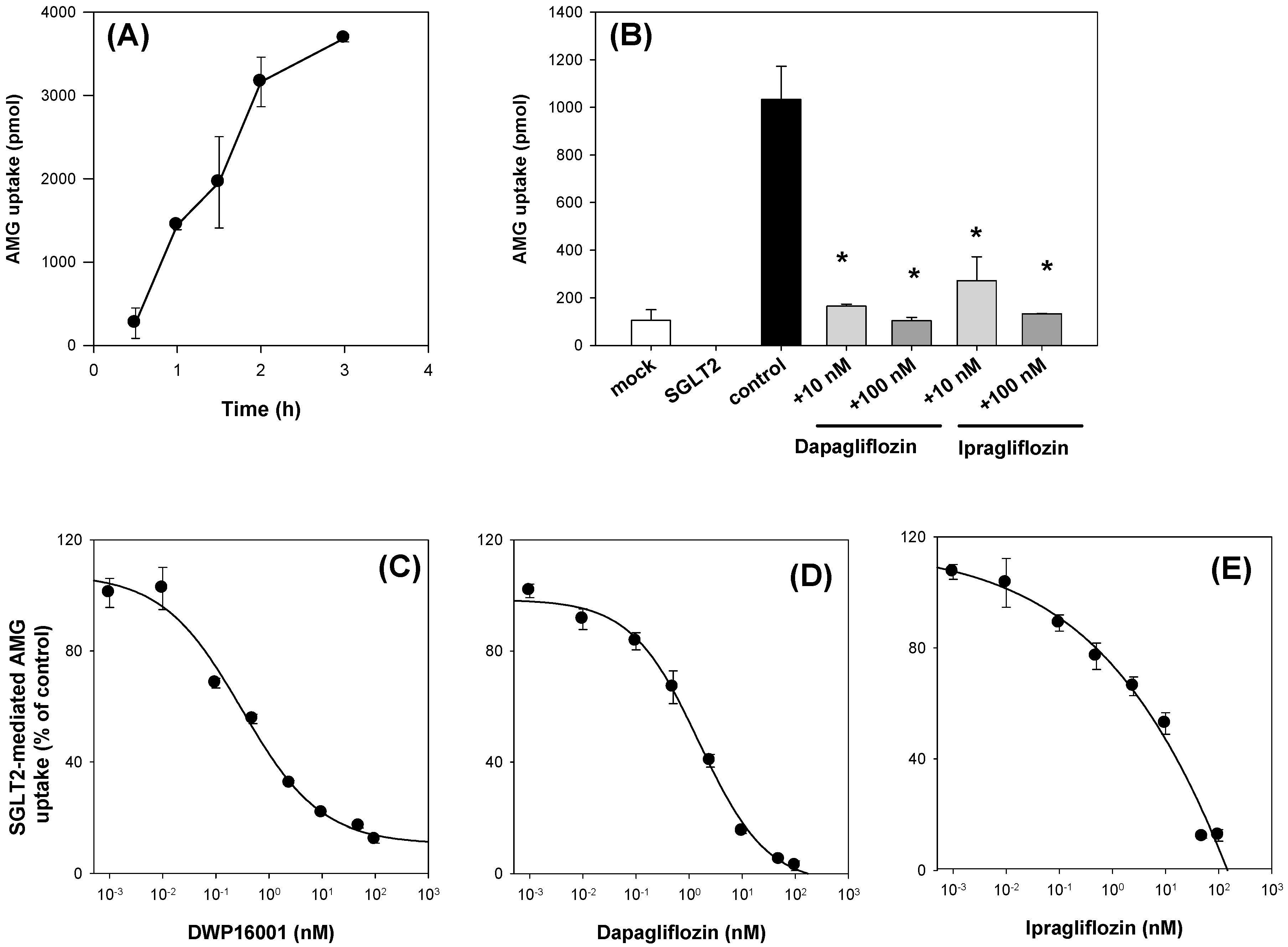
CHO cells overexpressing SGLT1 and SGLT2 cells (CHO-SGLT1 and -SGLT2) and CHO-mock cells were characterized as previously described [
11]. Cells were maintained in RPMI1640 medium supplemented with 10% fetal bovine serum and 200 µg/mL G418 at 37 °C in 5% CO
2 conditions. CHO-SGLT1 and -SGLT2 cells were seeded at a density of 1 × 10
5 cells/well in 96-well plates. After 24 h, the growth medium was discarded from the cells, and the cells were washed with pre-warmed Na
+-free buffer (10 mM HEPES, 5 mM Tris, 140 mM choline chloride, 2 mM KCl, 1 mM CaCl
2, 1 mM MgCl
2, pH7.4) and incubated for 1 h in Na
+-free buffer. After replacing Na
+-free buffer with Na
+ gradient buffer (10 mM HEPES, 5 mM Tris, 140 mM NaCl, 2 mM KCl, 1 mM CaCl
2, 1 mM MgCl
2, pH7.4) containing 10 µM [
14C]AMG, the uptake of [
14C]AMG into the CHO-mock cells and CHO-SGLT1 and -SLGT2 cells was measured for 0.5, 1, 1.5, 2, and 3 h. After a predetermined incubation time, cells were washed three times with 200 µL of ice-cold Na
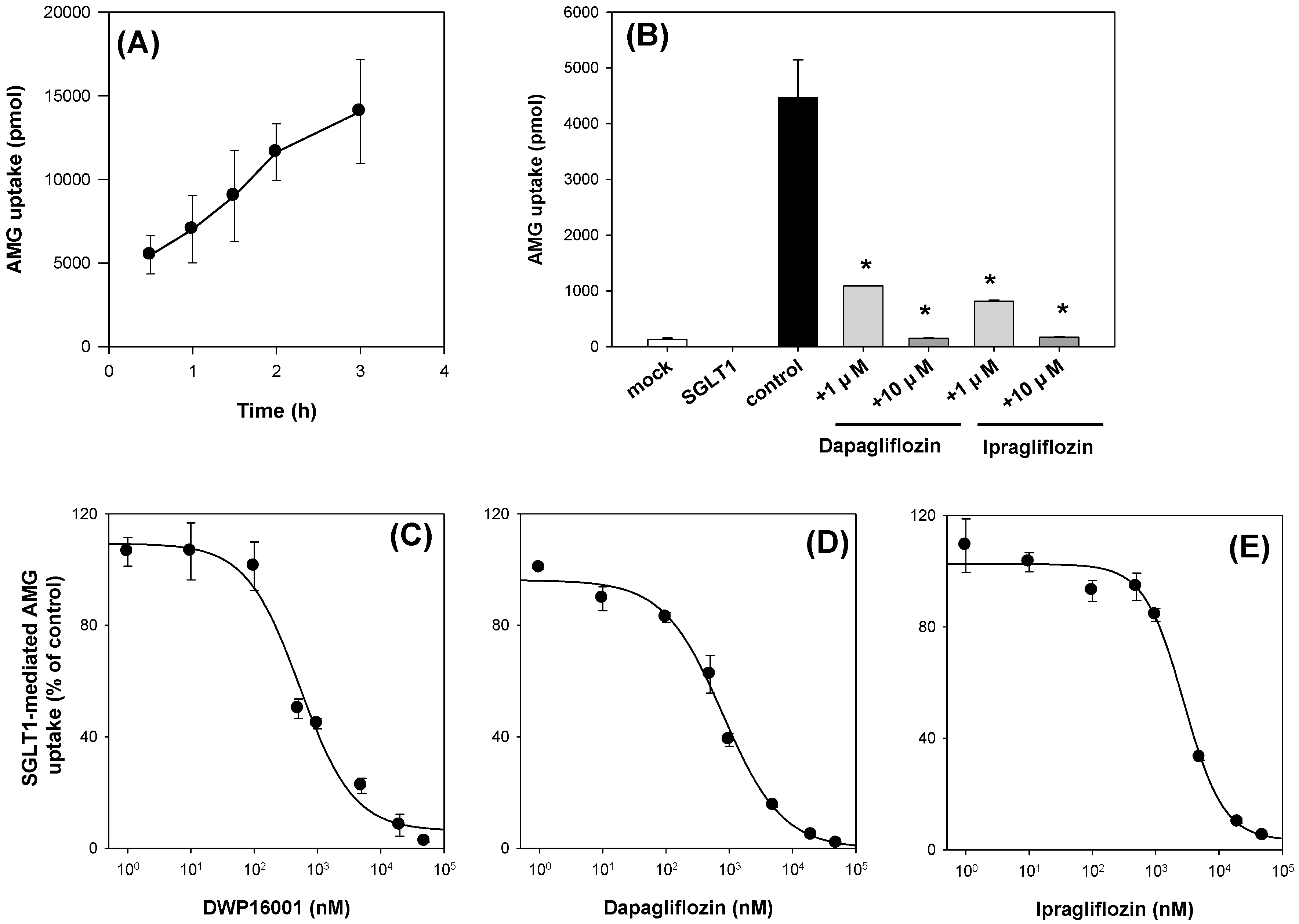
+-free buffer immediately after placing the plates on ice. Then, the cells were lysed with 10% SDS, and the cell lysates were mixed with Optiphase cocktail solution. Thereafter, the radioactivity of the cell lysates was measured using a liquid scintillation counter.
The inhibitory effect of known inhibitors, such as dapagliflozin and ipragliflozin, on [
14C]AMG uptake in the CHO-mock cells and CHO-SGLT1 and -SLGT2 cells was measured in the presence or absence of dapagliflozin and ipragliflozin (1, 10 μM for SGLT1; 10, 100 nM for SGLT2) for 2 h. For the calculation of IC
50 values, the uptake of 10 µM [
14C]AMG in the CHO-mock cells and CHO-SGLT1 and -SLGT2 cells was measured for 2 h with or without DWP16001, dapagliflozin, or ipragliflozin (1 nM–50 µM for SGLT1; 0.01 nM–1 µM for SGLT2). After 2 h, cells were washed three times with 200 µL of ice-cold Na
+-free buffer and the cells were lysed with 10% SDS (40 µL), followed by adding Optiphase cocktail solution (200 µL). The radioactivity of the cell lysates was measured using a liquid scintillation counter. The SGLT1 or SGLT2-mediated uptake of [
14C]AMG was calculated by the subtraction of the uptake rates of [
14C]AMG into the mock cells from those of the CHO-SGLT1 and -SLGT2 cells. In the inhibition studies, the percentages of the transport rate of AMG with or without SGLT2 inhibitors were calculated and the data were fitted to an inhibitory effect model. The IC
50 (the concentration of the inhibitor to show half-maximal inhibition) values were calculated using Sigma Plot ver.10.0 (Systat Software, Inc.; San Jose, CA, USA) [
12].
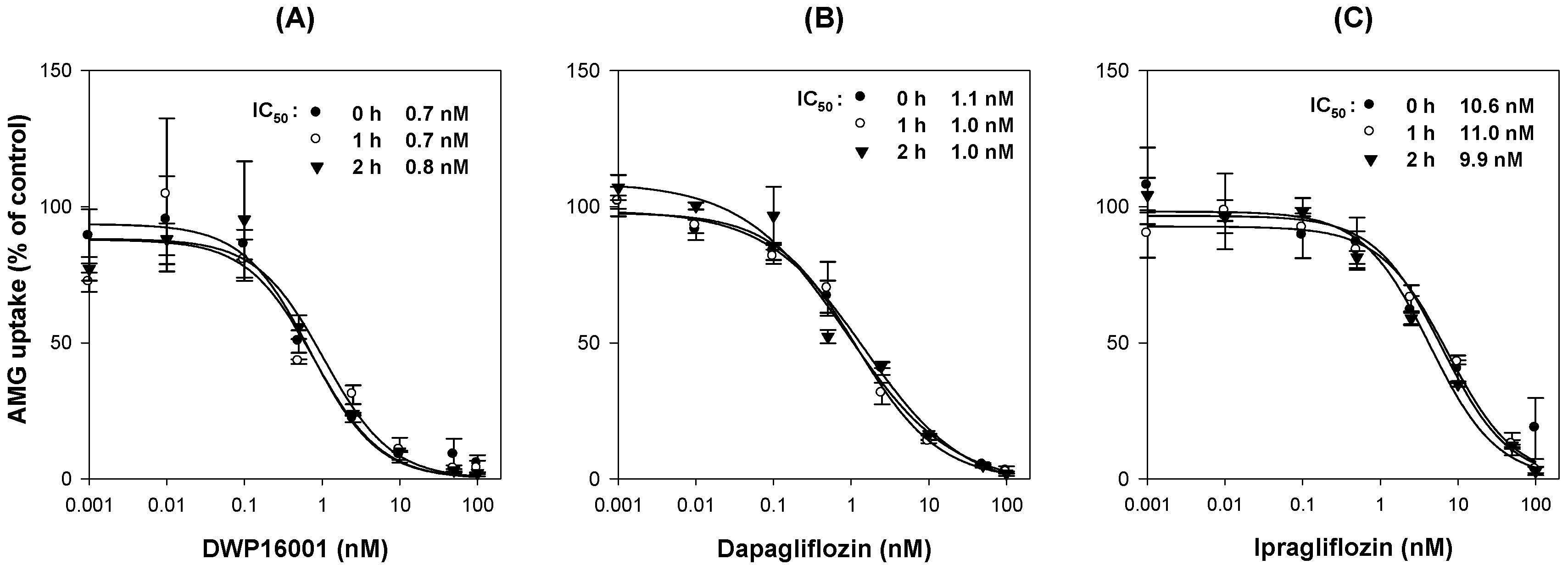
To investigate time dependency in the inhibition of SGLT2, CHO-mock and -SGLT2 cells were seeded at a density of 1 × 105 cells/well in 96-well plates. After 24 h, the growth medium was discarded from the cells, and the cells were washed with pre-warmed Na+-free buffer and pre-incubated with Na+-free buffer containing various concentrations of DWP16001, dapagliflozin, or ipragliflozin (0.001 nM–100 nM) for 1 and 2 h. Then, after replacing Na+-free buffer with Na+ gradient buffer containing 10 µM [14C]AMG and various concentrations of DWP16001, dapagliflozin, or ipragliflozin (0.001 nM–100 nM), the uptake of [14C]AMG into the CHO-mock and -SGLT2 cells was measured for 2 h. After 2 h of incubation, the radioactivity of the cell lysate was measured following the same sample preparation method described above.
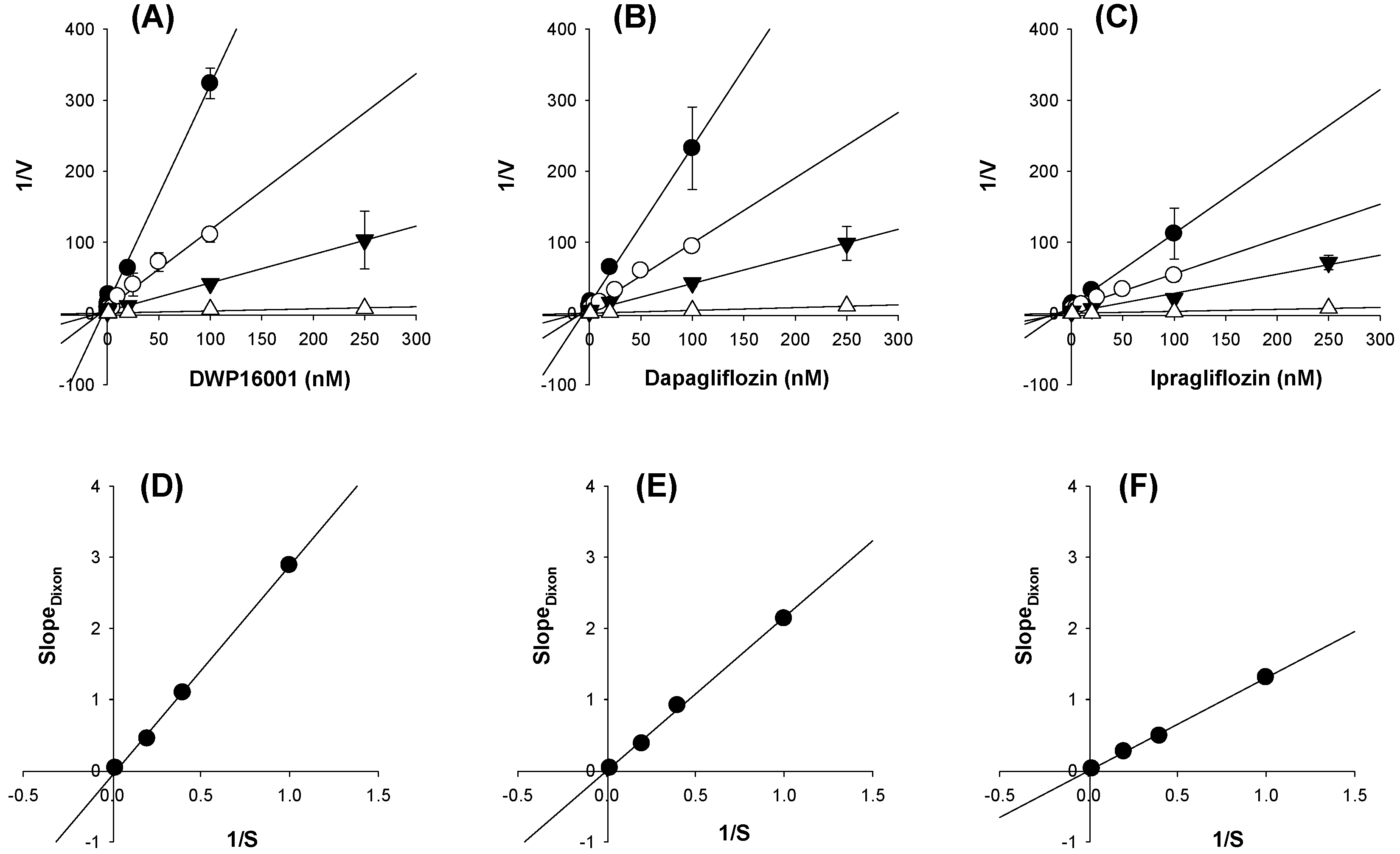
To investigate the mode of inhibition of the three SGLT2 inhibitors, the inhibition experiments were initiated by replacing Na
+-free buffer with Na
+ gradient buffer containing 1, 2.5, 5, and 50 µM [
14C]AMG and various concentrations of DWP16001, dapagliflozin, or ipragliflozin (0.001 nM–250 nM) and the uptake of [
14C]AMG into the CHO-mock and -SGLT2 cells was measured for 2 h. The radioactivity of the cell lysate was measured following the same sample preparation method described above. Uptake rate of AMG and concentrations of DWP16001, dapagliflozin, or ipragliflozin were plotted to Dixon plots to identify the mode of inhibition [
13,
14].
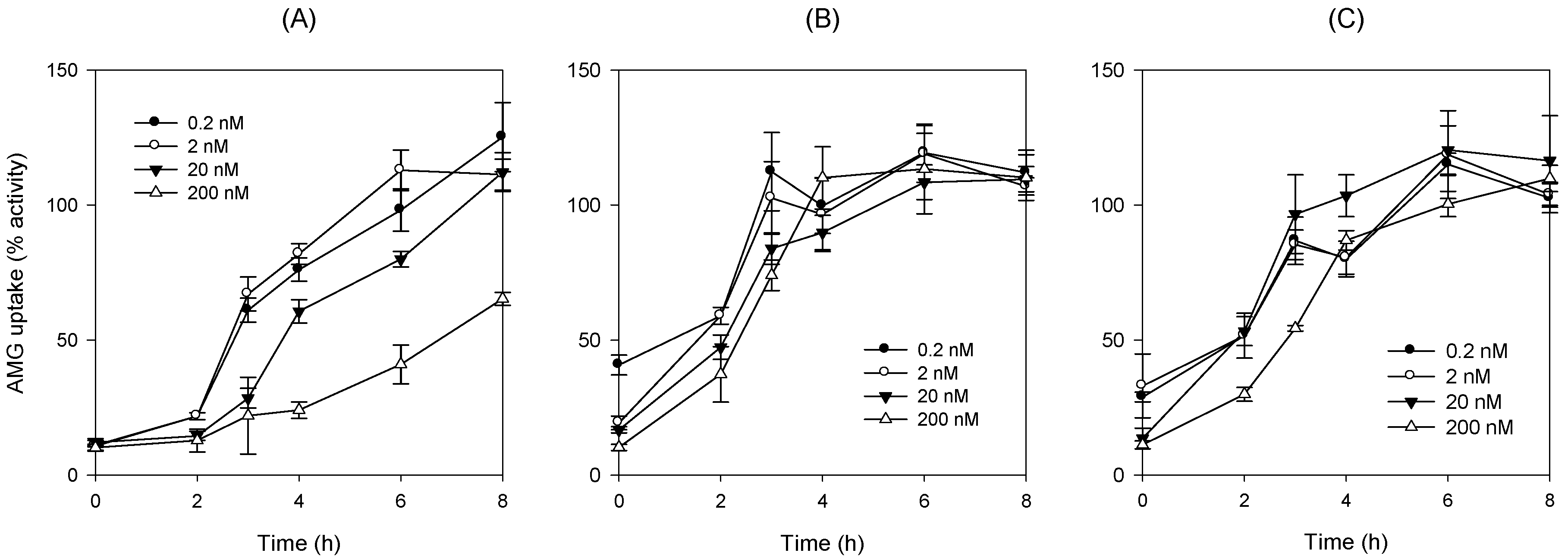
To investigate the recovery of SGLT2 activity depending on the washout period after 24 h exposure of DWP16001, dapagliflozin, or ipragliflozin, CHO-SGLT2 cells were seeded at a density of 1 × 105 cells/well in 96-well plates. After 24 h, the growth medium was discarded from the cells, and the cells were treated with RPMI1640 medium containing DWP16001, dapagliflozin, or ipragliflozin (0.2, 2, 20, and 200 nM) for 24 h. After 24 h, the RPMI1640 medium was replaced with pre-warmed fresh RPMI1640 medium and incubated for 1, 2, 3, 5, and 6 h, and proceeded another pre-incubation with Na+-free buffer for 1 h. Then, after replacing Na+-free buffer with Na+ gradient buffer containing 10 µM [14C]AMG, the uptake of [14C]AMG into the CHO-SGLT2 cells was measured for 2 h. After 2 h of incubation, the radioactivity of the cell lysate was measured following the same sample preparation method described above.
2.8. Statistics
The statistical significance was assessed by t-test using SPSS for Windows (version 24.0; IBM Corp., Armonk, NY, USA).
4. Discussion
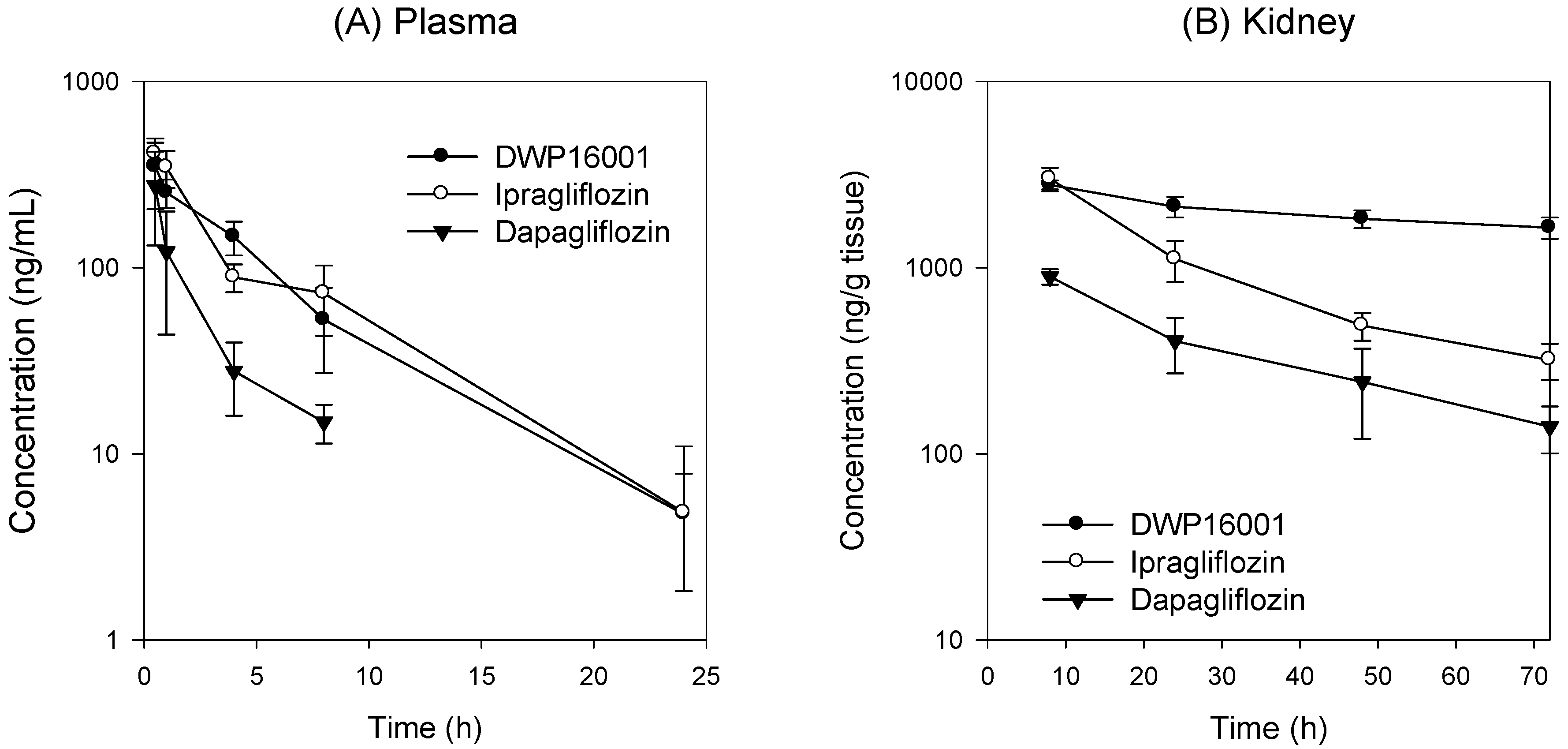
DWP16001 is a candidate SGLT2 inhibitor that is currently under development. As a first step, the pharmacokinetic properties and in vitro SGLT2 inhibition were compared with currently used SGLT2 inhibitors. Dapagliflozin and ipragliflozin were selected based on their high kidney distributions and long elimination half-lives (t
1/2) in the kidney [
5], which are thought to be important for the efficacy and duration of action of SGLT2 inhibitors. DWP16001 showed higher kidney distributions compared with dapagliflozin and ipragliflozin. DWP16001 also had longer t
1/2 in the kidney than dapagliflozin and ipragliflozin, as well as a comparable plasma profile with ipragliflozin (
Table 1). Moreover, the kidney concentration of DWP16001 was maintained over 72 h following oral administration of 1 mg/kg DWP16001 (
Figure 4). Taken together, these pharmacokinetic results show that the duration of action of DWP16001 is greater than that of dapagliflozin and ipragliflozin and that the oral therapeutic dose of DWP16001 could be reduced compared with both dapagliflozin and ipragliflozin.
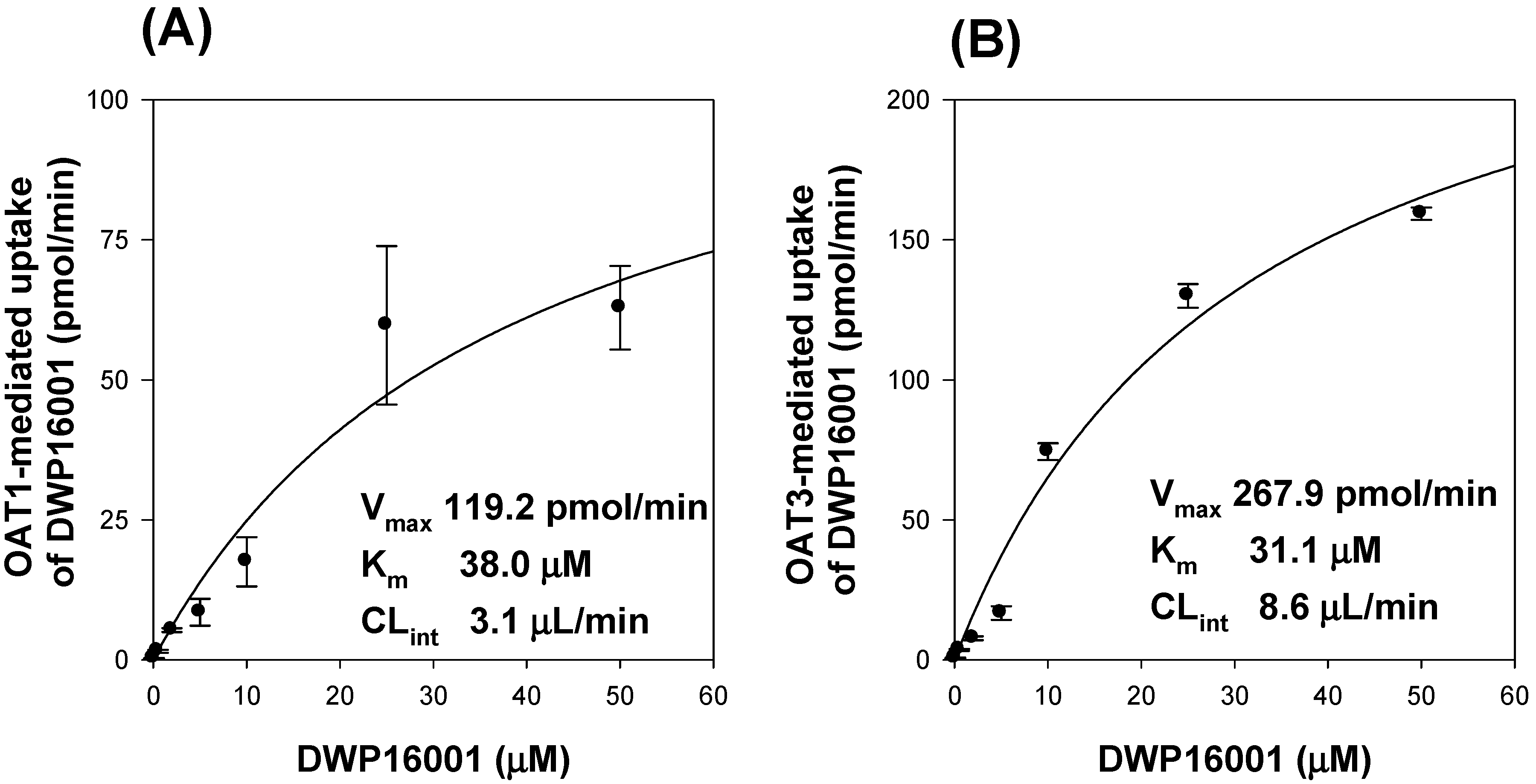
To investigate the underlying mechanisms of highest kidney distribution and maintained concentration of DWP16001 in the kidney, we measured the kidney tissue binding and involvement of OAT1 and OAT3 transporters of the three SGLT2 inhibitors. All three SGLT2 inhibitors showed high protein binding, but the kidney tissue binding performances of these three SGLT2 inhibitors were not different from their plasma protein binding capabilities (
Table 2). However, DWP16001 was a substrate for both OAT1 and OAT3, which are dominantly expressed in the kidney, whereas dapagliflozin and ipragliflozin were not (
Figure 5 and
Figure 6). Although this could not solely explain the high kidney distribution, OAT1- and OAT3-mediated transport process may contribute to the high kidney distribution.
Next, we compared the in vitro SGLT2 inhibition and selectivity of SGLT2 inhibition over SGLT1. All three SGLT2 inhibitors inhibited SGLT2 and SGLT1 activity in a concentration-dependent manner and IC
50 values of dapagliflozin and ipragliflozin were in the range of previous reports (1.0–1.3 nM for dapagliflozin; 6.75–8.07 nM for ipragliflozin) [
21,
22]. IC
50 values of DWP16001 to SGLT2 and SGLT1 were lower than those of dapagliflozin and ipragliflozin, suggesting a greater affinity to SGLT2 inhibition for DWP16001 with a higher selectivity over SGLT1 than dapagliflozin and ipragliflozin. The mode of inhibition of DWP16001 was not different from the other SGLT2 inhibitors. They all showed reversible and competitive inhibition (
Figure 10), which is consistent with other SGLT2 inhibitors [
23,
24]. However, the affinity to SGLT2 inhibition seemed to be different among the three SGLT2 inhibitors (
Table 3). In addition, the recovery of SGLT2 transport activity following the pretreatment of DWP16001, dapagliflozin, and ipragliflozin for 24 h was retained at a higher concentration (200 nM) of DWP16001 compared with dapagliflozin and ipragliflozin. These results suggested that DWP16001 had the highest SGLT2 inhibition potential and that this inhibition potential retained for a longer time compared with dapagliflozin and ipragliflozin. Combined with the higher kidney distribution of DWP16001, retained SGLT2 inhibition with a high concentration of DWP16001 could also potentiate the efficacy of DWP16001 compared with dapagliflozin and ipragliflozin.
The comparative pharmacokinetics and in vitro SGLT2 inhibition findings suggest that DWP16001 might be a superior alternative to dapagliflozin and ipragliflozin; however, we should note that comparisons of the in vivo pharmacologic properties of these agents using therapeutic doses in animals and humans need to be further undertaken.















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}