Cell Penetrating Peptides, Novel Vectors for Gene Therapy



Abstract

1. Introduction
2. Types of CPPs
2.1. Non-Cell-Specific CPPs
2.2. Cell-Specific CPPs
3. Identification of Tissue Specific CPPs
4. Mechanisms of Transduction
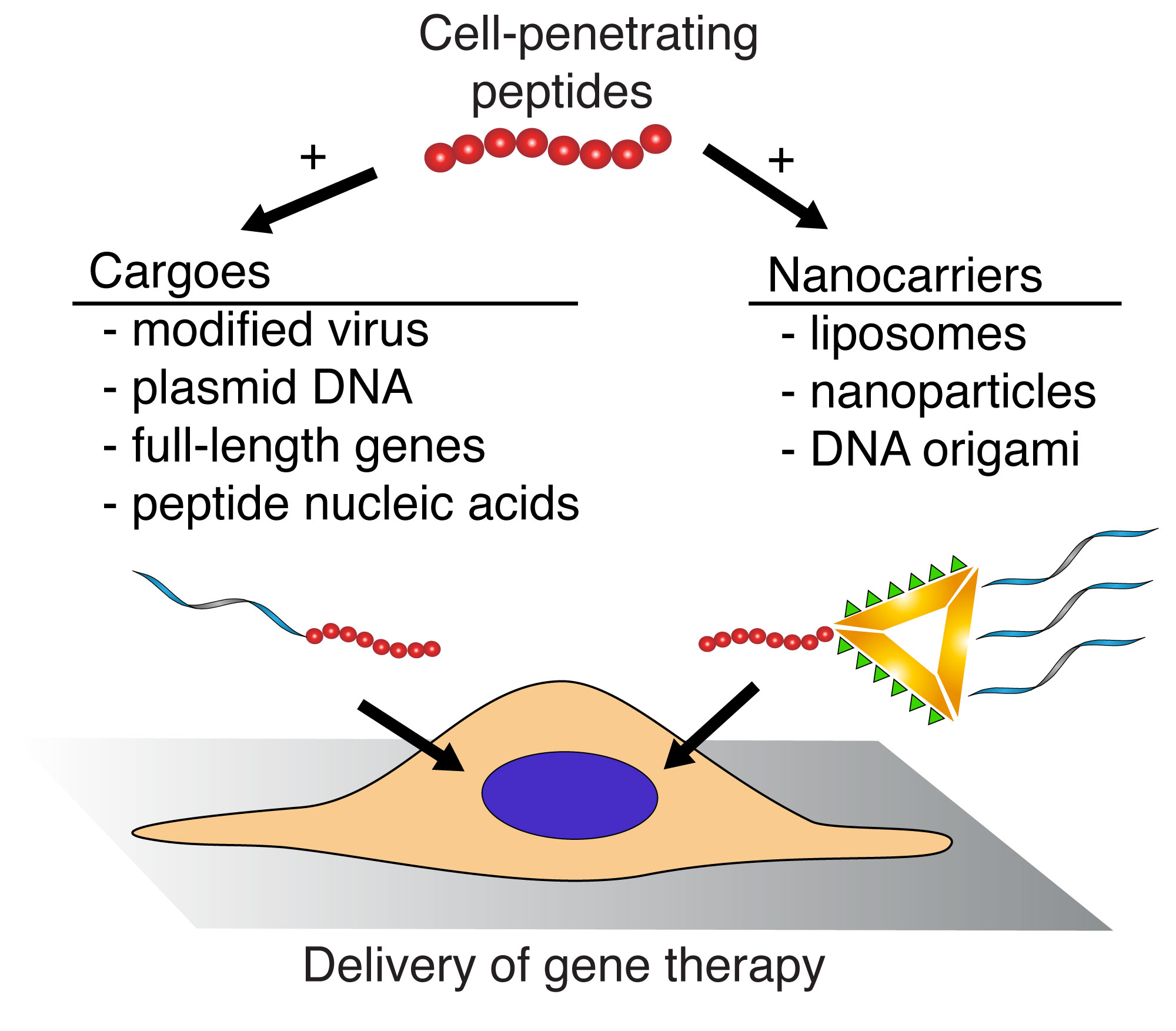
5. Cell Penetrating Peptides as Gene Delivery Vectors
5.1. Viral Vectors
5.2. Plasmid DNA
5.3. Small Interfering RNA
5.4. Nanoparticles
5.5. Liposomes
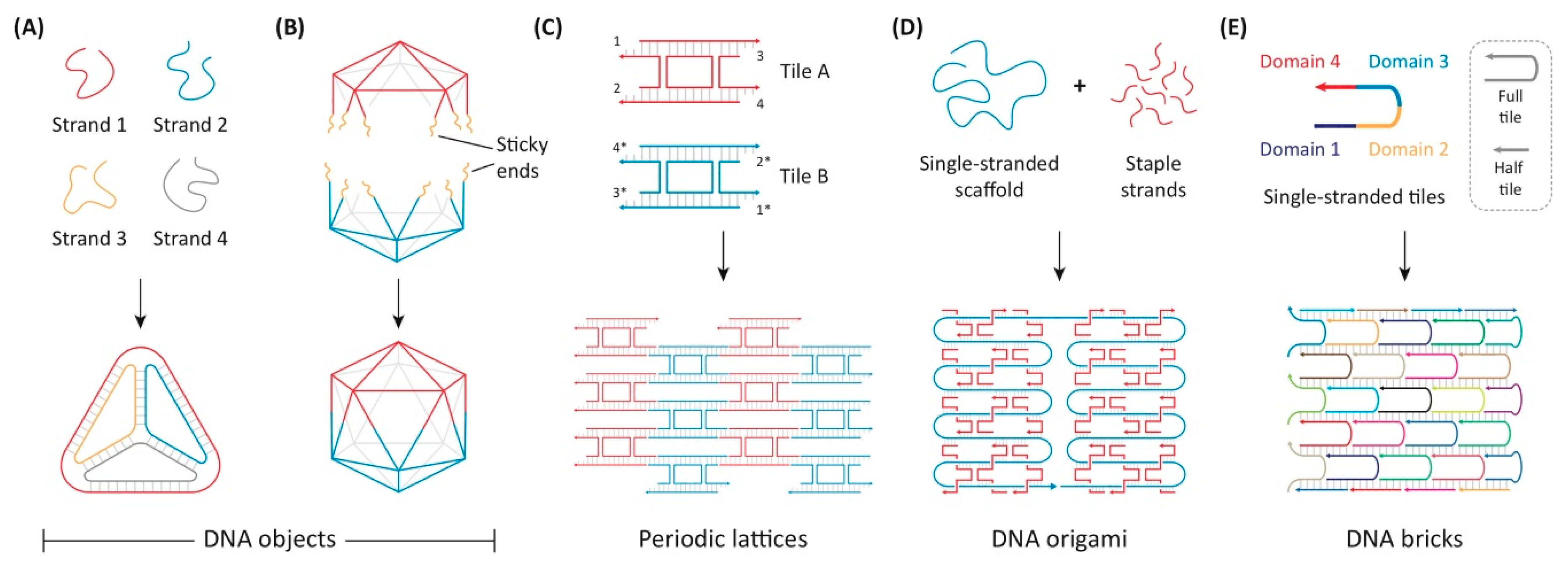
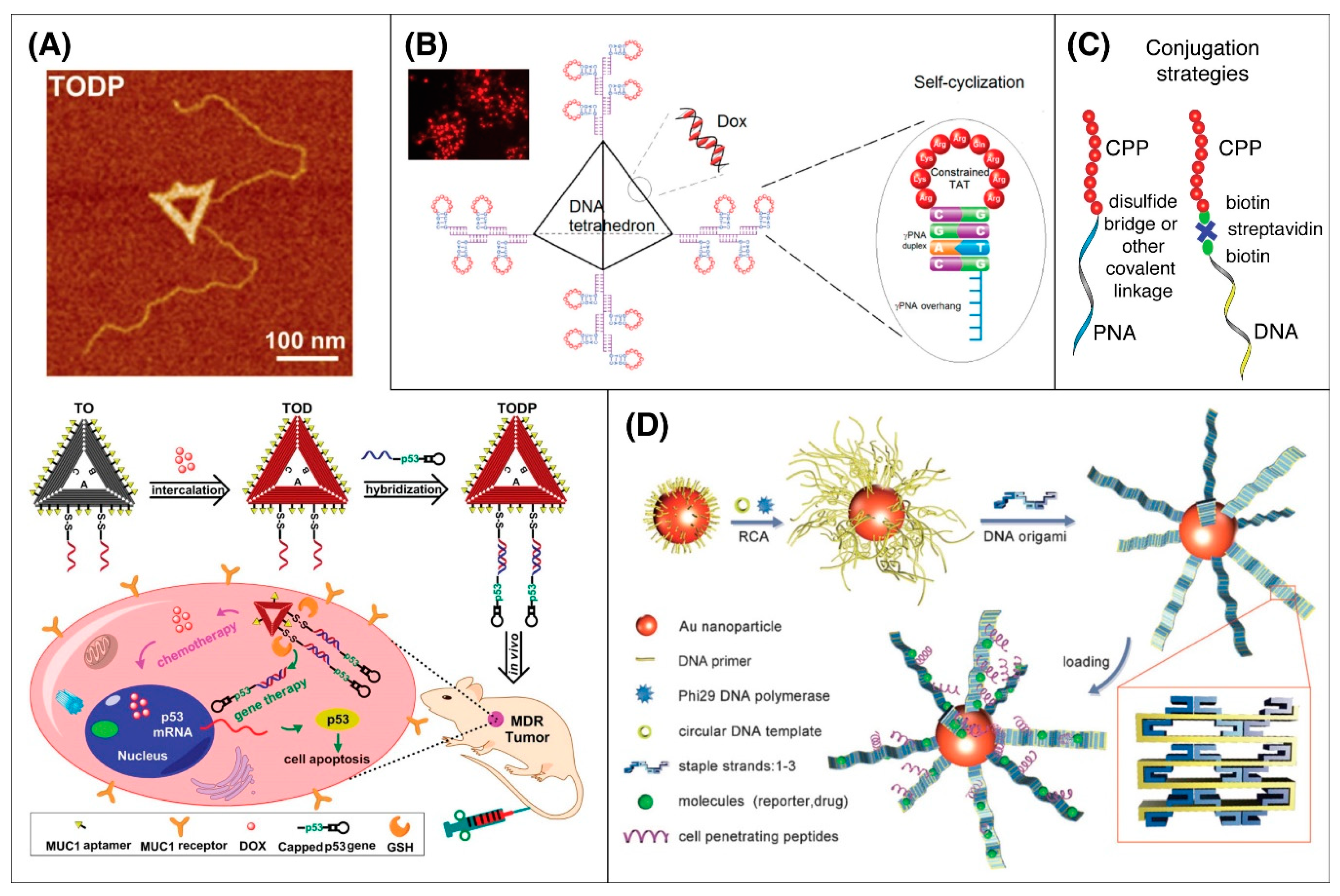
5.6. DNA Origami
5.7. Peptide Nucleic Acids
5.8. CRISPR-Cas
6. Hurdles to Clinical Application
7. Summary
8. Patents
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef]
- Green, M.; Loewenstein, P.M. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell 1988, 55, 1179–1188. [Google Scholar] [CrossRef]
- Fawell, S.; Seery, J.; Daikh, Y.; Moore, C.; Chen, L.L.; Pepinsky, B.; Barsoum, J. Tat-mediated delivery of heterologous proteins into cells. Proc. Natl. Acad. Sci. USA 1994, 91, 664–668. [Google Scholar] [CrossRef] [PubMed]
- Joliot, A.; Pernelle, C.; Deagostini-Bazin, H.; Prochiantz, A. Antennapedia homeobox peptide regulates neural morphogenesis. Proc. Natl. Acad. Sci. USA 1991, 88, 1864–1868. [Google Scholar] [CrossRef] [PubMed]
- Green, M.; Ishino, M.; Loewenstein, P.M. Mutational analysis of HIV-1 Tat minimal domain peptides: Identification of trans-dominant mutants that suppress HIV-LTR-driven gene expression. Cell 1989, 58, 215–223. [Google Scholar] [CrossRef]
- DeRossi, D.; Joliot, A.H.; Chassaing, G.; Prochiantz, A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J. Biol. Chem. 1994, 269, 10444–10450. [Google Scholar]
- Schwarze, S.R. In Vivo Protein Transduction: Delivery of a Biologically Active Protein into the Mouse. Science 1999, 285, 1569–1572. [Google Scholar] [CrossRef]
- Zahid, M.; Robbins, P.D. Cell-Type Specific Penetrating Peptides: Therapeutic Promises and Challenges. Molecules 2015, 20, 13055–13070. [Google Scholar] [CrossRef]
- Feni, L.; Neundorf, I.; Sunna, A.; Care, A.; Bergquist, P.L. The Current Role of Cell-Penetrating Peptides in Cancer Therapy. Adv. Exp. Med. Biol. 2017, 1030, 279–295. [Google Scholar]
- Guidotti, G.; Brambilla, L.; Rossi, D. Cell-Penetrating Peptides: From Basic Research to Clinics. Trends Pharmacol. Sci. 2017, 38, 406–424. [Google Scholar] [CrossRef]
- Kalafatovic, D.; Giralt, E. Cell-Penetrating Peptides: Design Strategies beyond Primary Structure and Amphipathicity. Molecules 2017, 22, 1929. [Google Scholar] [CrossRef] [PubMed]
- Wender, P.A.; Mitchell, D.J.; Pattabiraman, K.; Pelkey, E.T.; Steinman, L.; Rothbard, J.B. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: Peptoid molecular transporters. Proc. Natl. Acad. Sci. USA 2000, 97, 13003–13008. [Google Scholar] [CrossRef] [PubMed]
- Mai, J.C.; Shen, H.; Watkins, S.C.; Cheng, T.; Robbins, P.D. Efficiency of Protein Transduction Is Cell Type-dependent and Is Enhanced by Dextran Sulfate. J. Biol. Chem. 2002, 277, 30208–30218. [Google Scholar] [CrossRef]
- Madani, F.; Lindberg, S.; Langel, Ü.; Futaki, S.; Gräslund, A. Mechanisms of Cellular Uptake of Cell-Penetrating Peptides. J. Biophys. 2011, 2011, 1–10. [Google Scholar] [CrossRef]
- Fei, L.; Yap, L.-P.; Conti, P.S.; Shen, W.-C.; Zaro, J.L. Tumor targeting of a cell penetrating peptide by fusing with a pH-sensitive histidine-glutamate co-oligopeptide. Biomaterials 2014, 35, 4082–4087. [Google Scholar] [CrossRef]
- Tunnemann, G.; Ter-Avetisyan, G.; Martin, R.M.; Stockl, M.; Herrmann, A.; Cardoso, M.C. Live-cell analysis of cell penetration ability and toxicity of oligo-arginines. J. Pept. Sci. 2008, 14, 469–476. [Google Scholar] [CrossRef]
- Mi, Z.; Mai, J.; Lu, X.; Robbins, P.D. Characterization of a Class of Cationic Peptides Able to Facilitate Efficient Protein Transduction in Vitro and in Vivo. Mol. Ther. 2000, 2, 339–347. [Google Scholar] [CrossRef]
- Xu, Y.; Liang, W.; Qiu, Y.; Cespi, M.; Palmieri, G.F.; Mason, A.J.; Lam, J.K. Incorporation of a Nuclear Localization Signal in pH Responsive LAH4-L1 Peptide Enhances Transfection and Nuclear Uptake of Plasmid DNA. Mol. Pharm. 2016, 13, 3141–3152. [Google Scholar] [CrossRef]
- Nakayama, F.; Yasuda, T.; Umeda, S.; Asada, M.; Imamura, T.; Meineke, V.; Akashi, M. Fibroblast growth factor-12 (FGF12) translocation into intestinal epithelial cells is dependent on a novel cell-penetrating peptide domain: Involvement of internalization in the in vivo role of exogenous FGF12. J. Biol. Chem. 2011, 286, 25823–25834. [Google Scholar] [CrossRef]
- Höfling, S.; Scharnert, J.; Cromme, C.; Bertrand, J.; Pap, T.; Schmidt, M.A.; Rüter, C. Manipulation of pro-inflammatory cytokine production by the bacterial cell-penetrating effector protein YopM is independent of its interaction with host cell kinases RSK1 and PRK2. Virulence 2014, 5, 761–771. [Google Scholar] [CrossRef][Green Version]
- Gomarasca, M.; Martins, T.F.C.; Greune, L.; Hardwidge, P.R.; Schmidt, M.A.; Rüter, C. Bacterium-Derived Cell-Penetrating Peptides Deliver Gentamicin to Kill Intracellular Pathogens. Antimicrob. Agents Chemother. 2017, 61, e02545-16. [Google Scholar] [CrossRef]
- Norkowski, S.; Körner, B.; Greune, L.; Stolle, A.-S.; Lubos, M.-L.; Hardwidge, P.R.; Schmidt, M.A.; Rüter, C. Bacterial LPX motif-harboring virulence factors constitute a species-spanning family of cell-penetrating effectors. Cell. Mol. Life Sci. 2017, 75, 2273–2289. [Google Scholar] [CrossRef]
- Braun, G.B.; Sugahara, K.N.; Yu, O.M.; Kotamraju, V.R.; Mölder, T.; Lowy, A.M.; Ruoslahti, E.; Teesalu, T. Urokinase-controlled tumor penetrating peptide. J. Control. Release 2016, 232, 188–195. [Google Scholar] [CrossRef]
- De Cogan, F.; Hill, L.J.; Lynch, A.; Morgan-Warren, P.J.; Lechner, J.; Berwick, M.R.; Peacock, A.; Chen, M.; Scott, R.; Xu, H.; et al. Topical Delivery of Anti-VEGF Drugs to the Ocular Posterior Segment Using Cell-Penetrating Peptides. Investig. Opthalmol. Vis. Sci. 2017, 58, 2578. [Google Scholar] [CrossRef]
- Tai, L.; Liu, C.; Jiang, K.; Chen, X.; Wei, G.; Lu, W.; Pan, W. Noninvasive delivery of oligonucleotide by penetratin-modified polyplexes to inhibit protein expression of intraocular tumor. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 2091–2100. [Google Scholar] [CrossRef]
- Ildefonso, C.J.; Jaime, H.; Brown, E.E.; Iwata, R.L.; Ahmed, C.M.; Massengill, M.T.; Biswal, M.R.; Boye, S.E.; Hauswirth, W.; Ash, J.D.; et al. Targeting the Nrf2 Signaling Pathway in the Retina WIth a Gene-Delivered Secretable and Cell-Penetrating Peptide. Investig. Opthalmol. Vis. Sci. 2016, 57, 372–386. [Google Scholar] [CrossRef]
- Mi, Z.; Lu, X.; Mai, J.C.; Ng, B.G.; Wang, G.; Lechman, E.R.; Watkins, S.C.; Rabinowich, H.; Robbins, P.D. Identification of a synovial fibroblast-specific protein transduction domain for delivery of apoptotic agents to hyperplastic synovium. Mol. Ther. 2003, 8, 295–305. [Google Scholar] [CrossRef]
- Mai, J.C.; Mi, Z.; Kim, S.H.; Ng, B.; Robbins, P.D. A proapoptotic peptide for the treatment of solid tumors. Cancer Res. 2001, 61, 7709–7712. [Google Scholar]
- Smith, G. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef]
- Zahid, M.; Robbins, P.D. Identification and Characterization of Tissue-Specific Protein Transduction Domains Using Peptide Phage Display. Adv. Struct. Saf. Stud. 2010, 683, 277–289. [Google Scholar]
- Zahid, M.; Phillips, B.E.; Albers, S.M.; Giannoukakis, N.; Watkins, S.C.; Robbins, P.D. Identification of a Cardiac Specific Protein Transduction Domain by In Vivo Biopanning Using a M13 Phage Peptide Display Library in Mice. PLoS ONE 2010, 5, e12252. [Google Scholar] [CrossRef] [PubMed]
- Hyvönen, M.; Laakkonen, P. Identification and Characterization of Homing Peptides Using In Vivo Peptide Phage Display. Adv. Struct. Saf. Stud. 2015, 1324, 205–222. [Google Scholar]
- Nicklin, S.A.; White, S.; Watkins, S.J.; Hawkins, R.E.; Baker, A.H. Selective targeting of gene transfer to vascular endothelial cells by use of peptides isolated by phage display. Circulation 2000, 102, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Chamarthy, S.P.; Jia, L.; Kovacs, J.R.; Anderson, K.R.; Shen, H.; Firestine, S.M.; Meng, W.S. Gene delivery to dendritic cells facilitated by a tumor necrosis factor alpha-competing peptide. Mol. Immunol. 2004, 41, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Rehman, K.K.; Bai, X.-Y.; Miao, D.; Goltzman, D.; Karaplis, A.C. Protection of Islets byin SituPeptide-mediated Transduction of the Ikappa Kinase Inhibitor Nemo-binding Domain Peptide. J. Biol. Chem. 2003, 278, 9862–9868. [Google Scholar] [CrossRef] [PubMed]
- Arap, W.; Pasqualini, R.; Ruoslahti, E. Cancer Treatment by Targeted Drug Delivery to Tumor Vasculature in a Mouse Model. Science 1998, 279, 377–380. [Google Scholar] [CrossRef]
- Magzoub, M.; Eriksson, L.E.G.; Gräslund, A. Conformational states of the cell-penetrating peptide penetratin when interacting with phospholipid vesicles: Effects of surface charge and peptide concentration. Biochim. Biophys. Acta 2002, 1563, 53–63. [Google Scholar] [CrossRef]
- Jiao, C.-Y.; Delaroche, D.; Burlina, F.; Alves, I.; Chassaing, G.; Sagan, S. Translocation and Endocytosis for Cell-penetrating Peptide Internalization. J. Biol. Chem. 2009, 284, 33957–33965. [Google Scholar] [CrossRef]
- Gump, J.M.; June, R.K.; Dowdy, S.F. Revised role of glycosaminoglycans in TAT protein transduction domain-mediated cellular transduction. J. Biol. Chem. 2009, 285, 1500–1507. [Google Scholar] [CrossRef]
- Backlund, C.; Takeuchi, T.; Futaki, S.; Tew, G.N. Relating structure and internalization for ROMP-based protein mimics. Biochim. Biophys. Acta 2016, 1858, 1443–1450. [Google Scholar] [CrossRef]
- Illien, F.; Rodriguez, N.; Amoura, M.; Joliot, A.; Pallerla, M.; Cribier, S.; Burlina, F.; Sagan, S. Quantitative fluorescence spectroscopy and flow cytometry analyses of cell-penetrating peptides internalization pathways: Optimization, pitfalls, comparison with mass spectrometry quantification. Sci. Rep. 2016, 6, 36938. [Google Scholar] [CrossRef]
- Åmand, H.L.; Rydberg, H.A.; Fornander, L.H.; Lincoln, P.; Nordén, B.; Esbjörner, E.K. Cell surface binding and uptake of arginine- and lysine-rich penetratin peptides in absence and presence of proteoglycans. Biochim. Biophys. Acta (BBA) Biomembr. 2012, 1818, 2669–2678. [Google Scholar] [CrossRef]
- Takechi, Y.; Tanaka, H.; Kitayama, H.; Yoshii, H.; Tanaka, M.; Saito, H. Comparative study on the interaction of cell-penetrating polycationic polymers with lipid membranes. Chem. Phys. Lipids 2012, 165, 51–58. [Google Scholar] [CrossRef]
- Rusnati, M.; Coltrini, D.; Oreste, P.; Zoppetti, G.; Albini, A.; Noonan, D.; d’Adda di Fagagna, F.; Giacca, M.; Presta, M. Interaction of HIV-1 Tat protein with heparin. Role of the backbone structure, sulfation, and size. J. Biol. Chem. 1997, 272, 11313–11320. [Google Scholar] [CrossRef]
- Tyagi, M.; Rusnati, M.; Presta, M.; Giacca, M. Internalization of HIV-1 Tat Requires Cell Surface Heparan Sulfate Proteoglycans. J. Biol. Chem. 2000, 276, 3254–3261. [Google Scholar] [CrossRef]
- Ma, D.-X.; Shi, N.-Q.; Qi, X. Distinct transduction modes of arginine-rich cell-penetrating peptides for cargo delivery into tumor cells. Int. J. Pharm. 2011, 419, 200–208. [Google Scholar] [CrossRef]
- Li, Y.; Rosal, R.V.; Brandt-Rauf, P.W.; Fine, R.L. Correlation between hydrophobic properties and efficiency of carrier-mediated membrane transduction and apoptosis of a p53 C-terminal peptide. Biochem. Biophys. Res. Commun. 2002, 298, 439–449. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Yang, X.-Z.; Du, X.; Wang, J.-W.; Zhang, R.; Zhao, J.; Wang, F.-J.; Dong, Y.; Li, P.-F. Enhancing tumor-specific intracellular delivering efficiency of cell-penetrating peptide by fusion with a peptide targeting to EGFR. Amino Acids 2015, 47, 997–1006. [Google Scholar] [CrossRef]
- Yan, C.; Gu, J.; Hou, D.; Jing, H.; Wang, J.; Guo, Y.; Katsumi, H.; Sakane, T.; Yamamoto, A. Improved tumor targetability of Tat-conjugated PAMAM dendrimers as a novel nanosized anti-tumor drug carrier. Drug Dev. Ind. Pharm. 2014, 41, 617–622. [Google Scholar] [CrossRef]
- De La Torre, B.G.; Hornillos, V.; Luque-Ortega, J.R.; Abengozar, M.A.; Amat-Guerri, F.; Acuña, A.U.; Rivas, L.; Andreu, D. A BODIPY-embedding miltefosine analog linked to cell-penetrating Tat(48-60) peptide favors intracellular delivery and visualization of the antiparasitic drug. Amino Acids 2014, 46, 1047–1058. [Google Scholar] [CrossRef]
- Zhang, P.; Cheetham, A.G.; Lin, Y.-A.; Cui, H. Self-Assembled Tat Nanofibers as Effective Drug Carrier and Transporter. ACS Nano 2013, 7, 5965–5977. [Google Scholar] [CrossRef]
- Fu, B.; Long, W.; Zhang, Y.; Zhang, A.; Miao, F.; Shen, Y.; Pan, N.; Gan, G.; Nie, F.; He, Y.; et al. Enhanced antitumor effects of the BRBP1 compound peptide BRBP1-TAT-KLA on human brain metastatic breast cancer. Sci. Rep. 2015, 5, 8029. [Google Scholar] [CrossRef]
- Davoudi, Z.; Akbarzadeh, A.; Rahmatiyamchi, M.; Movassaghpour, A.; Alipour, M.; Nejati-Koshki, K.; Sadeghi, Z.; Dariushnejad, H.; Zarghami, N. Molecular target therapy of AKT and NF-kB signaling pathways and multidrug resistance by specific cell penetrating inhibitor peptides in HL-60 cells. Asian Pac. J. Cancer Prev. 2014, 15, 4353–4358. [Google Scholar] [CrossRef]
- Xie, X.; Kerrigan, J.E.; Minko, T.; Garbuzenko, O.; Lee, K.-C.; Scarborough, A.; Abali, E.E.; Budak-Alpdogan, T.; Johnson-Farley, N.; Banerjee, D.; et al. Antitumor and modeling studies of a penetratin-peptide that targets E2F-1 in small cell lung cancer. Cancer Biol. Ther. 2013, 14, 742–751. [Google Scholar] [CrossRef]
- Liu, R.; Xi, L.; Luo, D.; Ma, X.; Yang, W.; Xi, Y.; Wang, H.; Qian, M.; Fan, L.; Xia, X.; et al. Enhanced targeted anticancer effects and inhibition of tumor metastasis by the TMTP1 compound peptide TMTP1-TAT-NBD. J. Control. Release 2012, 161, 893–902. [Google Scholar] [CrossRef]
- Hotchkiss, R.S.; McConnell, K.W.; Bullok, K.; Davis, C.G.; Chang, K.C.; Schwulst, S.J.; Dunne, J.C.; Dietz, G.P.; Bähr, M.; McDunn, J.; et al. TAT-BH4 and TAT-Bcl-xL peptides protect against sepsis-induced lymphocyte apoptosis in vivo. J. Immunol. 2006, 176, 5471–5477. [Google Scholar] [CrossRef]
- Tan, M.; Lan, K.-H.; Yao, J.; Lu, C.-H.; Sun, M.; Neal, C.L.; Lu, J.; Yu, D. Selective Inhibition of ErbB2-Overexpressing Breast CancerIn vivoby a Novel TAT-Based ErbB2-Targeting Signal Transducers and Activators of Transcription 3–Blocking Peptide. Cancer Res. 2006, 66, 3764–3772. [Google Scholar] [CrossRef]
- Bidwell, G. Application of thermally responsive polypeptides directed against c-Myc transcriptional function for cancer therapy. Mol. Cancer Ther. 2005, 4, 1076–1085. [Google Scholar] [CrossRef]
- Perea, S.E.; Puchades, Y.; Mendoza, O.; Vispo, N.S.; Torrens, I.; Santos, A.; Acevedo, B.; Falcón, V.; Reyes, O.; Silva, R.; et al. Antitumor Effect of a Novel Proapoptotic Peptide that Impairs the Phosphorylation by the Protein Kinase 2 (Casein Kinase 2). Cancer Res. 2004, 64, 7127–7129. [Google Scholar] [CrossRef]
- Wang, R.; Wang, H.Y. Enhancement of antitumor immunity by prolonging antigen presentation on dendritic cells. Nat. Biotechnol. 2002, 20, 149–154. [Google Scholar] [CrossRef]
- Zhang, T.; Wu, K.; Ding, C.; Sun, K.; Guan, Z.; Wang, X.; Hsieh, J.-T.; He, D.; Fan, J. Inhibiting bladder tumor growth with a cell penetrating R11 peptide derived from the p53 C-terminus. Oncotarget 2015, 6, 37782–37791. [Google Scholar] [CrossRef]
- Chu, X.; Wu, B.; Fan, H.; Hou, J.; Hao, J.; Hu, J.; Wang, B.; Liu, G.; Li, C.; Meng, S. PTD-fused p53 as a potential antiviral agent directly suppresses HBV transcription and expression. Antivir. Res. 2016, 127, 41–49. [Google Scholar] [CrossRef]
- Wu, Y.; Xie, G.; Xu, Y.; Ma, L.; Tong, C.; Fan, D.; Du, F.; Yu, H. PEP-1-MsrA ameliorates inflammation and reduces atherosclerosis in apolipoprotein E deficient mice. J. Transl. Med. 2015, 13, 316. [Google Scholar] [CrossRef]
- Dietz, G.P.; Valbuena, P.C.; Dietz, B.; Meuer, K.; Mueller, P.; Weishaupt, J.H.; Bahr, M. Application of a blood-brain-barrier-penetrating form of GDNF in a mouse model for Parkinson’s disease. Brain Res. 2006, 1082, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Jung, E.; Park, J.; Park, D. Transdermal delivery of interferon-gamma (IFN-gamma) mediated by penetratin, a cell-permeable peptide. Biotechnol. Appl. Biochem. 2005, 42, 169–173. [Google Scholar] [PubMed]
- Zahid, M.; Feldman, K.S.; Garcia-Borrero, G.; Feinstein, T.N.; Pogodzinski, N.; Xu, X.; Yurko, R.; Czachowski, M.; Wu, Y.; Mason, N.S.; et al. Cardiac Targeting Peptide, a Novel Cardiac Vector: Studies in Bio-Distribution, Imaging Application, and Mechanism of Transduction. Biomolecules 2018, 8, 147. [Google Scholar] [CrossRef] [PubMed]
- Stangl, S.; Tei, L.; De Rose, F.; Reder, S.; Martinelli, J.; Sievert, W.; Shevtsov, M.; Öllinger, R.; Rad, R.; Schwaiger, M.; et al. Preclinical Evaluation of the Hsp70 Peptide Tracer TPP-PEG24-DFO[89Zr] for Tumor-Specific PET/CT Imaging. Cancer Res. 2018, 78, 6268–6281. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Yang, W.; Zhang, M.; Li, G.; Wang, S.; Wang, Z.; Ma, X.; Kang, F.; Wang, J. Evaluation of 68Ga-labeled iNGR peptide with tumor-penetrating motif for microPET imaging of CD13-positive tumor xenografts. Tumor Biol. 2016, 37, 12123–12131. [Google Scholar] [CrossRef]
- Dong, P.; Cai, H.; Chen, L.; Li, Y.; Yuan, C.; Wu, X.; Shen, G.; Zhou, H.; Zhang, W.; Li, L. Biodistribution and evaluation of131I-labeled neuropilin-binding peptide for targeted tumor imaging. Contrast Media Mol. Imaging 2016, 11, 467–474. [Google Scholar] [CrossRef]
- Falanga, A.; Vitiello, M.T.; Cantisani, M.; Tarallo, R.; Guarnieri, D.; Mignogna, E.; Netti, P.A.; Pedone, C.; Galdiero, M.; Galdiero, S. A peptide derived from herpes simplex virus type 1 glycoprotein H: Membrane translocation and applications to the delivery of quantum dots. Nanomed. Nanotechnol. Biol. Med. 2011, 7, 925–934. [Google Scholar] [CrossRef]
- Brunetti, J.; Riolo, G.; Gentile, M.; Bernini, A.; Paccagnini, E.; Falciani, C.; Lozzi, L.; Scali, S.; Depau, L.; Pini, A.; et al. Near-infrared quantum dots labelled with a tumor selective tetrabranched peptide for in vivo imaging. J. Nanobiotechnol. 2018, 16, 21. [Google Scholar] [CrossRef] [PubMed]
- Avula, U.M.; Yoon, H.K.; Lee, C.H.; Kaur, K.; Ramirez, R.J.; Takemoto, Y.; Ennis, S.R.; Morady, F.; Herron, T.; Berenfeld, O.; et al. Cell-selective arrhythmia ablation for photomodulation of heart rhythm. Sci. Transl. Med. 2015, 7, 311ra172. [Google Scholar] [CrossRef] [PubMed]
- Bourré, L.; Giuntini, F.; Eggleston, I.; Mosse, C.A.; MacRobert, A.J.; Wilson, M. Effective photoinactivation of Gram-positive and Gram-negative bacterial strains using an HIV-1 Tat peptide–porphyrin conjugate. Photochem. Photobiol. Sci. 2010, 9, 1613–1620. [Google Scholar] [CrossRef]
- Eto, Y.; Yoshioka, Y.; Asavatanabodee, R.; Kida, S.; Maeda, M.; Mukai, Y.; Mizuguchi, H.; Kawasaki, K.; Okada, N.; Nakagawa, S. Transduction of adenovirus vectors modified with cell-penetrating peptides. Peptides 2009, 30, 1548–1552. [Google Scholar] [CrossRef]
- Yu, D.; Jin, C.; Ramachandran, M.; Xu, J.; Nilsson, B.; Korsgren, O.; Le Blanc, K.; Uhrbom, L.; Forsberg-Nilsson, K.; Westermark, B.; et al. Adenovirus Serotype 5 Vectors with Tat-PTD Modified Hexon and Serotype 35 Fiber Show Greatly Enhanced Transduction Capacity of Primary Cell Cultures. PLoS ONE 2013, 8, e54952. [Google Scholar] [CrossRef]
- Kida, S.; Eto, Y.; Yoshioka, Y.; Nakagawa, S.; Kawasaki, K.; Maeda, M. Evaluation of synthetic cell-penetrating peptides, Pro-rich peptide and octaargine derivatives, as adenovirus vector carrier. Protein Pept. Lett. 2010, 17, 164–167. [Google Scholar] [CrossRef]
- Nigatu, A.S.; Vupputuri, S.; Flynn, N.; Neely, B.J.; Ramsey, J.D. Evaluation of Cell-Penetrating Peptide/Adenovirus Particles for Transduction of CAR-Negative Cells. J. Pharm. Sci. 2013, 102, 1981–1993. [Google Scholar] [CrossRef][Green Version]
- Jin, C.; Yu, D.; Cancer, M.; Nilsson, B.; Leja, J.; Essand, M. Tat-PTD-modified oncolytic adenovirus driven by the SCG3 promoter and ASH1 enhancer for neuroblastoma therapy. Hum. Gene Ther. 2013, 24, 766–775. [Google Scholar] [CrossRef]
- Ma, X.-C.; Liu, P.; Zhang, X.-L.; Jiang, W.-H.; Jia, M.; Wang, C.-X.; Dong, Y.-Y.; Dang, Y.-H.; Gao, C.-G. Intranasal Delivery of Recombinant AAV Containing BDNF Fused with HA2TAT: A Potential Promising Therapy Strategy for Major Depressive Disorder. Sci. Rep. 2016, 6, 22404. [Google Scholar] [CrossRef]
- Wang, G.; Jia, T.; Xu, X.; Chang, L.; Zhang, R.; Fu, Y.; Li, Y.; Yang, X.; Zhang, K.; Lin, G.; et al. Novel miR-122 delivery system based on MS2 virus like particle surface displaying cell-penetrating peptide TAT for hepatocellular carcinoma. Oncotarget 2016, 7, 59402–59416. [Google Scholar] [CrossRef]
- Sun, Y.; Sun, Y.; Zhao, R. Establishment of MicroRNA delivery system by PP7 bacteriophage-like particles carrying cell-penetrating peptide. J. Biosci. Bioeng. 2017, 124, 242–249. [Google Scholar] [CrossRef]
- Tian, Y.; Zhou, M.; Shi, H.; Gao, S.; Xie, G.; Zhu, M.; Wu, M.; Chen, J.; Niu, Z.-W. Integration of Cell-Penetrating Peptides with Rod-like Bionanoparticles: Virus-Inspired Gene-Silencing Technology. Nano Lett. 2018, 18, 5453–5460. [Google Scholar] [CrossRef]
- Ignatovich, I.A.; Dizhe, E.B.; Pavlotskaya, A.V.; Akifiev, B.N.; Burov, S.V.; Orlov, S.; Perevozchikov, A. Complexes of Plasmid DNA with Basic Domain 47–57 of the HIV-1 Tat Protein Are Transferred to Mammalian Cells by Endocytosis-mediated Pathways. J. Biol. Chem. 2003, 278, 42625–42636. [Google Scholar] [CrossRef]
- Rudolph, C.; Plank, C.; Lausier, J.; Schillinger, U.; Müller, R.H.; Rosenecker, J. Oligomers of the Arginine-rich Motif of the HIV-1 TAT Protein Are Capable of Transferring Plasmid DNA into Cells. J. Biol. Chem. 2003, 278, 11411–11418. [Google Scholar] [CrossRef]
- Lehto, T.; Simonson, O.E.; Mäger, I.; Ezzat, K.; Sork, H.; Copolovici, D.; Viola, J.R.; Zaghloul, E.; Lundin, P.; Moreno, P.; et al. A Peptide-based Vector for Efficient Gene Transfer in Vitro and in Vivo. Mol. Ther. 2011, 19, 1457–1467. [Google Scholar] [CrossRef]
- Zhang, W.; Song, J.; Liang, R.; Zheng, X.; Chen, J.; Li, G.; Zhang, B.; Wang, K.; Yan, X.; Wang, R. Stearylated antimicrobial peptide [D]-K6L9 with cell penetrating property for efficient gene transfer. Peptides 2013, 46, 33–39. [Google Scholar] [CrossRef]
- Cardoso, A.M.; Trabulo, S.; Cardoso, A.L.; Maia, S.; Gomes, P.A.C.; Jurado, A.S.; De Lima, M.C.P. Comparison of the Efficiency of Complexes Based on S413-PV Cell-Penetrating Peptides in Plasmid DNA and siRNA Delivery. Mol. Pharm. 2013, 10, 2653–2666. [Google Scholar] [CrossRef]
- Alhakamy, N.A.; Ishiguro, S.; Uppalapati, D.; Berkland, C.; Tamura, M. AT2R Gene Delivered by Condensed Polylysine Complexes Attenuates Lewis Lung Carcinoma after Intravenous Injection or Intratracheal Spray. Mol. Cancer Ther. 2015, 15, 209–218. [Google Scholar] [CrossRef]
- Qu, W.; Chen, W.-H.; Kuang, Y.; Zeng, X.; Cheng, S.-X.; Zhou, X.; Zhuo, R.-X.; Zhang, X.-Z. Avidin–Biotin Interaction Mediated Peptide Assemblies as Efficient Gene Delivery Vectors for Cancer Therapy. Mol. Pharm. 2012, 10, 261–269. [Google Scholar] [CrossRef]
- Gong, C.; Pan, D.; Qiu, F.; Sun, P.; Zhang, Y.-H. Selective DNA Delivery to Tumor Cells Using an Oligoarginine-LTVSPWY Peptide. PLoS ONE 2014, 9, e110632. [Google Scholar] [CrossRef]
- Wang, H.; Wang, H.; Liang, J.; Jiang, Y.; Guo, Q.; Peng, H.; Xu, Q.; Huang, Y. Cell-Penetrating Apoptotic Peptide/p53 DNA Nanocomplex as Adjuvant Therapy for Drug-Resistant Breast Cancer. Mol. Pharm. 2014, 11, 3352–3360. [Google Scholar] [CrossRef] [PubMed]
- Nam, H.Y.; Kim, J.; Kim, S.W.; Bull, D.A. Cell Targeting Peptide Conjugation to siRNA Polyplexes for Effective Gene Silencing in Cardiomyocytes. Mol. Pharm. 2012, 9, 1302–1309. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zheng, X.; Koren, V.; Vashist, Y.K.; Tsui, T.Y. Highly efficient delivery of siRNA to a heart transplant model by a novel cell penetrating peptide-dsRNA binding domain. Int. J. Pharm. 2014, 469, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Ruan, W.; Zhai, Y.; Yu, K.; Wu, C.; Xu, Y. Coated microneedles mediated intradermal delivery of octaarginine/BRAF siRNA nanocomplexes for anti-melanoma treatment. Int. J. Pharm. 2018, 553, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Uchida, T.; Kanazawa, T.; Kawai, M.; Takashima, Y.; Okada, H. Therapeutic Effects on Atopic Dermatitis by Anti-RelA Short Interfering RNA Combined with Functional Peptides Tat and AT1002. J. Pharmacol. Exp. Ther. 2011, 338, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Freire, J.M.; De Figueiredo, I.R.; Valle, J.; Veiga, A.S.; Andreu, D.; Enguita, F.J.; Castanho, M.A.R.B. siRNA-cell-penetrating peptides complexes as a combinatorial therapy against chronic myeloid leukemia using BV173 cell line as model. J. Control. Release 2017, 245, 127–136. [Google Scholar] [CrossRef]
- Suh, J.S.; Lee, H.J.; Nam, H.; Jo, B.S.; Lee, D.W.; Kim, J.-H.; Lee, J.Y.; Chung, C.P.; Lee, G.; Park, Y.J. Control of cancer stem cell like population by intracellular target identification followed by the treatment with peptide-siRNA complex. Biochem. Biophys. Res. Commun. 2017, 491, 827–833. [Google Scholar] [CrossRef]
- Tuttolomondo, M.; Casella, C.; Hansen, P.L.; Polo, E.; Herda, L.M.; Dawson, K.A.; Ditzel, H.J.; Mollenhauer, J. Human DMBT1-Derived Cell-Penetrating Peptides for Intracellular siRNA Delivery. Mol. Ther. Nucleic Acids 2017, 8, 264–276. [Google Scholar] [CrossRef]
- Morais, C.; Cardoso, A.M.; Cunha, P.; Aguiar, L.; Vale, N.; Lage, E.V.; Pinheiro, M.; Nunes, C.; Gomes, P.A.C.; Reis, S.; et al. Acylation of the S413-PV cell-penetrating peptide as a means of enhancing its capacity to mediate nucleic acid delivery: Relevance of peptide/lipid interactions. Biochim. Biophys. Acta (BBA) Biomembr. 2018, 1860, 2619–2634. [Google Scholar] [CrossRef]
- Tai, Z.; Wang, X.; Tian, J.; Gao, Y.; Zhang, L.; Yao, C.; Wu, X.; Zhang, W.; Zhu, Q.; Gao, S. Biodegradable Stearylated Peptide with Internal Disulfide Bonds for Efficient Delivery of siRNA In Vitro and In Vivo. Biomacromolecules 2015, 16, 1119–1130. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, X.; Gao, Y.; Zhang, J.; Zhang, D.; Gu, S.; Zhu, G.; Liu, G.-L.; Li, X.-Y. Aptamer-functionalized peptide H3CR5C as a novel nanovehicle for codelivery of fasudil and miRNA-195 targeting hepatocellular carcinoma. Int. J. Nanomed. 2016, 11, 3891–3905. [Google Scholar] [CrossRef]
- Lo, J.H.; Hao, L.; Muzumdar, M.; Raghavan, S.; Kwon, E.J.; Pulver, E.M.; Hsu, F.; Aguirre, A.J.; Wolpin, B.M.; Fuchs, C.S.; et al. iRGD-guided Tumor-penetrating Nanocomplexes for Therapeutic siRNA Delivery to Pancreatic Cancer. Mol. Cancer Ther. 2018, 17, 2377–2388. [Google Scholar] [CrossRef] [PubMed]
- Alexander-Bryant, A.A.; Zhang, H.; Attaway, C.C.; Pugh, W.; Eggart, L.; Sansevere, R.M.; Andino, L.M.; Dinh, L.; Cantini, L.P.; Jakymiw, A. Dual peptide-mediated targeted delivery of bioactive siRNAs to oral cancer cells in vivo. Oral Oncol. 2017, 72, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Guo, H.; Lu, Y.; Feng, W.; Sun, X.; Tang, C.; Wang, X.; Shen, M. Blocking hepatic metastases of colon cancer cells using an shRNA against Rac1 delivered by activatable cell-penetrating peptide. Oncotarget 2016, 7, 77183–77195. [Google Scholar] [CrossRef][Green Version]
- Yang, Y.; Xia, X.; Dong, W.; Wang, H.; Li, L.; Ma, P.; Sheng, W.; Xu, X.; Liu, Y. Acid Sensitive Polymeric Micelles Combining Folate and Bioreducible Conjugate for Specific Intracellular siRNA Delivery. Macromol. Biosci. 2016, 16, 759–773. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.-Y.; Lai, L.-H.; Shen, J.; Wang, Q.-Q.; Xu, F.-J.; Tang, G. Gene delivery to tumor cells by cationic polymeric nanovectors coupled to folic acid and the cell-penetrating peptide octaarginine. Biomaterials 2011, 32, 7253–7262. [Google Scholar] [CrossRef]
- Ye, S.; Tian, M.-M.; Wang, T.-X.; Ren, L.; Wang, N.; Shen, L.; Shang, T. Synergistic effects of cell-penetrating peptide Tat and fusogenic peptide HA2-enhanced cellular internalization and gene transduction of organosilica nanoparticles. Nanomed. Nanotechnol. Biol. Med. 2012, 8, 833–841. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Shao, K.; Liu, Y.; Kuang, Y.; Li, J.; An, S.; Guo, Y.; Ma, H.; Jiang, C. Tumor-Targeting and Microenvironment-Responsive Smart Nanoparticles for Combination Therapy of Antiangiogenesis and Apoptosis. ACS Nano 2013, 7, 2860–2871. [Google Scholar] [CrossRef]
- Yang, Y.; Xie, X.; Yang, Y.; Li, Z.; Yu, F.; Gong, W.; Li, Y.; Zhang, H.; Wang, Z.; Mei, X. Polymer Nanoparticles Modified with Photo- and pH-Dual-Responsive Polypeptides for Enhanced and Targeted Cancer Therapy. Mol. Pharm. 2016, 13, 1508–1519. [Google Scholar] [CrossRef]
- Osman, G.; Rodriguez, J.; Chan, S.Y.; Chisholm, J.; Duncan, G.; Kim, N.; Tatler, A.L.; Shakesheff, K.M.; Hanes, J.; Suk, J.S.; et al. PEGylated enhanced cell penetrating peptide nanoparticles for lung gene therapy. J. Control. Release 2018, 285, 35–45. [Google Scholar] [CrossRef]
- Dowaidar, M.; Abdelhamid, H.N.; Hällbrink, M.; Freimann, K.; Kurrikoff, K.; Zou, X.; Langel, Ü. Magnetic Nanoparticle Assisted Self-assembly of Cell Penetrating Peptides-Oligonucleotides Complexes for Gene Delivery. Sci. Rep. 2017, 7, 9159. [Google Scholar] [CrossRef] [PubMed]
- Ben Djemaa, S.; David, S.; Aubert, K.H.; Falanga, A.; Galdiero, S.; Allard-Vannier, E.; Chourpa, I.; Munnier, E. Formulation and in vitro evaluation of a siRNA delivery nanosystem decorated with gH625 peptide for triple negative breast cancer theranosis. Eur. J. Pharm. Biopharm. 2018, 131, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.-H.; Niu, J.; Zhang, C.-Z.; Yu, W.; Wu, J.-H.; Shan, Y.-H.; Wang, X.-R.; Shen, Y.; Mao, Z.; Liang, W.; et al. TAT conjugated cationic noble metal nanoparticles for gene delivery to epidermal stem cells. Biomaterials 2014, 35, 5605–5618. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Tsutsumi, H.; Mihara, H. Cell penetration and cell-selective drug delivery using α-helix peptides conjugated with gold nanoparticles. Biomaterials 2013, 34, 4872–4879. [Google Scholar] [CrossRef] [PubMed]
- Wonder, E.; Simón-Gracia, L.; Scodeller, P.; Majzoub, R.N.; Kotamraju, V.R.; Ewert, K.; Teesalu, T.; Safinya, C. Competition of charge-mediated and specific binding by peptide-tagged cationic liposome–DNA nanoparticles in vitro and in vivo. Biomaterials 2018, 166, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.K.; Mattern-Schain, S.I.; Best, M.D.; Kirkpatrick, S.S.; Freeman, M.B.; Grandas, O.H.; Mountain, D.J. Improving the efficacy of liposome-mediated vascular gene therapy via lipid surface modifications. J. Surg. Res. 2017, 219, 136–144. [Google Scholar] [CrossRef]
- Fu, H.; Shi, K.; Hu, G.; Yang, Y.; Kuang, Q.; Lu, L.; Zhang, L.; Chen, W.; Dong, M.; Chen, Y.; et al. Tumor-Targeted Paclitaxel Delivery and Enhanced Penetration Using TAT-Decorated Liposomes Comprising Redox-Responsive Poly(Ethylene Glycol). J. Pharm. Sci. 2015, 104, 1160–1173. [Google Scholar] [CrossRef]
- Liu, Z.; Xiong, M.; Gong, J.; Zhang, Y.; Bai, N.; Luo, Y.; Li, L.; Wei, Y.; Liu, Y.; Tan, X.; et al. Legumain protease-activated TAT-liposome cargo for targeting tumours and their microenvironment. Nat. Commun. 2014, 5, 4280. [Google Scholar] [CrossRef]
- Seeman, N.C. Nucleic acid junctions and lattices. J. Theor. Biol. 1982, 99, 237–247. [Google Scholar] [CrossRef]
- Rothemund, P.W.K. Folding DNA to create nanoscale shapes and patterns. Nature 2006, 440, 297–302. [Google Scholar] [CrossRef]
- Yin, P.; Hariadi, R.; Sahu, S.; Choi, H.; Park, S.H.; LaBean, T.; Reif, J.H. Programming DNA Tube Circumferences. Science 2008, 321, 824–826. [Google Scholar] [CrossRef] [PubMed]
- Rothemund, P.W.K.; Ekani-Nkodo, A.; Papadakis, N.; Kumar, A.; Fygenson, D.K.; Winfree, E. Design and Characterization of Programmable DNA Nanotubes. J. Am. Chem. Soc. 2004, 126, 16344–16352. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Ong, L.L.; Sun, W.; Song, J.; Dong, M.; Shih, W.M.; Yin, P. DNA brick crystals with prescribed depths. Nat. Chem. 2014, 6, 994–1002. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Ong, L.L.; Shih, W.M.; Yin, P. Three-Dimensional Structures Self-Assembled from DNA Bricks. Science 2012, 338, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Sacca, B.; Niemeyer, C.M. Functionalization of DNA nanostructures with proteins. Chem. Soc. Rev. 2011, 40, 5910. [Google Scholar] [CrossRef] [PubMed]
- Sacca, B.; Meyer, R.; Erkelenz, M.; Kiko, K.; Arndt, A.; Schroeder, H.; Rabe, K.S.; Niemeyer, C.M. Orthogonal protein decoration of DNA origami. Angew. Chem. Int. Ed. Engl. 2010, 49, 9378–9383. [Google Scholar] [CrossRef]
- Schaffert, D.H.; Okholm, A.H.; Sørensen, R.S.; Nielsen, J.S.; Tørring, T.; Rosen, C.B.; Kodal, A.L.B.; Mortensen, M.R.; Gothelf, K.V.; Kjems, J. Intracellular Delivery of a Planar DNA Origami Structure by the Transferrin-Receptor Internalization Pathway. Small 2016, 12, 2634–2640. [Google Scholar] [CrossRef]
- Liu, J.; Song, L.; Liu, S.; Jiang, Q.; Liu, Q.; Li, N.; Wang, Z.-G.; Ding, B. A DNA-Based Nanocarrier for Efficient Gene Delivery and Combined Cancer Therapy. Nano Lett. 2018, 18, 3328–3334. [Google Scholar] [CrossRef]
- Madhanagopal, B.R.; Zhang, S.; Demirel, E.; Wady, H.; Chandrasekaran, A.R. DNA Nanocarriers: Programmed to Deliver. Trends Biochem. Sci. 2018, 43, 997–1013. [Google Scholar] [CrossRef]
- Yan, J.; Hu, C.; Wang, P.; Zhao, B.; Ouyang, X.; Zhou, J.; Liu, R.; He, D.; Fan, C.; Song, S. Growth and origami folding of DNA on nanoparticles for high-efficiency molecular transport in cellular imaging and drug delivery. Angew. Chem. Int. Ed. Engl. 2015, 54, 2431–2435. [Google Scholar] [CrossRef]
- Guo, X.-L.; Yuan, D.-D.; Song, T.; Li, X.-M. DNA nanopore functionalized with aptamer and cell-penetrating peptide for tumor cell recognition. Anal. Bioanal. Chem. 2017, 4, 713–797. [Google Scholar] [CrossRef]
- Lee, H.; Lytton-Jean, A.; Chen, Y.; Love, K.T.; Park, A.I.; Karagiannis, E.; Sehgal, A.; Querbes, W.; Zurenko, C.S.; Jayaraman, M.; et al. Molecularly self-assembled nucleic acid nanoparticles for targeted in vivo siRNA delivery. Nat. Nanotechnol. 2012, 7, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Bruchez, M.P.; Armitage, B. Closing the Loop: Constraining TAT Peptide by γPNA Hairpin for Enhanced Cellular Delivery of Biomolecules. Bioconjug. Chem. 2018, 29, 2892–2898. [Google Scholar] [CrossRef]
- Liu, Y.; Kumar, S.; Taylor, R. Mix-and-match nanobiosensor design: Logical and spatial programming of biosensors using self-assembled DNA nanostructures. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2018, 10, e1518. [Google Scholar] [CrossRef] [PubMed]
- Ijäs, H.; Nummelin, S.; Shen, B.; Kostiainen, M.A.; Linko, V. Dynamic DNA Origami Devices: From Strand-Displacement Reactions to External-Stimuli Responsive Systems. Int. J. Mol. Sci. 2018, 19, 2114. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, P.; Egholm, M.; Berg, R.; Buchardt, O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 1991, 254, 1497–1500. [Google Scholar] [CrossRef]
- Demidov, V.V.; Potaman, V.N.; Frank-Kamenetskil, M.; Egholm, M.; Buchard, O.; Sönnichsen, S.H.; Nlelsen, P.E. Stability of peptide nucleic acids in human serum and cellular extracts. Biochem. Pharmacol. 1994, 48, 1310–1313. [Google Scholar] [CrossRef]
- Takiya, T.; Seto, Y.; Yasuda, H.; Suzuki, T.; Kawai, K. An empirical approach for thermal stability (Tm) prediction of PNA/DNA duplexes. Nucleic Acids Symp. Ser. 2004, 48, 131–132. [Google Scholar] [CrossRef]
- Uhlmann, E.; Peyman, A.; Breipohl, G.; Will, D.W. PNA. Synthetic Polyamide Nucleic Acids with Unusual Binding Properties. Angew. Chem. Int. Ed. Engl. 1998, 37, 2796–2823. [Google Scholar] [CrossRef]
- Eriksson, M.; Nielsen, P.E. PNA-nucleic acid complexes. Structure, stability and dynamics. Q. Rev. Biophys. 1996, 29, 369–394. [Google Scholar] [CrossRef]
- Sahu, B.; Sacui, I.; Rapireddy, S.; Zanotti, K.J.; Bahal, R.; Armitage, B.A.; Ly, D.H. Synthesis and characterization of conformationally preorganized, (R)-diethylene glycol-containing gamma-peptide nucleic acids with superior hybridization properties and water solubility. J. Org. Chem. 2011, 76, 5614–5627. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, P.E. Addressing the challenges of cellular delivery and bioavailability of peptide nucleic acids (PNA). Q. Rev. Biophys. 2005, 38, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Koppelhus, U.; Awasthi, S.K.; Zachar, V.; Holst, H.U.; Ebbesen, P.; Nielsen, P.E. Cell-Dependent Differential Cellular Uptake of PNA, Peptides, and PNA-Peptide Conjugates. Antisense Nucleic Acid Drug Dev. 2002, 12, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Egholm, M.; Buchardt, O.; Christensen, L.; Behrens, C.; Freier, S.M.; Driver, D.A.; Berg, R.H.; Kim, S.K.; Norden, B.; Nielsen, P.E. PNA hybridizes to complementary oligonucleotides obeying the Watson–Crick hydrogen-bonding rules. Nature 1993, 365, 566–568. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, T.; Nielsen, P.E. Cellular Delivery of Peptide Nucleic Acids (PNAs). Adv. Struct. Saf. Stud. 2013, 1050, 193–205. [Google Scholar]
- Quijano, E.; Bahal, R.; Ricciardi, A.; Saltzman, W.M.; Glazer, P.M. Therapeutic Peptide Nucleic Acids: Principles, Limitations, and Opportunities. Yale J. Biol. Med. 2017, 90, 583–598. [Google Scholar]
- Pooga, M.; Soomets, U.; Hällbrink, M.; Valkna, A.; Saar, K.; Rezaei, K.; Kahl, U.; Hao, J.-X.; Xu, X.-J.; Wiesenfeld-Hallin, Z.; et al. Cell penetrating PNA constructs regulate galanin receptor levels and modify pain transmission in vivo. Nat. Biotechnol. 1998, 16, 857–861. [Google Scholar] [CrossRef]
- Aldrian-Herrada, G.; Desarmenien, M.G.; Orcel, H.; Boissin-Agasse, L.; Mery, J.; Brugidou, J.; Rabie, A. A peptide nucleic acid (PNA) is more rapidly internalized in cultured neurons when coupled to a retro-inverso delivery peptide. The antisense activity depresses the target mRNA and protein in magnocellular oxytocin neurons. Nucleic Acids Res. 1998, 26, 4910–4916. [Google Scholar] [CrossRef]
- Dragulescu-Andrasi, A.; Rapireddy, S.; He, G.; Bhattacharya, B.; Hyldig-Nielsen, J.J.; Zon, G.; Ly, D.H. Cell-permeable peptide nucleic acid designed to bind to the 5’-untranslated region of E-cadherin transcript induces potent and sequence-specific antisense effects. J. Am. Chem. Soc. 2006, 128, 16104–16112. [Google Scholar] [CrossRef]
- Dragulescu-Andrasi, A.; Rapireddy, S.; Frezza, B.M.; Gayathri, C.; Gil, R.R.; Ly, D.H. A Simple γ-Backbone Modification Preorganizes Peptide Nucleic Acid into a Helical Structure. J. Am. Chem. Soc. 2006, 128, 10258–10267. [Google Scholar] [CrossRef]
- Rogers, F.A.; Lin, S.S.; Hegan, D.C.; Krause, D.S.; Glazer, P.M. Targeted Gene Modification of Hematopoietic Progenitor Cells in Mice Following Systemic Administration of a PNA-peptide Conjugate. Mol. Ther. 2011, 20, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, T.; Pankratova, S.; Nielsen, P.E. Calcium Ions Effectively Enhance the Effect of Antisense Peptide Nucleic Acids Conjugated to Cationic Tat and Oligoarginine Peptides. Chem. Biol. 2005, 12, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Babar, I.A.; Cheng, C.J.; Booth, C.J.; Liang, X.; Weidhaas, J.; Saltzman, W.M.; Slack, F.J. Nanoparticle-based therapy in an in vivo microRNA-155 (miR-155)-dependent mouse model of lymphoma. Proc. Natl. Acad. Sci. USA 2012, 109, E1695–E1704. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-X.; Song, Z.; Lao, Y.-H.; Xu, X.; Gong, J.; Cheng, D.; Chakraborty, S.; Park, J.S.; Li, M.; Huang, D.; et al. Nonviral gene editing via CRISPR/Cas9 delivery by membrane-disruptive and endosomolytic helical polypeptide. Proc. Natl. Acad. Sci. USA 2018, 115, 4903–4908. [Google Scholar] [CrossRef]
- Lostalé-Seijo, I.; Louzao, I.; Juanes, M.; Montenegro, J. Peptide/Cas9 nanostructures for ribonucleoprotein cell membrane transport and gene edition. Chem. Sci. 2017, 8, 7923–7931. [Google Scholar]
- Ramakrishna, S.; Dad, A.-B.K.; Beloor, J.; Gopalappa, R.; Lee, S.-K.; Kim, H.H. Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Res. 2014, 24, 1020–1027. [Google Scholar] [CrossRef]
- Del’Guidice, T.; Lepetit-Stoffaes, J.P.; Bordeleau, L.J.; Roberge, J.; Theberge, V.; Lauvaux, C.; Barbeau, X.; Trottier, J.; Dave, V.; Roy, D.C.; et al. Membrane permeabilizing amphiphilic peptide delivers recombinant transcription factor and CRISPR-Cas9/Cpf1 ribonucleoproteins in hard-to-modify cells. PLoS ONE 2018, 13, e0195558. [Google Scholar] [CrossRef]
- Suresh, B.; Ramakrishna, S.; Kim, H.H. Cell-Penetrating Peptide-Mediated Delivery of Cas9 Protein and Guide RNA for Genome Editing. Adv. Struct. Saf. Stud. 2016, 1507, 81–94. [Google Scholar]
- Coriat, R.; Faivre, S.; Mir, O.; Dreyer, C.; Ropert, S.; Bouattour, M.; Desjardins, R.; Goldwasser, F.; Raymond, E. Pharmacokinetics and safety of DTS-108, a human oligopeptide bound to SN-38 with an esterase-sensitive cross-linker in patients with advanced malignancies: A Phase I study. Int. J. Nanomed. 2016, 11, 6207–6216. [Google Scholar] [CrossRef]
- Lulla, R.R.; Goldman, S.; Yamada, T.; Beattie, C.W.; Bressler, L.; Pacini, M.; Pollack, I.F.; Fisher, P.G.; Packer, R.J.; Dunkel, I.J.; et al. Phase I trial of p28 (NSC745104), a non-HDM2-mediated peptide inhibitor of p53 ubiquitination in pediatric patients with recurrent or progressive central nervous system tumors: A Pediatric Brain Tumor Consortium Study. Neuro Oncol. 2016, 18, 1319–1325. [Google Scholar] [CrossRef]
- Warso, M.A.; Richards, J.M.; Mehta, D.; Christov, K.; Schaeffer, C.; Bressler, L.R.; Yamada, T.; Majumdar, D.; Kennedy, S.; Beattie, C.W.; et al. A first-in-class, first-in-human, phase I trial of p28, a non-HDM2-mediated peptide inhibitor of p53 ubiquitination in patients with advanced solid tumours. Br. J. Cancer 2013, 108, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Unkart, J.T.; Wapnir, I.L.; E González, J.; Harootunian, A.; Wallace, A.M.; Chen, S.L. Intraoperative Tumor Detection Using a Ratiometric Activatable Fluorescent Peptide: A First-in-Human Phase 1 Study. Ann. Surg. Oncol. 2017, 24, 3167–3173. [Google Scholar] [CrossRef] [PubMed]
- Gurbel, P.A.; Bliden, K.P.; Turner, S.E.; Tantry, U.S.; Gesheff, M.G.; Barr, T.P.; Covic, L.; Kuliopulos, A. Cell-Penetrating Pepducin Therapy Targeting PAR1 in Subjects with Coronary Artery Disease. Arter. Thromb. Vasc. Biol. 2016, 36, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Martyna, A.; Hard, R.L.; Wang, J.; Appiah-Kubi, G.; Coss, C.; Phelps, M.; Rossman, J.; Pei, D. Discovery and Mechanism of Highly Efficient Cyclic Cell-Penetrating Peptides. Biochemistry 2016, 55, 2601–2612. [Google Scholar] [CrossRef]
- Pooga, M.; Hallbrink, M.; Zorko, M.; Langle, U. Cell penetration by transportan. FASEB J. 1998, 12, 67–77. [Google Scholar] [CrossRef]
- Robbins, P.F.; A Kantor, J.; Salgaller, M.; Hand, P.H.; Fernsten, P.D.; Schlom, J. Transduction and expression of the human carcinoembryonic antigen gene in a murine colon carcinoma cell line. Cancer Res. 1991, 51, 3657–3662. [Google Scholar]
- Jones, S.; Farquhar, M.; Martin, A.; Howl, J. Intracellular translocation of the decapeptide carboxyl terminal of Gi3 alpha induces the dual phosphorylation of p42/p44 MAP kinases. Biochim. Biophys. Acta 2005, 1745, 207–214. [Google Scholar] [CrossRef]
- Gao, C.; Mao, S.; Ditzel, H.J.; Farnaes, L.; Wirsching, P.; Lerner, R.A.; Janda, K.D. A cell-penetrating peptide from a novel pVII-pIX phage-displayed random peptide library. Bioorg. Med. Chem. 2002, 10, 4057–4065. [Google Scholar] [CrossRef]
- Lim, J.; Kim, J.; Duong, T.; Lee, G.; Yoon, J.; Kim, H.; Ruley, H.E.; El-Rifai, W.; Jo, D. Antitumor activity of cell-permeable p18(INK4c) with enhanced membrane and tissue penetration. Mol. Ther. 2012, 20, 1540–1549. [Google Scholar] [CrossRef]
- Lim, J.; Duong, T.; Do, N.; Do, P.; Kim, J.; Kim, H.; El-Rifai, W.; Ruley, H.E.; Jo, D. Antitumor activity of cell-permeable RUNX3 protein in gastric cancer cells. Clin. Cancer Res. 2013, 19, 680–690. [Google Scholar] [CrossRef]
- Findlay, D.M.; Houssami, S.; Lin, H.Y.; Myers, D.E.; Brady, C.L.; Darcy, P.K.; Ikeda, K.; Martin, T.J.; Sexton, P.M. Truncation of the porcine calcitonin receptor cytoplasmic tail inhibits internalization and signal transduction but increases receptor affinity. Mol. Endocrinol. 1994, 8, 1691–1700. [Google Scholar] [PubMed]
- Sadler, K.; Eom, K.D.; Yang, J.-L.; Dimitrova, Y.; Tam, J.P. Translocating Proline-Rich Peptides from the Antimicrobial Peptide Bactenecin 7. Biochemistry 2002, 41, 14150–14157. [Google Scholar] [CrossRef] [PubMed]
- McConnell, S.J.; Thon, V.J.; Spinella, D.G. Isolation of fibroblast growth factor receptor binding sequences using evolved phage display libraries. Comb. Chem. High Throughput Screen. 1999, 2, 155–163. [Google Scholar] [PubMed]
- Zong, X.; Jiang, D.-Y.; Li, G.-J.; Cai, J.-L. Construction of keratinocyte growth factor phage active peptides for the promotion of epidermal cell proliferation. Zhonghua Yi Xue Za Zhi 2013, 1058–1062. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CPPs-Non-Tissue Specific | Peptide Sequence | Origin |
| Cationic | ||
| Tat [5] | GRKKRRQRRRPPQ | HIV Tat Protein |
| Ant [6] | RQIKIWFQNRRMKWKK | Antennapedia homeodomain |
| 8-Arginine [12] | RRRRRRRR | n/a |
| 8-Lysine [13] | KKKKKKKK | n/a |
| PTD-5 [17] | RRQRRTSKLMKR | Phage display |
| Hydrophobic | ||
| Transportan [165] | GWTLNSAGYLLGKINLKALAALAKKIL | Galanin and mastoparan |
| MAP [166] | KLALKLALKALKAALKLA | Galanin and mastoparan |
| TP10 [167] | AGYLLGKINLKALAALAKKIL | |
| Pep-7 [168] | SDLWEMMMVSLACQY | CHL8 peptide |
| Amphipathic | ||
| Azurin p18 [169] | LSTAADMQGVVTDGMASG | Azurin |
| Azurin p28 [170] | LSTAADMQGVVTDGMASGLDKDYLKPDD | Azurin |
| hCT18-32 [171] | KFHTFPQTAIGVGAP | Calcitonin |
| Bac 7 [172] | RRIRPRPPRLPRPRPRPLPFPRPG | Bactenecin |
| CPPs-Tissue Specific | Peptide Sequence | Origin |
| CTP [31,66] | APWHLSSQYSRT | Phage display |
| K5-FGF [173] | AAVALLPAVLLALLP | Phage display |
| HAP-1 [27] | SFHQFARATLAS | Phage display |
| 293P-1 [174] | SNNNVRPIHIWP | Phage display |
| Vascular Endothelium [33] | SIGYPLP | Phage display |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taylor, R.E.; Zahid, M. Cell Penetrating Peptides, Novel Vectors for Gene Therapy. Pharmaceutics 2020, 12, 225. https://doi.org/10.3390/pharmaceutics12030225
Taylor RE, Zahid M. Cell Penetrating Peptides, Novel Vectors for Gene Therapy. Pharmaceutics. 2020; 12(3):225. https://doi.org/10.3390/pharmaceutics12030225
Chicago/Turabian StyleTaylor, Rebecca E., and Maliha Zahid. 2020. "Cell Penetrating Peptides, Novel Vectors for Gene Therapy" Pharmaceutics 12, no. 3: 225. https://doi.org/10.3390/pharmaceutics12030225
APA StyleTaylor, R. E., & Zahid, M. (2020). Cell Penetrating Peptides, Novel Vectors for Gene Therapy. Pharmaceutics, 12(3), 225. https://doi.org/10.3390/pharmaceutics12030225