Sphingomyelin-Based Nanosystems (SNs) for the Development of Anticancer miRNA Therapeutics


Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
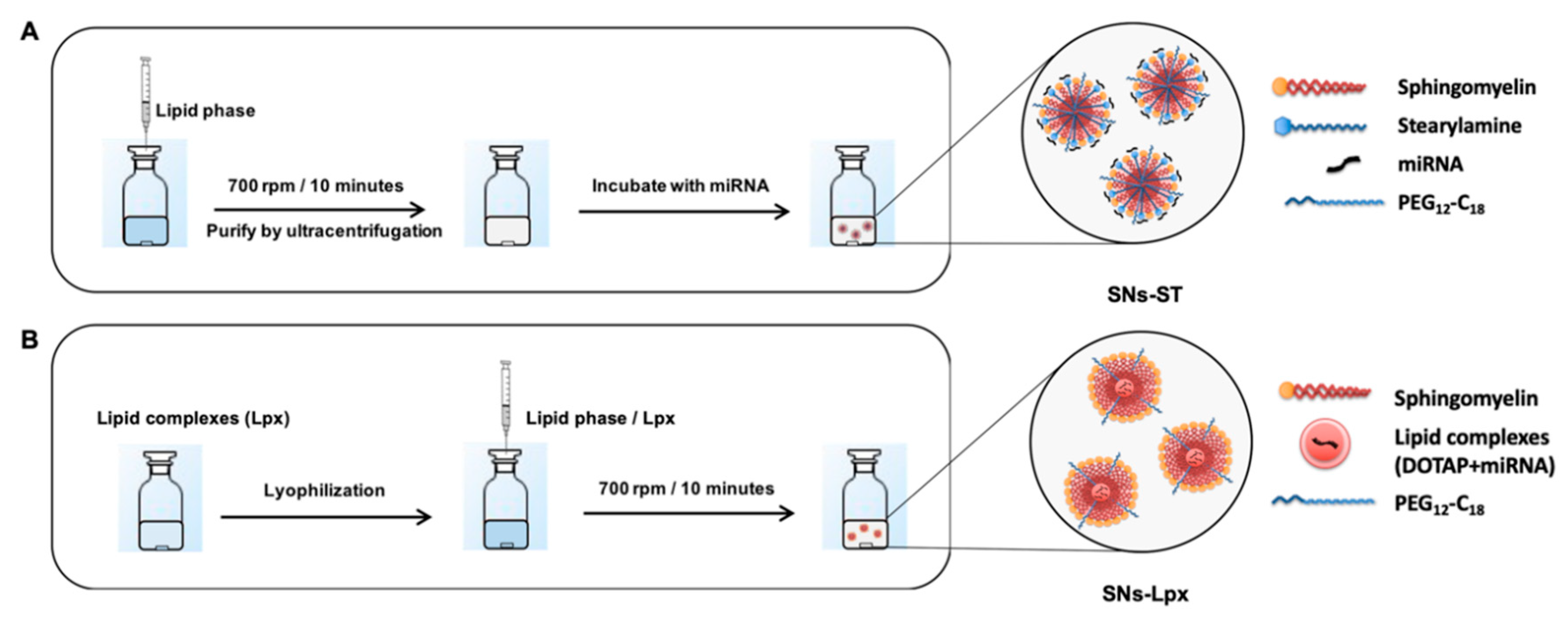
2.2. Preparation and Characterization of SNs
2.3. Preparation and Characterization of Cationic Stearylamine SNs (SNs-ST) and Association of miRNA (miR)
2.4. Preparation and Characterization of Lipid Complexes of miRNA with Cationic Lipids
2.5. Loading of Lpx into SNs (SNs-Lpx)
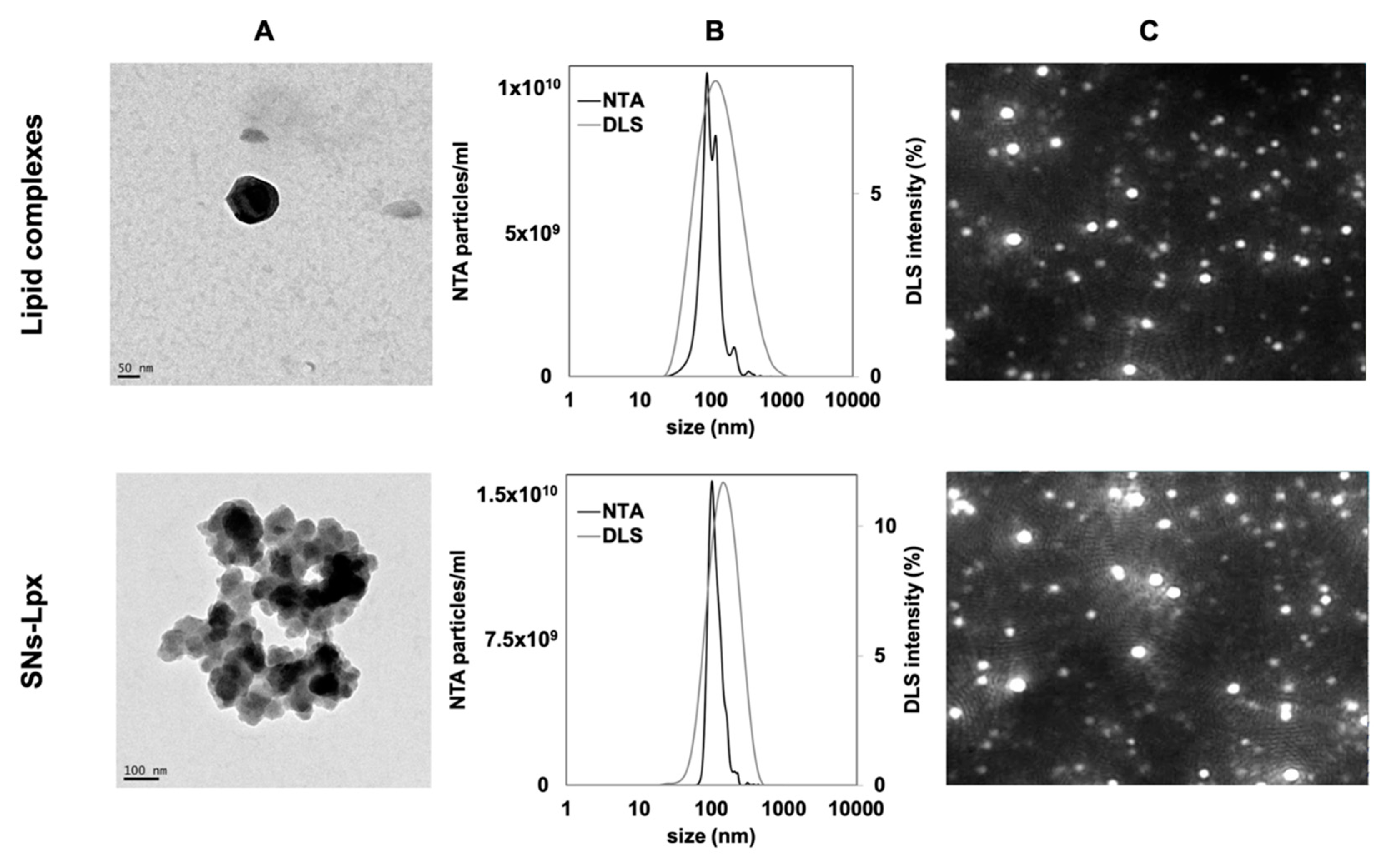
2.6. Characterization of Nanosystems (Size and Zeta-Potential Measurements, Transmission Electron Microscopic (TEM), and Nanoparticle Tracking Analysis (NTA))
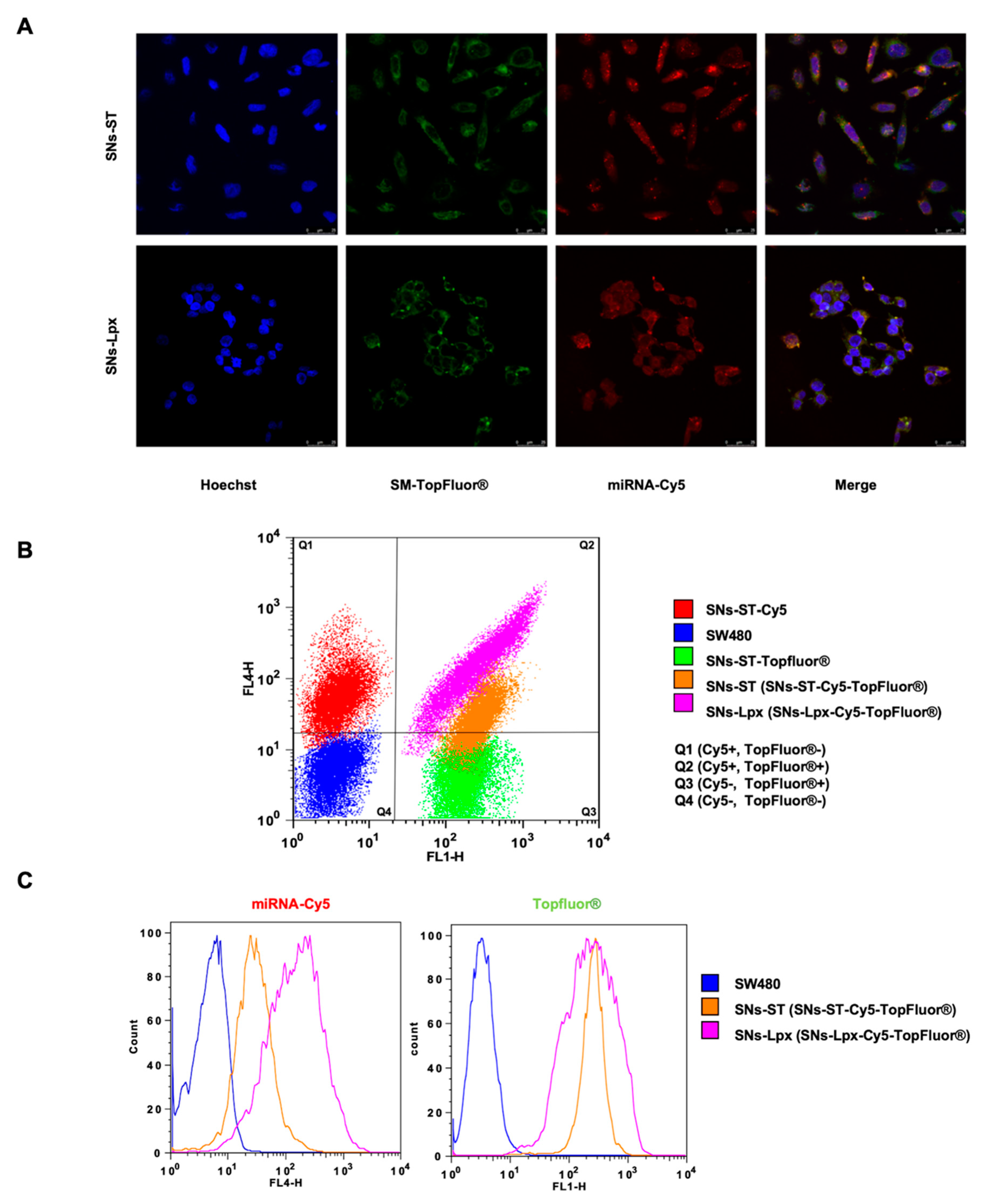
2.7. In Vitro Cell Uptake
2.8. Transfection Efficiency
2.9. Functional Assays
2.10. Statistical Analysis
3. Results and Discussion
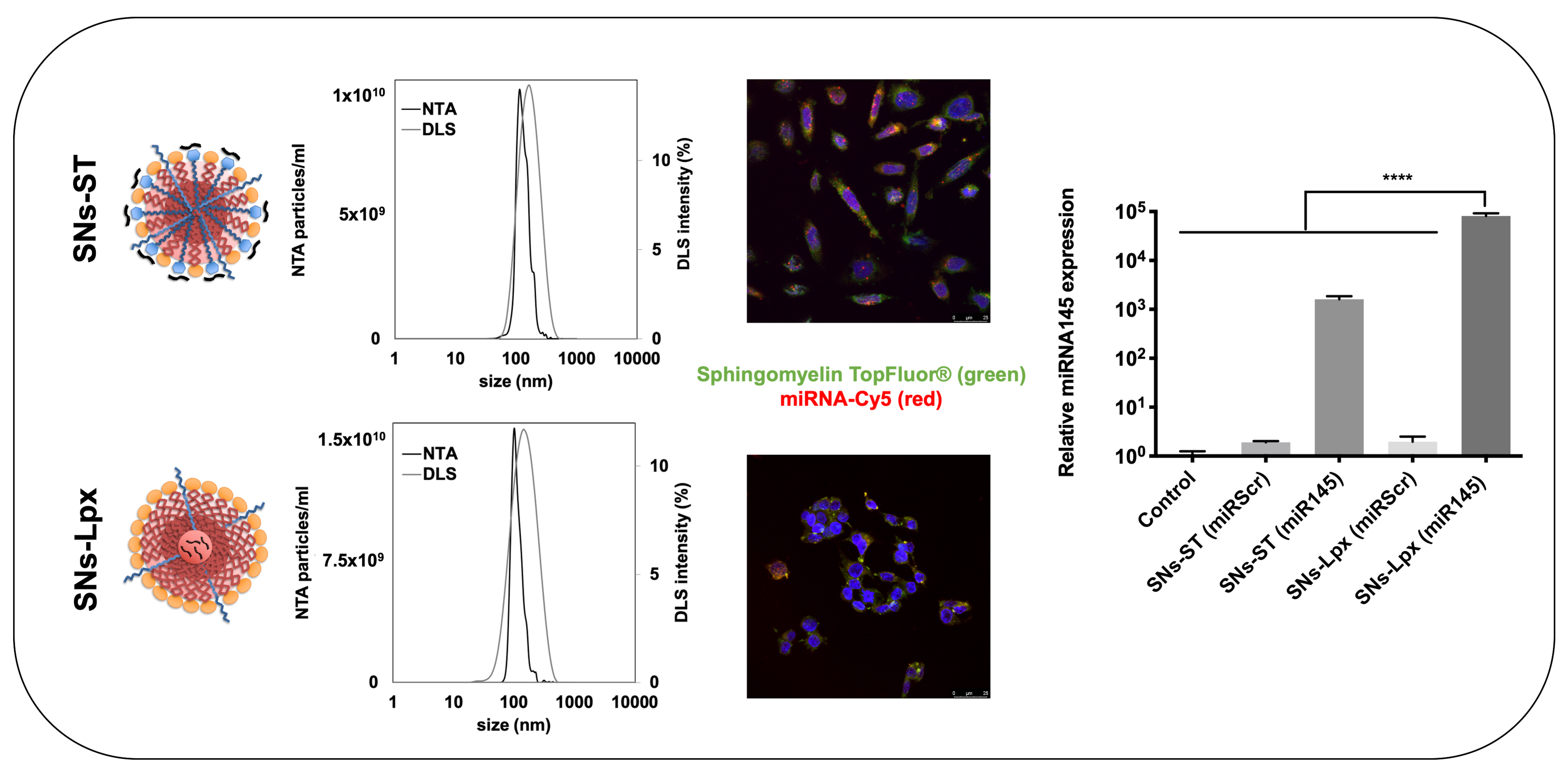
3.1. Development and Characterizations of miRNA-Loaded SNs
3.1.1. Development and Characterization of SNs-ST
3.1.2. Development and Characterization of SNs-Lpx
3.2. In Vitro Cell Uptake
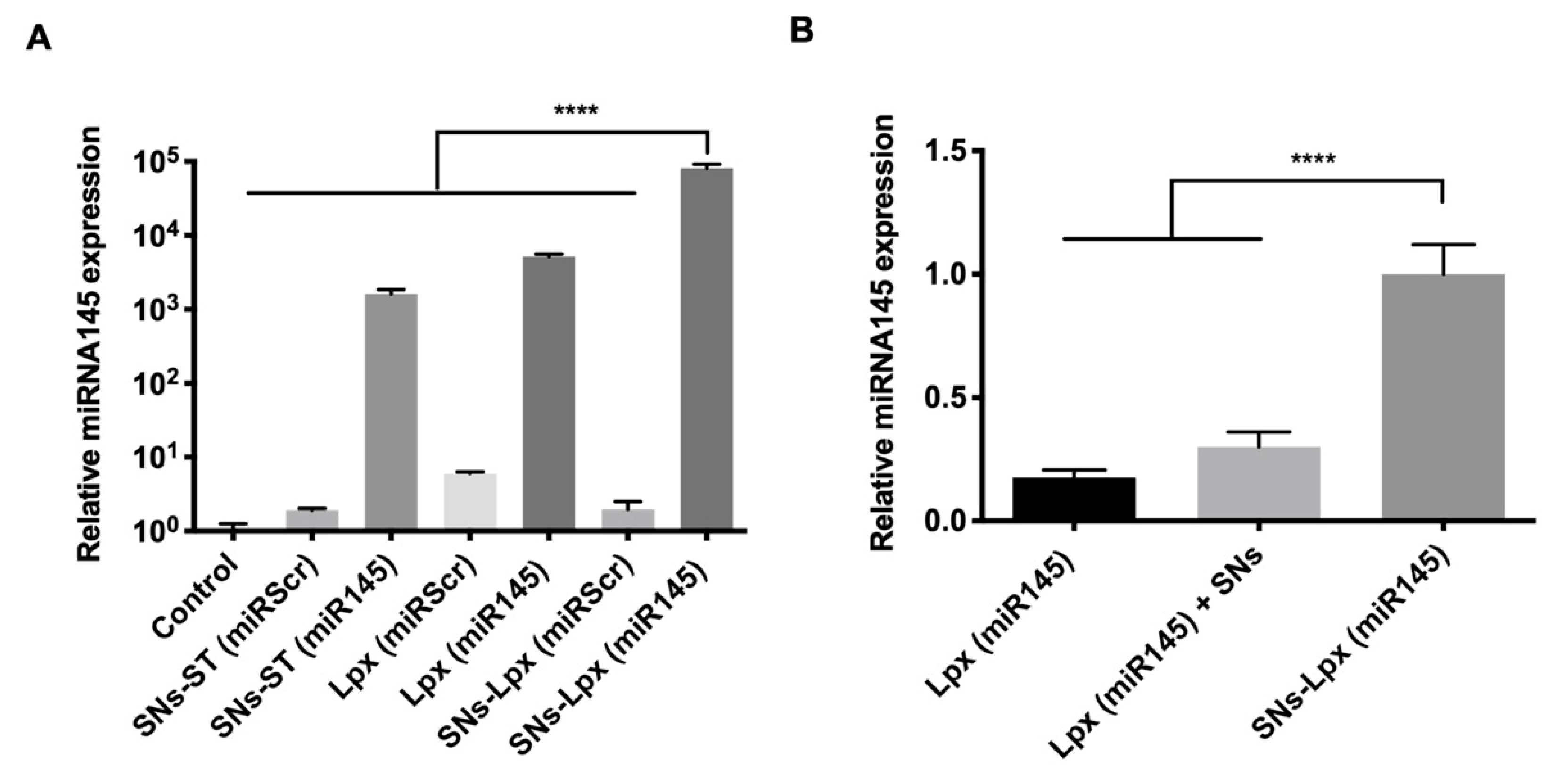
3.3. Transfection Efficiency
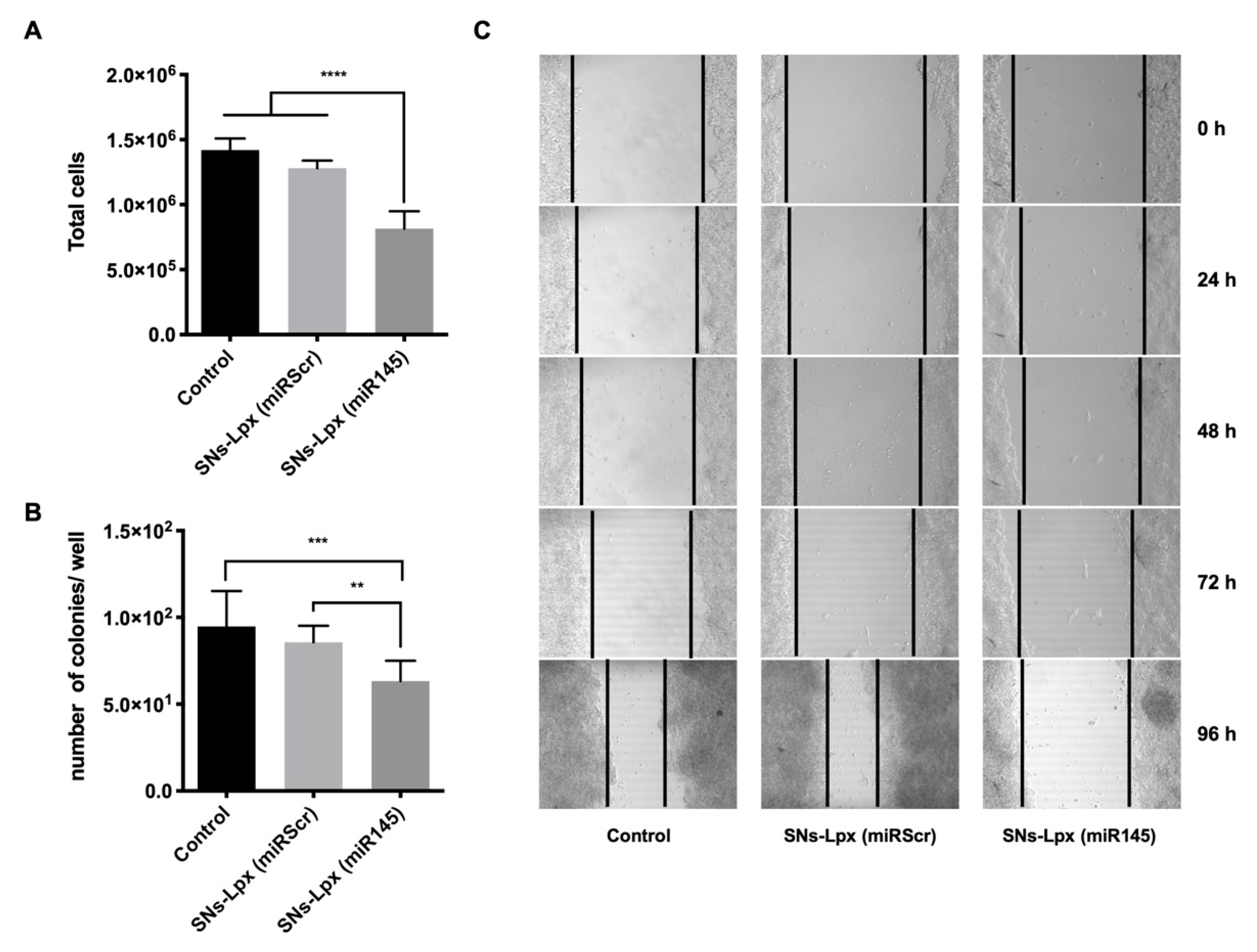
3.4. Anticancer Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2019. CA. Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, W.; Zhu, G.; Xie, J.; Chen, X. Rethinking cancer nanotheranostics. Nat. Rev. Mater. 2017, 2, 17024. [Google Scholar] [CrossRef]
- Jasinski, D.; Haque, F.; Binzel, D.W.; Guo, P. Advancement of the Emerging Field of RNA Nanotechnology. ACS Nano 2017, 11, 1142–1164. [Google Scholar] [CrossRef]
- Hare, J.I.; Lammers, T.; Ashford, M.B.; Puri, S.; Storm, G.; Barry, S.T. Challenges and strategies in anti-cancer nanomedicine development: An industry perspective. Adv. Drug Deliv. Rev. 2017, 108, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Roemeling, C.V.; Jiang, W.; Chan, C.K.; Weissman, I.L.; Kim, B.Y.S. Breaking Down the Barriers to Precision Cancer Nanomedicine. Trends Biotechnol. 2017, 35, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; An, K.; Duan, X.; Xu, H.; Li, F.; Xu, F. Recent advances in siRNA delivery for cancer therapy using smart nanocarriers. Drug Discov. Today 2018, 23, 900–911. [Google Scholar] [CrossRef] [PubMed]
- Ganju, A.; Khan, S.; Hafeez, B.B.; Behrman, S.W.; Yallapu, M.M.; Chauhan, S.C.; Jaggi, M. miRNA nanotherapeutics for cancer. Drug Discov. Today 2017, 22, 424–432. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–221. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, Y.; Wu, H.; Li, Y.; Zhang, Y.; Liu, M.; Li, X.; Tang, H. miR-10a suppresses colorectal cancer metastasis by modulating the epithelial-to-mesenchymal transition and anoikis. Cell Death Dis. 2017, 8, 2739. [Google Scholar] [CrossRef]
- Xie, Y.; Zong, P.; Wang, W.; Liu, D.; Li, B.; Wang, Y.; Hu, J.; Ren, Y.; Qi, Y.; Cui, X.; et al. Hypermethylation of potential tumor suppressor miR-34b/c is correlated with late clinical stage in patients with soft tissue sarcomas. Exp. Mol. Pathol. 2015, 98, 446–454. [Google Scholar] [CrossRef]
- Dong, J.; Xiao, D.; Zhao, Z.; Ren, P.; Li, C.; Hu, Y.; Shi, J.; Su, H.; Wang, L.; Liu, H.; et al. Epigenetic silencing of microRNA-137 enhances ASCT2 expression and tumor glutamine metabolism. Oncogenesis 2017, 6, 356. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Z.; Chen, M.; Peng, L.; Wang, X.; Ma, Q.; Ma, F.; Jiang, B. MicroRNA-143 Targets MACC1 to Inhibit Cell Invasion and Migration in Colorectal cancer. Mol. Cancer 2012, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Slaby, O.; Svoboda, M.; Fabian, P.; Smerdova, T.; Knoflickova, D.; Bednarikova, M.; Nenutil, R.; Vyzula, R. Altered expression of miR-21, miR-31, miR-143 and miR-145 is related to clinicopathologic features of colorectal cancer. Oncology 2008, 72, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Zeshan, B.; Sadaf, A.; Othman, N.H. Recent Progress in Delivery of Cancer Related MicroRNAs. J. Nanosci. Nanotechnol. 2016, 16, 6622–6633. [Google Scholar] [CrossRef]
- Liang, G.; Zhu, Y.; Jing, A.; Wang, J.; Hu, F.; Feng, W.; Xiao, Z.; Chen, B. Cationic microRNA-delivering nanocarriers for efficient treatment of colon carcinoma in xenograft model. Gene Ther. 2016, 23, 829–838. [Google Scholar] [CrossRef]
- Setua, S.; Khan, S.; Yallapu, M.M.; Behrman, S.W.; Sikander, M.; Khan, S.S.; Jaggi, M.; Chauhan, S.C. Restitution of Tumor Suppressor MicroRNA-145 Using Magnetic Nanoformulation for Pancreatic Cancer Therapy. J. Gastrointest. Surg. 2017, 21, 94–105. [Google Scholar] [CrossRef]
- Reimondez-Troitiño, S.; González-Aramundiz, J.V.; Ruiz-Bañobre, J.; López-López, R.; Alonso, M.J.; Csaba, N.; de la Fuente, M. Versatile protamine nanocapsules to restore miR-145 levels and interfere tumor growth in colorectal cancer cells. Eur. J. Pharm. Biopharm. 2019, 142, 449–459. [Google Scholar] [CrossRef]
- De la Fuente, M.; López, R.L.; Bouzo, B.L.; Vázquez ríos, A.J.; Nocelo, M.A. Nanosystems as Selective Vehicles. European Patent WO2019138139, 15 January 2018. [Google Scholar]
- Bouzo, B.L.; Calvelo, M.; Martin, M.; García-Fandiño, R.; de la Fuente, M. Design and characterization of novel delivery systems for advanced personalized medicine following an in vitro/in silico approach. submitted.
- Griesser, J.; Hetényi, G.; Federer, C.; Steinbring, C.; Ellemunter, H.; Niedermayr, K.; Bernkop-Schnürch, A. Highly mucus permeating and zeta potential changing self-emulsifying drug delivery systems: A potent gene delivery model for causal treatment of cystic fibrosis. Int. J. Pharm. 2018, 557, 124–134. [Google Scholar] [CrossRef]
- Mahmood, A.; Prüfert, F.; Efiana, N.A.; Ashraf, M.I.; Hermann, M.; Hussain, S.; Bernkop-Schnürch, A. Cell-penetrating self-nanoemulsifying drug delivery systems (SNEDDS) for oral gene delivery. Expert Opin. Drug Deliv. 2016, 13, 1503–1512. [Google Scholar] [CrossRef]
- Abraham, M.-K.; Peter, K.; Michel, T.; Wendel, H.P.; Krajewski, S.; Wang, X. Nanoliposomes for Safe and Efficient Therapeutic mRNA Delivery: A Step Toward Nanotheranostics in Inflammatory and Cardiovascular Diseases as well as Cancer. Nanotheranostics 2017, 1, 154–165. [Google Scholar] [CrossRef][Green Version]
- Kim, B.K.; Hwang, G.B.; Seu, Y.B.; Choi, J.S.; Jin, K.S.; Doh, K.O. DOTAP/DOPE ratio and cell type determine transfection efficiency with DOTAP-liposomes. Biochim. Biophys. Acta Biomembr. 2015, 1848, 1996–2001. [Google Scholar] [CrossRef]
- Matsumoto, M.; Kishikawa, R.; Kurosaki, T.; Nakagawa, H.; Ichikawa, N.; Hamamoto, T.; To, H.; Kitahara, T.; Sasaki, H. Hybrid vector including polyethylenimine and cationic lipid, DOTMA, for gene delivery. Int. J. Pharm. 2008, 363, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.L.; Alexandrino, F.; Verissimo, L.M.; Agnez-Lima, L.F.; Egito, L.C.M.; de Oliveira, A.G.; do Egito, E.S.T. Physical factors affecting plasmid DNA compaction in stearylamine-containing nanoemulsions intended for gene delivery. Pharmaceuticals 2012, 5, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Singh, Y.; Meher, J.G.; Raval, K.; Khan, F.A.; Chaurasia, M.; Jain, N.K.; Chourasia, M.K. Nanoemulsion: Concepts, development and applications in drug delivery. J. Control. Release 2017, 252, 28–49. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Fan, H.; Levorse, D.A.; Crocker, L.S. Ionization behavior of amino lipids for siRNA delivery: Determination of ionization constants, SAR, and the impact of lipid p K a on cationic lipid-biomembrane interactions. Langmuir 2011, 27, 1907–1914. [Google Scholar] [CrossRef] [PubMed]
- Jeong, M.W.; Oh, S.G.; Kim, Y.C. Effects of amine and amine oxide compounds on the zeta-potential of emulsion droplets stabilized by phosphatidylcholine. Colloids Surf. A Physicochem. Eng. Asp. 2001, 181, 247–253. [Google Scholar] [CrossRef]
- Silva, A.L.; Marcelino, H.R.; Veríssimo, L.M.; Araujo, I.B.; Agnez-Lima, L.F.; do Egito, E.S.T. Stearylamine-Containing Cationic Nanoemulsion as a Promising Carrier for Gene Delivery. J. Nanosci. Nanotechnol. 2016, 16, 1339–1345. [Google Scholar] [CrossRef]
- Teixeira, H.F.; Bruxel, F.; Fraga, M.; Schuh, R.S.; Zorzi, G.K.; Matte, U.; Fattal, E. Cationic nanoemulsions as nucleic acids delivery systems. Int. J. Pharm. 2017, 534, 356–367. [Google Scholar] [CrossRef]
- Junquera, E.; Aicart, E. Recent progress in gene therapy to deliver nucleic acids with multivalent cationic vectors. Adv. Colloid Interface Sci. 2016, 233, 161–175. [Google Scholar] [CrossRef]
- Prabha, S.; Arya, G.; Chandra, R.; Ahmed, B.; Nimesh, S. Effect of size on biological properties of nanoparticles employed in gene delivery. Artif. Cells Nanomed. Biotechnol. 2016, 44, 83–91. [Google Scholar] [CrossRef]
- Duan, X.; Li, Y. Physicochemical characteristics of nanoparticles affect circulation, biodistribution, cellular internalization, and trafficking. Small 2013, 9, 1521–1532. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Meng, T.; Yuan, M.; Wen, L.J.; Cheng, B.L.; Liu, N.; Huang, X.; Hong, Y.; Yuan, H.; Hu, F.Q. MicroRNA-200c delivered by solid lipid nanoparticles enhances the effect of paclitaxel on breast cancer stem cell. Int. J. Nanomed. 2016, 11, 6713–6725. [Google Scholar] [CrossRef] [PubMed]
- Martini, É.; Fattal, E.; de Oliveira, M.C.; Teixeira, H. Effect of cationic lipid composition on properties of oligonucleotide/emulsion complexes: Physico-chemical and release studies. Int. J. Pharm. 2008, 352, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Vader, P.; Van Der Aa, L.J.; Engbersen, J.F.J.; Storm, G.; Schiffelers, R.M. Physicochemical and biological evaluation of siRNA polyplexes based on PEGylated poly(amido amine)s. Pharm. Res. 2012, 29, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Delgado, D.; Del Pozo-Rodríguez, A.; Angeles Solinís, M.; Bartkowiak, A.; Rodríguez-Gascón, A. New gene delivery system based on oligochitosan and solid lipid nanoparticles: “In vitro” and “in vivo” evaluation. Eur. J. Pharm. Sci. 2013, 50, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Zhang, W.; Du, Y.Z.; Hu, F.Q. Ternary nanoparticles of anionic lipid nanoparticles/protamine/DNA for gene delivery. Int. J. Pharm. 2010, 392, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Hauptstein, S.; Prüfert, F.; Bernkop-Schnürch, A. Self-nanoemulsifying drug delivery systems as novel approach for pDNA drug delivery. Int. J. Pharm. 2015, 487, 25–31. [Google Scholar] [CrossRef]
- Hintzen, F.; Perera, G.; Hauptstein, S.; Müller, C.; Laffleur, F.; Bernkop-Schnürch, A. In vivo evaluation of an oral self-microemulsifying drug delivery system (SMEDDS) for leuprorelin. Int. J. Pharm. 2014, 472, 20–26. [Google Scholar] [CrossRef]
- Brito, L.A.; Chan, M.; Shaw, C.A.; Hekele, A.; Carsillo, T.; Schaefer, M.; Archer, J.; Seubert, A.; Otten, G.R.; Beard, C.W.; et al. A cationic nanoemulsion for the delivery of next-generation RNA vaccines. Mol. Ther. 2014, 22, 2118–2129. [Google Scholar] [CrossRef]
- Fraga, M.; Laux, M.; Rejane Dos Santos, G.; Zandoná, B.; Dos Santos Giuberti, C.; De Oliveira, M.C.; Da Silveira Matte, U.; Ferreira Teixera, H. Evaluation of the toxicity of oligonucleotide/cationic nanoemulsion complexes on Hep G2 cells through MTT assay. Pharmazie 2008, 63, 667–670. [Google Scholar]
- Hillaireau, H.; Couvreur, P. Nanocarriers’ entry into the cell: Relevance to drug delivery. Cell. Mol. Life Sci. 2009, 66, 2873–2896. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Li, B.; Qi, S.; Liu, Y.; Gai, Y.; Ye, P.; Yang, G.; Zhang, W.; Zhang, P.; He, X.; et al. Co-delivery of doxorubicin and Bmil siRNA by folate receptor targeted liposomes exhibits enhanced anti-tumor effects in vitro and in vivo. Theranostics 2014, 4, 1096–1111. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Dai, X.; Zhao, Y.; Guo, Q.; Wu, Z.; Zhang, X.; Li, C. A biodegradable and fluorescent nanovehicle with enhanced selective uptake by tumor cells. Polym. Chem. 2015, 6, 6529–6542. [Google Scholar] [CrossRef]
- Shin, H.; Park, S.-J.; Yim, Y.; Kim, J.; Choi, C.; Won, C.; Min, D.-H. Recent Advances in RNA Therapeutics and RNA Delivery Systems Based on Nanoparticles. Adv. Ther. 2018, 1, 1800065. [Google Scholar] [CrossRef]
- Mahmood, A.; Bernkop-Schnürch, A. SEDDS: A game changing approach for the oral administration of hydrophilic macromolecular drugs. Adv. Drug Deliv. Rev. 2018, 142, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, H.; Dubernet, C.; Chacun, H.; Rabinovich, L.; Boutet, V.; Deverre, J.R.; Benita, S.; Couvreur, P. Cationic emulsions improves the delivery of oligonucleotides to leukemic P388/ADR cells in ascite. J. Control. Release 2003, 89, 473–482. [Google Scholar] [CrossRef]
- Tao, J.; Ding, W.F.; Che, X.H.; Chen, Y.C.; Chen, F.; Chen, X.D.; Ye, X.L.; Xiong, S. Bin Optimization of a cationic liposome-based gene delivery system for the application of miR-145 in anticancer therapeutics. Int. J. Mol. Med. 2016, 37, 1345–1354. [Google Scholar] [CrossRef]
- Li, C.; Xu, N.; Li, Y.Q.; Wang, Y.; Zhu, Z.T. Inhibition of SW620 human colon cancer cells by upregulating miRNA-145 Basic Study. World J. Gastroenterol. 2016, 22, 2771–2778. [Google Scholar] [CrossRef]
- Hidalgo, T.; Alonso-Nocelo, M.; Bouzo, B.L.; Reimondez-Troitiño, S.; Abuin-Redondo, C.; de la Fuente, M.; Horcajada, P. Biocompatible iron(III) carboxylate Metal-Organic Frameworks as promising RNA nanocarriers. Nanoscale 2020. [Google Scholar] [CrossRef]
- Wu, J.; Wang, J.; Su, Q.; Ding, W.; Li, T.; Yu, J.; Cao, B. Traditional Chinese medicine Astragalus polysaccharide enhanced antitumor effects of the angiogenesis inhibitor apatinib in pancreatic cancer cells on proliferation, invasiveness, and apoptosis. Onco. Targets Ther. 2018, 11, 2685–2698. [Google Scholar] [CrossRef]
- Augello, G.; Modica, M.; Azzolina, A.; Puleio, R.; Cassata, G.; Emma, M.R.; Di Sano, C.; Cusimano, A.; Montalto, G.; Cervello, M. Preclinical evaluation of antitumor activity of the proteasome inhibitor MLN2238 (ixazomib) in hepatocellular carcinoma cells. Cell Death Dis. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | a ST (w/w) | Size (nm) | b PDI | ζ-Potential (mV) |
|---|---|---|---|---|
| - | 131 ± 8 | 0.2 | −13 ± 7 | |
| SNs-ST | 0.1 | 104 ± 10 | 0.2 | +40 ± 12 |
| 0.5 | 115 ± 11 | 0.2 | +44 ± 4 | |
| 1 | 109 ± 11 | 0.2 | +46 ± 6 | |
| 1.5 | 99 ± 12 | 0.3 | +48 ± 11 | |
| 2 | 104 ± 22 | 0.3 | +51 ± 3 |
| Formulations | Mass Ratio (w/w) a miRNA:ST | Theoretical miRNA Loading (%) | Size (nm) | PDI b | ζ-Potential (mV) |
|---|---|---|---|---|---|
| SNs-ST (miR) | 1:10 | 0.8 | 158 ± 8 | 0.2 | +26 ± 3 |
| 1:7.5 | 1.1 | Precipitate | |||
| 1:5 | 1.6 | Precipitate | |||
| 1:2.5 | 3.2 | 172 ± 4 | 0.2 | −15 ± 2 | |
| 1:1 | 7.7 | 155 ± 2 | 0.2 | −30 ± 3 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagachinta, S.; Bouzo, B.L.; Vazquez-Rios, A.J.; Lopez, R.; Fuente, M.d.l. Sphingomyelin-Based Nanosystems (SNs) for the Development of Anticancer miRNA Therapeutics. Pharmaceutics 2020, 12, 189. https://doi.org/10.3390/pharmaceutics12020189
Nagachinta S, Bouzo BL, Vazquez-Rios AJ, Lopez R, Fuente Mdl. Sphingomyelin-Based Nanosystems (SNs) for the Development of Anticancer miRNA Therapeutics. Pharmaceutics. 2020; 12(2):189. https://doi.org/10.3390/pharmaceutics12020189
Chicago/Turabian StyleNagachinta, Surasa, Belen Lopez Bouzo, Abi Judit Vazquez-Rios, Rafael Lopez, and Maria de la Fuente. 2020. "Sphingomyelin-Based Nanosystems (SNs) for the Development of Anticancer miRNA Therapeutics" Pharmaceutics 12, no. 2: 189. https://doi.org/10.3390/pharmaceutics12020189
APA StyleNagachinta, S., Bouzo, B. L., Vazquez-Rios, A. J., Lopez, R., & Fuente, M. d. l. (2020). Sphingomyelin-Based Nanosystems (SNs) for the Development of Anticancer miRNA Therapeutics. Pharmaceutics, 12(2), 189. https://doi.org/10.3390/pharmaceutics12020189