Solid State Stability and Kinetics of Degradation for Candesartan—Pure Compound and Pharmaceutical Formulation

 ,
, 


 ,
, 
 and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples and Preparation
2.2. Spectroscopic Description of Samples
2.3. Thermal Stability Investigations
2.4. Kinetic Investigations
3. Results and Discussions
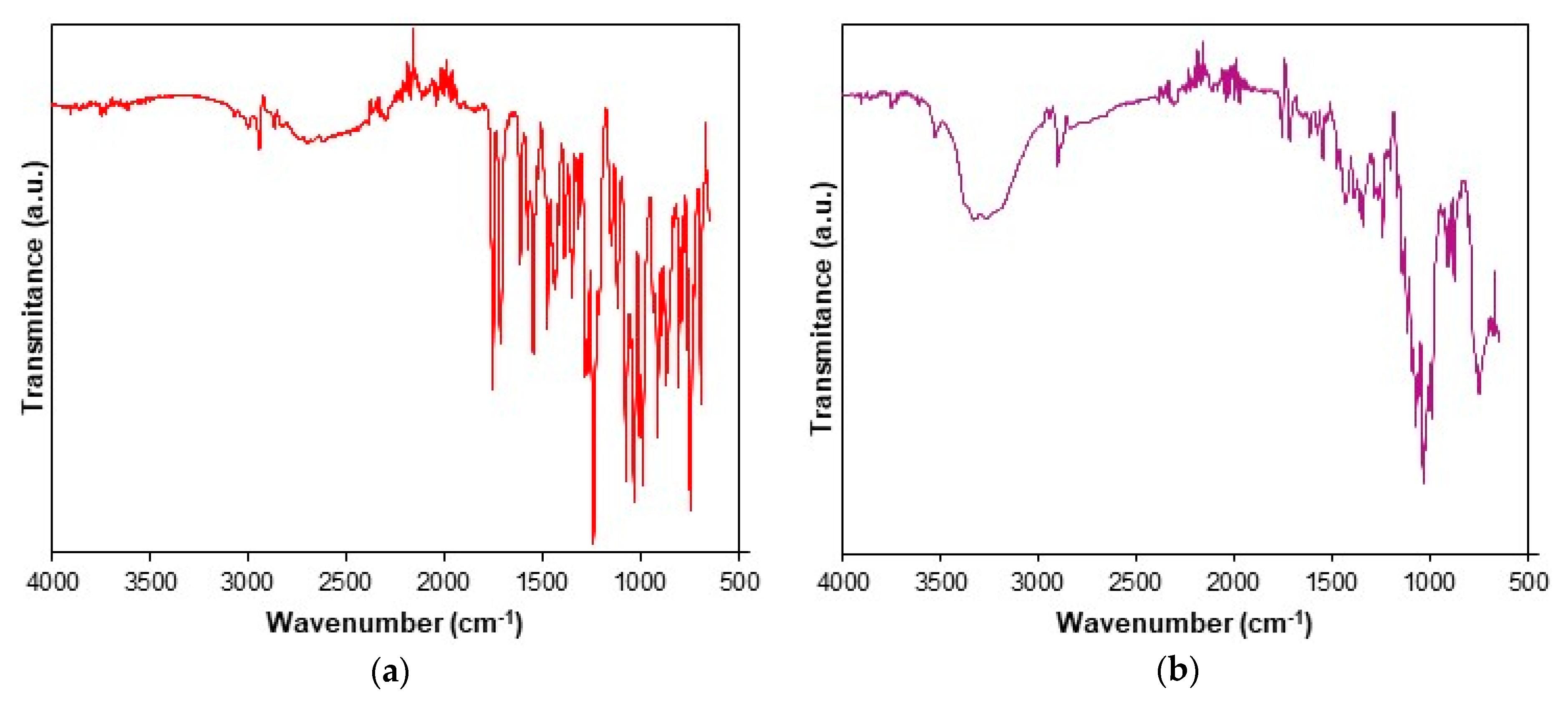
3.1. Spectroscopic Description of Samples
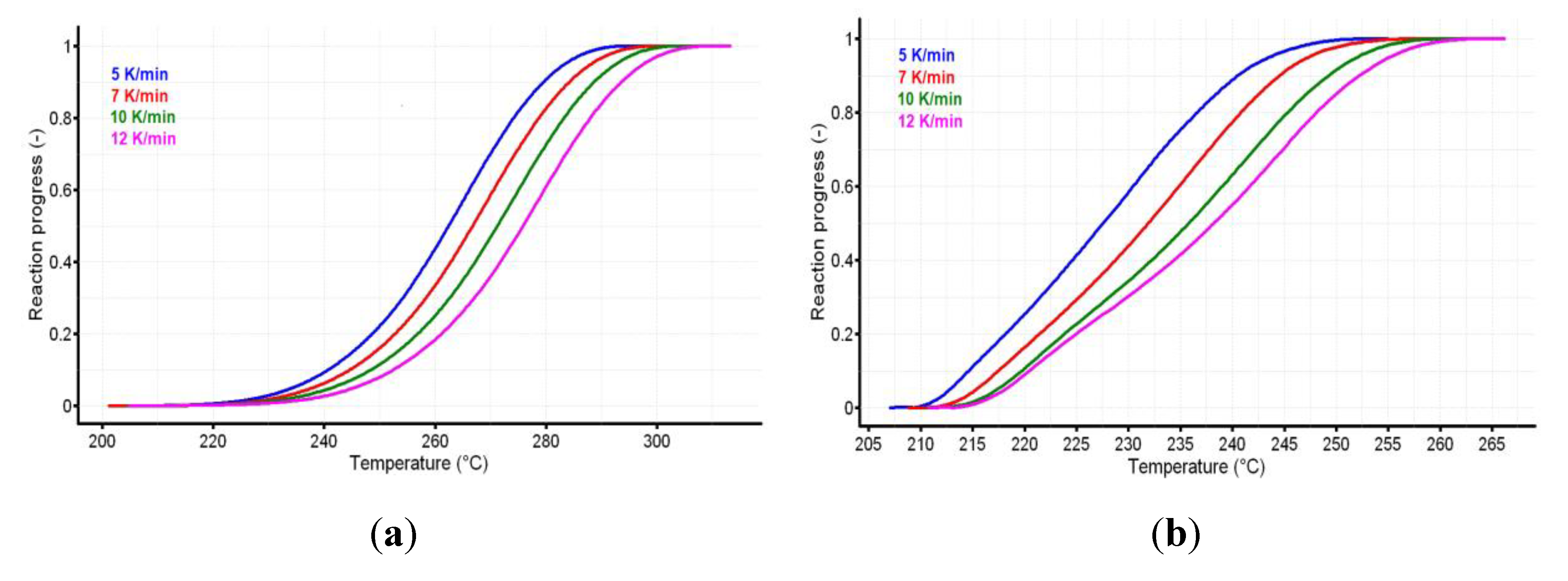
3.2. Thermal Stability Investigations
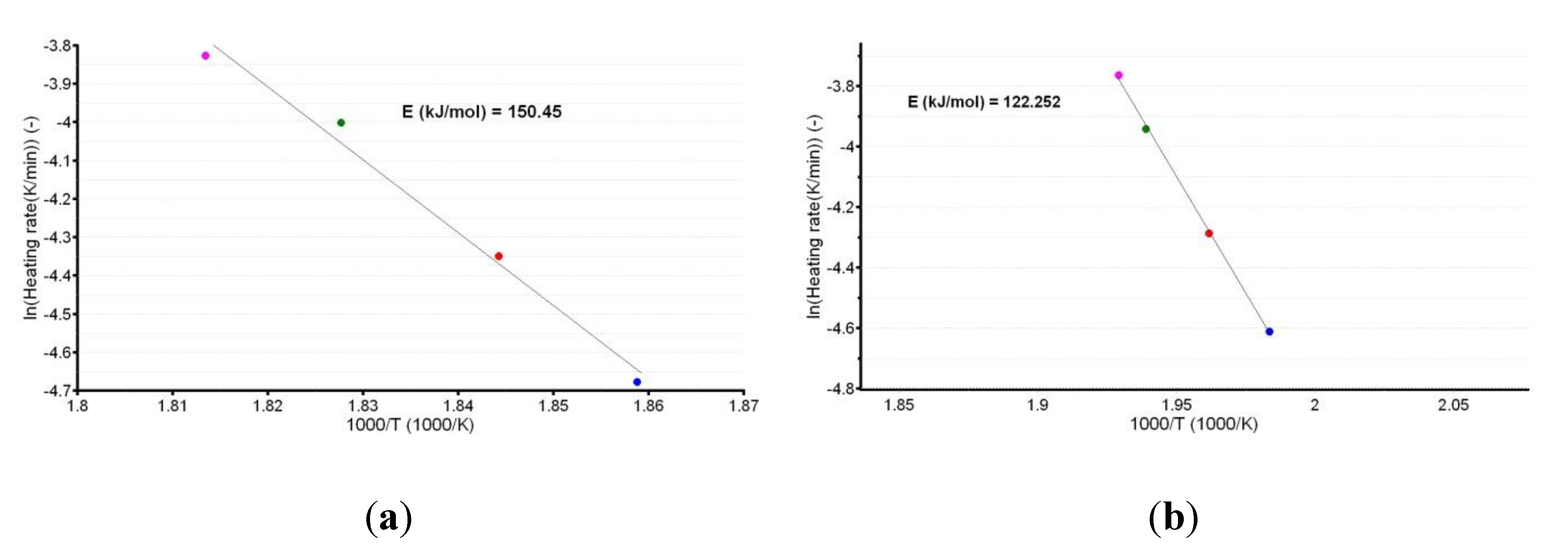
3.3. Kinetic Investigations
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bulsara, K.G.; Makaryus, A.N. Candesartan; StatPearls Publishing: Treasure Island, FL, USA, 2018. [Google Scholar]
- Takezako, T.; Unal, H.; Karnik, S.S.; Node, K. Current topics in angiotensin II type 1 receptor research: Focus on inverse agonism, receptor dimerization and biased agonism. Pharmacol. Res. 2017, 123, 40–50. [Google Scholar] [CrossRef]
- Gleiter, C.H.; Jägle, C.; Gresser, U.; Mörike, K. Candesartan. Cardiovasc. Drug Rev. 2004, 22, 263–284. [Google Scholar] [CrossRef]
- Gavras, H. Update on the clinical pharmacology of candesartan cilexetil. Am. J. Hypertens. 2000, 13, S25–S30. [Google Scholar] [CrossRef]
- Zhao, D.; Liu, H.; Dong, P. A Meta-analysis of antihypertensive effect of telmisartan versus candesartan in patients with essential hypertension. Clin. Exp. Hypertens. 2019, 41, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Isobe, N.; Taniguchi, K.; Oshima, S.; Ono, Z.; Adachi, H.; Toyama, T.; Naito, S.; Hoshizaki, H.; Kamiyama, H. Candesartan cilexetil improves left ventricular function, left ventricular hypertrophy, and endothelial function in patients with hypertensive heart disease. Circ. J. 2002, 66, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, M.A.; Swedberg, K.; Granger, C.B.; Held, P.; McMurray, J.J.V.; Michelson, E.L.; Olofsson, B.; Östergren, J.; Yusuf, S. Effects of candesartan on mortality and morbidity in patients with chronic heart failure: The CHARM-overall programme. Lancet 2003, 362, 759–766. [Google Scholar] [CrossRef]
- Donaire, J.A.G.; Ruilope, L.M. Angiotensin receptor blockade in diabetic renal disease-Focus on candesartan. Diabetes Res. Clin. Pract. 2007, 76, S22–S30. [Google Scholar] [CrossRef] [PubMed]
- Behl, T.; Kotwani, A. Potential of angiotensin II receptor blockers in the treatment of diabetic retinopathy. Life Sci. 2017, 176, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gullo, C.E.; de Almeida Zia, V.A.; Vilela-Martin, J.F. Blockade of renin angiotensin system in heart failure post-myocardial infarction: What is the best therapy? Recent Pat. Cardiovasc. Drug Discov. 2014, 9, 28–37. [Google Scholar] [CrossRef]
- Buda, V.; Andor, M.; Cristescu, C.; Voicu, M.; Cochera, F.; Tuduce, P.; Petrescu, L.; Tomescu, M.C. The effect of candesartan on pentraxin-3 plasma levels as marker of endothelial dysfunction in patients with essential arterial hypertension. Ir. J. Med. Sci. 2017, 186, 621–629. [Google Scholar] [CrossRef]
- Chen, T.; Yu, D.; Cornelius, V.; Qin, R.; Cai, Y.; Jiang, Z.; Zhao, Z. Potential health impact and cost-effectiveness of drug therapy for prehypertension. Int. J. Cardiol. 2017, 240, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Ducharme, A.; Swedberg, K.; Pfeffer, M.A.; Cohen-Solal, A.; Granger, C.B.; Maggioni, A.P.; Michelson, E.L.; McMurray, J.J.V.; Olsson, L.; Rouleau, J.L.; et al. Prevention of atrial fibrillation in patients with symptomatic chronic heart failure by candesartan in the Candesartan in Heart failure: Assessment of Reduction in Mortality and morbidity (CHARM) program. Am. Heart J. 2006, 152, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Stovner, L.J.; Linde, M.; Gravdahl, G.B.; Tronvik, E.; Aamodt, A.H.; Sand, T.; Hagen, K. A comparative study of candesartan versus propranolol for migraine prophylaxis: A randomised, triple-blind, placebo-controlled, double cross-over study. Cephalalgia 2014, 34, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Venkat, P.; Shen, Y.; Chopp, M.; Chen, J. Cell-based and pharmacological neurorestorative therapies for ischemic stroke. Neuropharmacology 2018, 134, 310–322. [Google Scholar] [CrossRef]
- Torika, N.; Asraf, K.; Apte, R.N.; Fleisher-Berkovich, S. Candesartan ameliorates brain inflammation associated with Alzheimer’s disease. CNS Neurosci. Ther. 2018, 24, 231–242. [Google Scholar] [CrossRef]
- Perez-Lloret, S.; Otero-Losada, M.; Toblli, J.E.; Capani, F. Renin-angiotensin system as a potential target for new therapeutic approaches in Parkinson’s disease. Expert Opin. Investig. Drugs 2017, 26, 1163–1173. [Google Scholar] [CrossRef]
- Kim, M.J.; Im, D.S. Suppressive effects of type I angiotensin receptor antagonists, candesartan and irbesartan on allergic asthma. Eur. J. Pharmacol. 2019, 852, 25–33. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, X. Candesartan targeting of angiotensin II type 1 receptor demonstrates benefits for hypertension in pregnancy via the NF-κB signaling pathway. Mol. Med. Rep. 2018, 18, 705–714. [Google Scholar] [CrossRef]
- Candesartan on Drugs.com. Available online: https://www.drugs.com/mtm/candesartan.html (accessed on 27 November 2019).
- Pinto, E.C.; do Carmo, F.A.; da Honório, T.S.; de da Barros, R.C.S.A.; Castro, H.C.R.; Rodrigues, C.R.; Esteves, V.S.D.; Rocha, H.V.A.; de Sousa, V.P.; Cabral, L.M. Influence of the Efavirenz Micronization on Tableting and Dissolution. Pharmaceutics 2012, 4, 430–441. [Google Scholar] [CrossRef]
- Viet Nguyen, K.; Laidmäe, I.; Kogermann, K.; Lust, A.; Meos, A.; Viet Ho, D.; Raal, A.; Heinämäki, J.; Thi Nguyen, H. Preformulation Study of Electrospun Haemanthamine-Loaded Amphiphilic Nanofibers Intended for a Solid Template for Self-Assembled Liposomes. Pharmaceutics 2019, 11, 499. [Google Scholar] [CrossRef]
- Modhave, D.; Barrios, B.; Paudel, A. PVP-H2O2 Complex as a New Stressor for the Accelerated Oxidation Study of Pharmaceutical Solids. Pharmaceutics 2019, 11, 457. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, L.J.; Stofella, N.C.F.; Veiga, A.; Federle, S.; da Toledo, M.G.T.; Bernardi, L.S.; Oliveira, P.R.; Carvalho Filho, M.A.S.; Andreazza, I.F.; Murakami, F.S. Physical-chemical characterization studies of ketoprofen for orodispersible tablets. J. Therm. Anal. Calorim. 2018, 133, 1521–1533. [Google Scholar] [CrossRef]
- Ledeţi, I.; Ledeţi, A.; Vlase, G.; Vlase, T.; Matusz, P.; Bercean, V.; Şuta, L.-M.; Piciu, D. Thermal stability of synthetic thyroid hormone l-thyroxine and l-thyroxine sodium salt hydrate both pure and in pharmaceutical formulations. J. Pharm. Biomed. Anal. 2016, 125, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Bandas (Ratiu), C.; Orha, C.; Misca, C.; Lazau, C.; Sfirloaga, P.; Olariu, S. Photocatalytical Inactivation of Enterococcus faecalis from Water Using Functional Materials Based on Natural Zeolite and Titanium Dioxide. Chin. J. Chem. Eng. 2014, 22, 38–43. [Google Scholar] [CrossRef]
- Faya, P.; Seaman, J.W.; Stamey, J.D. Using accelerated drug stability results to inform long-term studies in shelf life determination. Stat. Med. 2018, 37, 2599–2615. [Google Scholar] [CrossRef]
- Zilker, M.; Sörgel, F.; Holzgrabe, U. A systematic review of the stability of finished pharmaceutical products and drug substances beyond their labeled expiry dates. J. Pharm. Biomed. Anal. 2019, 166, 222–235. [Google Scholar] [CrossRef]
- Ledeti, I.; Pusztai, A.M.; Murariu, M.; Olariu, S.; Ivan, C.; Circioban, D.; Vlase, G.; Ledeti, A.; Vlase, T.; Matusz, P. Comparative instrumental investigations of some bile acids. J. Therm. Anal. Calorim. 2018, 134, 1345–1350. [Google Scholar] [CrossRef]
- Ledeti, A.; Vlase, G.; Vlase, T.; Circioban, D.; Dehelean, C.; Ledeti, I.; Suta, L.M. Binary adduct formation of desipramine with dicarboxylic acids: An instrumental study. J. Therm. Anal. Calorim. 2018, 131, 167–173. [Google Scholar] [CrossRef]
- Ledeti, I.; Bolintineanu, S.; Vlase, G.; Circioban, D.; Ledeti, A.; Vlase, T.; Suta, L.M.; Caunii, A.; Murariu, M. Compatibility study between antiparkinsonian drug Levodopa and excipients by FTIR spectroscopy, X-ray diffraction and thermal analysis. J. Therm. Anal. Calorim. 2017, 130, 433–441. [Google Scholar] [CrossRef]
- Ledeti, I.; Pusztai, A.M.; Muresan, C.M.; Circioban, D.; Vlase, G.; Murariu, M.; Suta, L.-M.; Vlase, T.; Ledeti, A.; Suciu, O.; et al. Study of solid-state degradation of prochlorperazine and promethazine. J. Therm. Anal. Calorim. 2018, 134, 731–740. [Google Scholar] [CrossRef]
- Wu, C.; You, J.; Wang, X. Study on the thermal decomposition of famciclovir. J. Therm. Anal. Calorim. 2018, 131, 1361–1371. [Google Scholar] [CrossRef]
- Dartora, P.C.; da Loureiro, M.R.; de Camargo Forte, M.M. Crystallization kinetics and morphology of poly(lactic acid) with polysaccharide as nucleating agent. J. Therm. Anal. Calorim. 2018, 134, 1705–1713. [Google Scholar] [CrossRef]
- Moisei, A.; Gligor, F.; Bojiţă, M.; Chiş, A.; Totan, M.; Vonica-Gligor, L.A.; Ciurba, A. Compatibility and stability studies of antihypertensive/excipients by thermal methods, Used in the preformulation phase. Farmacia 2014, 62, 1239–1248. [Google Scholar]
- Amer, A.M.; Allam, A.N.; Abdallah, O.Y. Comparative Pharmaceutical Evaluation of Candesartan and Candesartan Cilexetil: Physicochemical Properties, In Vitro Dissolution and Ex Vivo In Vivo Studies. AAPS PharmSciTech 2018, 19, 661–667. [Google Scholar] [CrossRef]
- Tita, I.C.; Marian, E.; Tita, B.; Toma, C.C.; Vicas, L. Thermal Behaviour of Candesartan Active substance and in pharmaceutical compounds. Rev. Chim. 2017, 68, 1895–1902. [Google Scholar]
- Sharma, G.; Beg, S.; Thanki, K.; Katare, O.P.; Jain, S.; Kohli, K.; Singh, B. Systematic development of novel cationic self-nanoemulsifying drug delivery systems of candesartan cilexetil with enhanced biopharmaceutical performance. RSC Adv. 2015, 5, 71500–71513. [Google Scholar] [CrossRef]
- Yuce, M.; Guven, O.; Karahan, S.; Saracbasi, O.; Capan, Y. Preformulation of Immediate Release Candesartan Cilexetil Tablets Using Full Factorial Experimental Design. Lat. Am. J. Pharm. 2016, 35, 921–930. [Google Scholar]
- Cui, P.; Yin, Q.; Guo, Y.; Gong, J. Polymorphic crystallization and transformation of candesartan cilexetil. Ind. Eng. Chem. Res. 2012, 51, 12910–12916. [Google Scholar] [CrossRef]
- Ledeti, I.; Murariu, M.; Vlase, G.; Vlase, T.; Doca, N.; Ledeti, A.; Suta, L.-M.; Olariu, T. Investigation of thermal-induced decomposition of iodoform. J. Therm. Anal. Calorim. 2017, 127, 565–570. [Google Scholar] [CrossRef]
- Nozela, W.C.; Nozela, C.F.V.; Silva, F.R.; Dias, D.S.; Almeida, S.; Ribeiro, C.A.; Crespi, M.S. Kinetic study of the energetic reuse from torrefied sewage sludge and urban pruning blends. J. Therm. Anal. Calorim. 2018, 134, 1285–1291. [Google Scholar] [CrossRef]
- El-Sadek, M.H.; Ahmed, H.M.; El-Barawy, K.; Morsi, M.B.; El-Didamony, H.; Bjorkman, B. Non-isothermal carbothermic reduction kinetics of mechanically activated ilmenite containing self-reducing mixtures. J. Therm. Anal. Calorim. 2018, 131, 2457–2465. [Google Scholar] [CrossRef]
- Mothe, C.G.; de Miranda, I.C. Decomposition through pyrolysis process of coconut fiber and rice husk and determination of kinetic parameters according isoconversional methods. J. Therm. Anal. Calorim. 2018, 131, 601–609. [Google Scholar] [CrossRef]
- Antar, K.; Jemal, M. A thermogravimetric study into the effects of additives and water vapor on the reduction of gypsum and Tunisian phosphogypsum with graphite or coke in a nitrogen atmosphere. J. Therm. Anal. Calorim. 2018, 132, 113–125. [Google Scholar] [CrossRef]
- Erceg, M.; Kresic, I.; Vrandecic, N.S.; Jakic, M. Different approaches to the kinetic analysis of thermal degradation of poly(ethylene oxide). J. Therm. Anal. Calorim. 2018, 131, 325–334. [Google Scholar] [CrossRef]
- Ledeţi, I.; Fuliaş, A.; Vlase, G.; Vlase, T.; Bercean, V.; Doca, N. Thermal behavior and kinetic study of some triazoles as potential anti-inflammatory agents. J. Therm. Anal. Calorim. 2013, 114, 1295–1305. [Google Scholar] [CrossRef]
- Ledeti, I.; Vlase, G.; Vlase, T.; Doca, N.; Bercean, V.; Fulias, A. Thermal decomposition, kinetic study and evolved gas analysis of 1,3,5-triazine-2,4,6-triamine. J. Therm. Anal. Calorim. 2014, 118, 1057–1063. [Google Scholar] [CrossRef]
- Ledeţi, I.; Vlase, G.; Vlase, T.; Fuliaş, A. Kinetic analysis of solid-state degradation of pure pravastatin versus pharmaceutical formulation. J. Therm. Anal. Calorim. 2015, 121, 1103–1110. [Google Scholar] [CrossRef]
- Circioban, D.; Ledeţi, I.; Vlase, G.; Ledeţi, A.; Axente, C.; Vlase, T.; Dehelean, C. Kinetics of heterogeneous-induced degradation for artesunate and artemether. J. Therm. Anal. Calorim. 2018, 134, 749–756. [Google Scholar] [CrossRef]
- Vlase, T.; Albu, P.; Ledeti, A.; Circioban, D.; Mateescu, M.; Mosoiu, C.; Vlase, G. Thermal behavior of entacapone, a catechol-O-methyltransferase inhibitor used in Parkinson’s disease. J. Therm. Anal. Calorim. 2018, 134, 711–720. [Google Scholar] [CrossRef]
- Ledeţi, I.; Vlase, G.; Vlase, T.; Bercean, V.; Fuliaş, A. Kinetic of solid-state degradation of transitional coordinative compounds containing functionalized 1,2,4-triazolic ligand. J. Therm. Anal. Calorim. 2015, 121, 1049–1057. [Google Scholar] [CrossRef]
- Ardiana, F.; Lestari, M.L.A.D.; Indrayanto, G. Candesartan Cilexetil. In Profiles of Drug Substances, Excipients and Related Methodology; Brittain, H.G., Ed.; Profiles of Drug Substances, Excipients and Related Methodology; Academic Press: Cambridge, MA, USA, 2012; Volume 37, pp. 79–112. [Google Scholar]
- Silverstein, R.M.; Webster, F.X.; Kiemle, D. Spectrometric Identification of Organic Compounds, 7th ed.; Wiley: Hoboken, NY, USA, 2005; pp. 72–125. ISBN 0-471-39362-2. [Google Scholar]
- Chi, Y.; Xu, W.; Yang, Y.; Yang, Z.; Lv, H.; Yang, S.; Lin, Z.; Li, J.; Gu, J.; Hill, C.L.; et al. Three Candesartan Salts with Enhanced Oral Bioavailability. Cryst. Growth Des. 2015, 15, 3707–3714. [Google Scholar] [CrossRef]
- Svetlik, J.; Martvon, A.; Lesko, J. Preparation and Spectral Properties of Tetrazoles. Chem. Zvesti 1979, 33, 521–527. [Google Scholar]
- Shaikh, S.M.; Avachat, A.M. Enhancement of Solubility and Permeability of Candesartan Cilexetil by Using Different Pharmaceutical Interventions. Curr. Drug Deliv. 2011, 8, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Buda, V.; Andor, M.; Ledeti, A.; Ledeti, I.; Vlase, G.; Vlase, T.; Cristescu, C.; Voicu, M.; Suciu, L.; Tomescu, M.C. Comparative solid-state stability of perindopril active substance vs. pharmaceutical formulation. Int. J. Mol. Sci. 2017, 18, 164. [Google Scholar] [CrossRef]
- Budrugeac, P.; Segal, E. On the Li and Tang’s isoconversional method for kinetic analysis of solid-state reactions from thermoanalytical data. J. Mater. Sci. 2001, 36, 2707–2710. [Google Scholar] [CrossRef]
- Budrugeac, P.; Homentcovschi, D.; Segal, E. Critical analysis of the isoconversional methods for evaluating the activation energy. I. Theoretical background. J. Therm. Anal. Calorim. 2001, 63, 457–463. [Google Scholar] [CrossRef]
- Budrugeac, P.; Segal, E. Applicability of the Kissinger equation in thermal analysis. J. Therm. Anal. Calorim. 2007, 88, 703–707. [Google Scholar] [CrossRef]
- Ledeţi, I.; Alexa, A.; Bercean, V.; Vlase, G.; Vlase, T.; Şuta, L.M.; Fuliaş, A. Synthesis and degradation of Schiff bases containing heterocyclic pharmacophore. Int. J. Mol. Sci. 2015, 16, 1711–1727. [Google Scholar] [CrossRef]
- Friedman, H.L. New methods for evaluating kinetic parameters from thermal analysis data. J. Polym. Sci. Part B Polym. Lett. 1969, 7, 41–46. [Google Scholar] [CrossRef]
- Ozawa, T. Estimation of activation energy by isoconversion methods. Thermochim. Acta 1992, 203, 159–165. [Google Scholar] [CrossRef]
- Ozawa, T. A New Method of Analyzing Thermogravimetric Data. Bull. Chem. Soc. Jpn. 1965, 38, 1881–1886. [Google Scholar] [CrossRef]
- Flynn, J.H. The isoconversional method for determination of energy of activation at constant heating rates-Corrections for the Doyle approximation. J. Therm. Anal. 1983, 27, 95–102. [Google Scholar] [CrossRef]
- Flynn, J.H.; Wall, L.A. A quick, direct method for the determination of activation energy from thermogravimetric data. J. Polym. Sci. Part B Polym. Lett. 1966, 4, 323–328. [Google Scholar] [CrossRef]
- Galwey, A.K. What is meant by the term ‘variable activation energy’ when applied in the kinetic analyses of solid state decompositions (crystolysis reactions)? Thermochim. Acta 2003, 397, 249–268. [Google Scholar] [CrossRef]
- Vyazovkin, S.V.; Lesnikovich, A.I. An approach to the solution of the inverse kinetic problem in the case of complex processes. Part 1. Methods employing a series of thermoanalytical curves. Thermochim. Acta 1990, 165, 273–280. [Google Scholar] [CrossRef]
- Vyazovkin, S.V.; Goryachko, V.I.; Lesnikovich, A.I. An approach to the solution of the inverse kinetic problem in the case of complex processes. Part III. Parallel independent reactions. Thermochim. Acta 1992, 197, 41–51. [Google Scholar] [CrossRef]
- Vyazovkin, S.; Goryachko, V. Potentialities of software for kinetic processing of thermoanalytical data by the isoconversion method. Thermochim. Acta 1992, 194, 221–230. [Google Scholar] [CrossRef]
- Vyazovkin, S.; Linert, W. Kinetic-analysis of reversible thermal-decomposition of solids. Int. J. Chem. Kinet. 1995, 27, 73–84. [Google Scholar] [CrossRef]
- Vyazovkin, S.; Wight, C.A. Kinetics in solids. Annu. Rev. Phys. Chem. 1997, 48, 125–149. [Google Scholar] [CrossRef]
- Serra, R.; Sempere, J.; Nomen, R. A new method for the kinetic study of thermoanalytical data: The non-parametric kinetics model. Thermochim. Acta 1998, 316, 37–45. [Google Scholar] [CrossRef]
- Serra, R.; Sempere, J.; Nomen, R. The non-parametric kinetics. A new method for the kinetic study of thermoanalytical data. J. Therm. Anal. 1998, 52, 933–943. [Google Scholar] [CrossRef]
- Sempere, J.; Nomen, R.; Serra, R. Progress in non-parametric kinetics. J. Therm. Anal. Calorim. 1999, 56, 843–849. [Google Scholar] [CrossRef]
- Wall, M.E.; Rechtsteiner, A.; Rocha, L.M. Singular value decomposition and principal component analysis. In A Practical Approach to Microarray Data Analysis; Berrar, D.P., Dunitzky, W., Granzow, M., Eds.; Kluwer: Alfen am Rhein, The Netherlands, 2003; pp. 91–109. ISBN 1402072600. [Google Scholar]
- Sestak, J.; Berggren, G. Study of the kinetics of the mechanism of solid-state reactions at increasing temperatures. Thermochim. Acta 1971, 3, 1–12. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
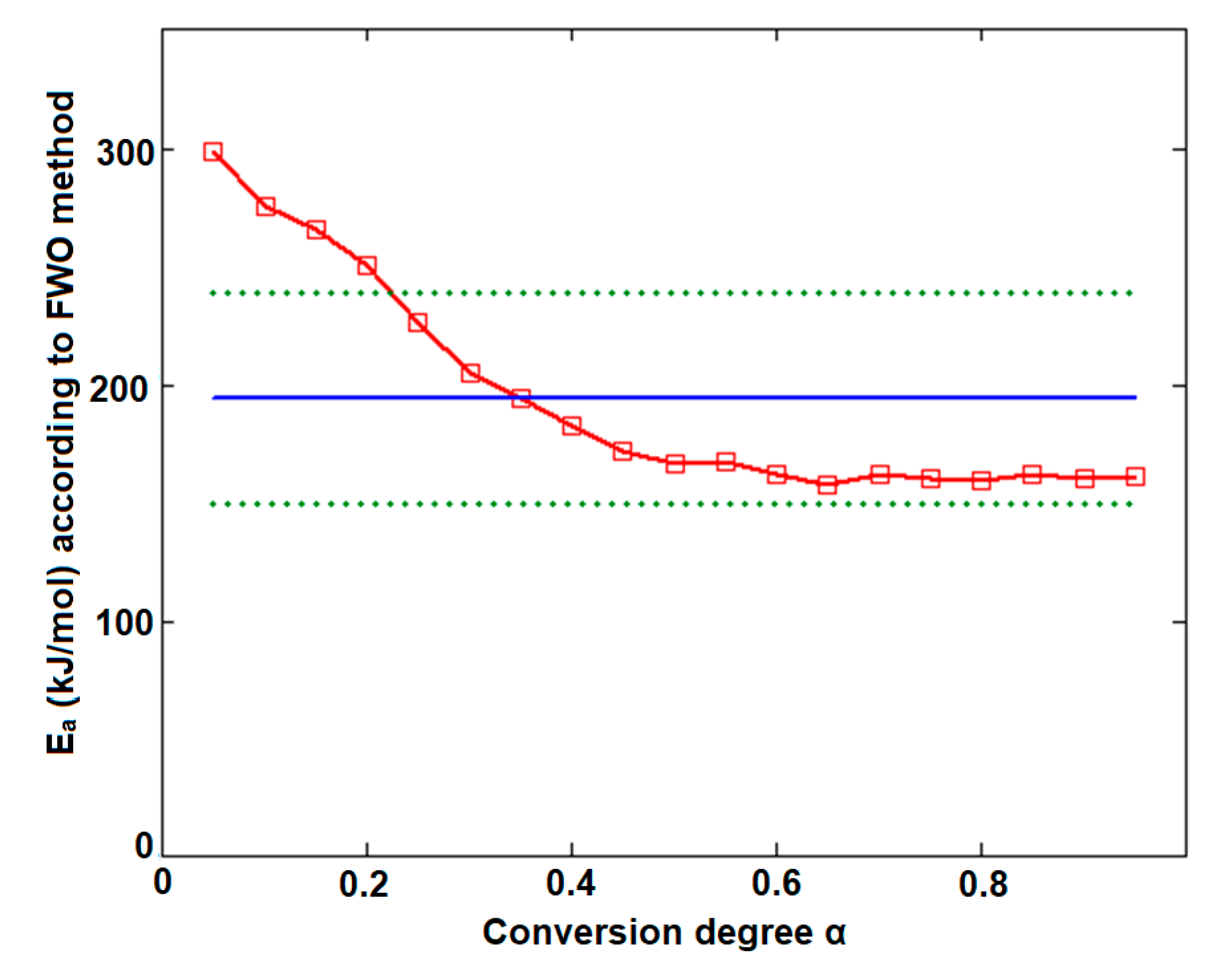
| Conversion Degree α | Ea (kJ/mol) vs. α for | |||
|---|---|---|---|---|
| CC | CCTAB | |||
| Fr | FWO | Fr | FWO | |
| 0.05 | 152.9 | 168.5 | 197.0 | 307.3 |
| 0.10 | 153.1 | 163.3 | 215.0 | 278.5 |
| 0.15 | 152.1 | 160.3 | 215.7 | 263.6 |
| 0.20 | 151.0 | 158.5 | 179.2 | 248.4 |
| 0.25 | 151.3 | 157.1 | 133.1 | 227.6 |
| 0.30 | 149.0 | 156.0 | 120.6 | 208.1 |
| 0.35 | 149.2 | 155.1 | 112.7 | 192.1 |
| 0.40 | 153.6 | 154.7 | 125.1 | 181.0 |
| 0.45 | 155.8 | 154.9 | 126.3 | 173.6 |
| 0.50 | 156.8 | 155.2 | 134.5 | 168.0 |
| 0.55 | 157.3 | 155.5 | 140.6 | 164.6 |
| 0.60 | 158.0 | 155.9 | 137.9 | 161.8 |
| 0.65 | 158.5 | 156.3 | 144.8 | 159.6 |
| 0.70 | 158.9 | 156.7 | 159.6 | 158.8 |
| 0.75 | 158.6 | 157.0 | 176.1 | 160.2 |
| 0.80 | 158.5 | 157.4 | 166.4 | 161.7 |
| 0.85 | 157.8 | 157.7 | 161.1 | 161.7 |
| 0.90 | 158.8 | 157.9 | 149.2 | 160.3 |
| 0.95 | 162.8 | 158.6 | 201.3 | 163.7 |
| (kJ/mol) | 155.5 ± 0.9 | 157.7 ± 0.7 | 157.7 ± 7.2 | 194.8 ± 10.5 |
| Sample | Step | λ (%) | A (s−1) | Ea (kJ/mol) | n | m | R2 | f(α) | (kJ/mol) |
|---|---|---|---|---|---|---|---|---|---|
| CC | 1 | 87.0 | 1.6 × 1016 ± 2.1 × 104 | 162.8 ± 9.1 | 1/3 | 0 | 0.996 | (1 − x)1/3 | 154.5 ± 11.1 |
| 2 | 8.5 | 8.6 × 1013 ± 4.4 × 109 | 147.2 ± 2.0 | 0 | 1/3 | 0.996 | x1/3 | ||
| CCTAB | 1 | 75.3 | 9.6 × 1020 ± 4.5 × 106 | 198.2 ± 11.5 | 1/4 | 0 | 0.993 | (1 − x)1/4 | 192.5 ± 16.6 |
| 2 | 24.5 | 4.6 × 1018 ± 7.8 × 108 | 176.1 ± 5.1 | 0 | 5/3 | 0.999 | x5/3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buda, V.; Baul, B.; Andor, M.; Man, D.E.; Ledeţi, A.; Vlase, G.; Vlase, T.; Danciu, C.; Matusz, P.; Peter, F.; et al. Solid State Stability and Kinetics of Degradation for Candesartan—Pure Compound and Pharmaceutical Formulation. Pharmaceutics 2020, 12, 86. https://doi.org/10.3390/pharmaceutics12020086
Buda V, Baul B, Andor M, Man DE, Ledeţi A, Vlase G, Vlase T, Danciu C, Matusz P, Peter F, et al. Solid State Stability and Kinetics of Degradation for Candesartan—Pure Compound and Pharmaceutical Formulation. Pharmaceutics. 2020; 12(2):86. https://doi.org/10.3390/pharmaceutics12020086
Chicago/Turabian StyleBuda, Valentina, Bianca Baul, Minodora Andor, Dana Emilia Man, Adriana Ledeţi, Gabriela Vlase, Titus Vlase, Corina Danciu, Petru Matusz, Francisc Peter, and et al. 2020. "Solid State Stability and Kinetics of Degradation for Candesartan—Pure Compound and Pharmaceutical Formulation" Pharmaceutics 12, no. 2: 86. https://doi.org/10.3390/pharmaceutics12020086
APA StyleBuda, V., Baul, B., Andor, M., Man, D. E., Ledeţi, A., Vlase, G., Vlase, T., Danciu, C., Matusz, P., Peter, F., & Ledeţi, I. (2020). Solid State Stability and Kinetics of Degradation for Candesartan—Pure Compound and Pharmaceutical Formulation. Pharmaceutics, 12(2), 86. https://doi.org/10.3390/pharmaceutics12020086