Combining Co-Amorphous-Based Spray Drying with Inert Carriers to Achieve Improved Bioavailability and Excellent Downstream Manufacturability

Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
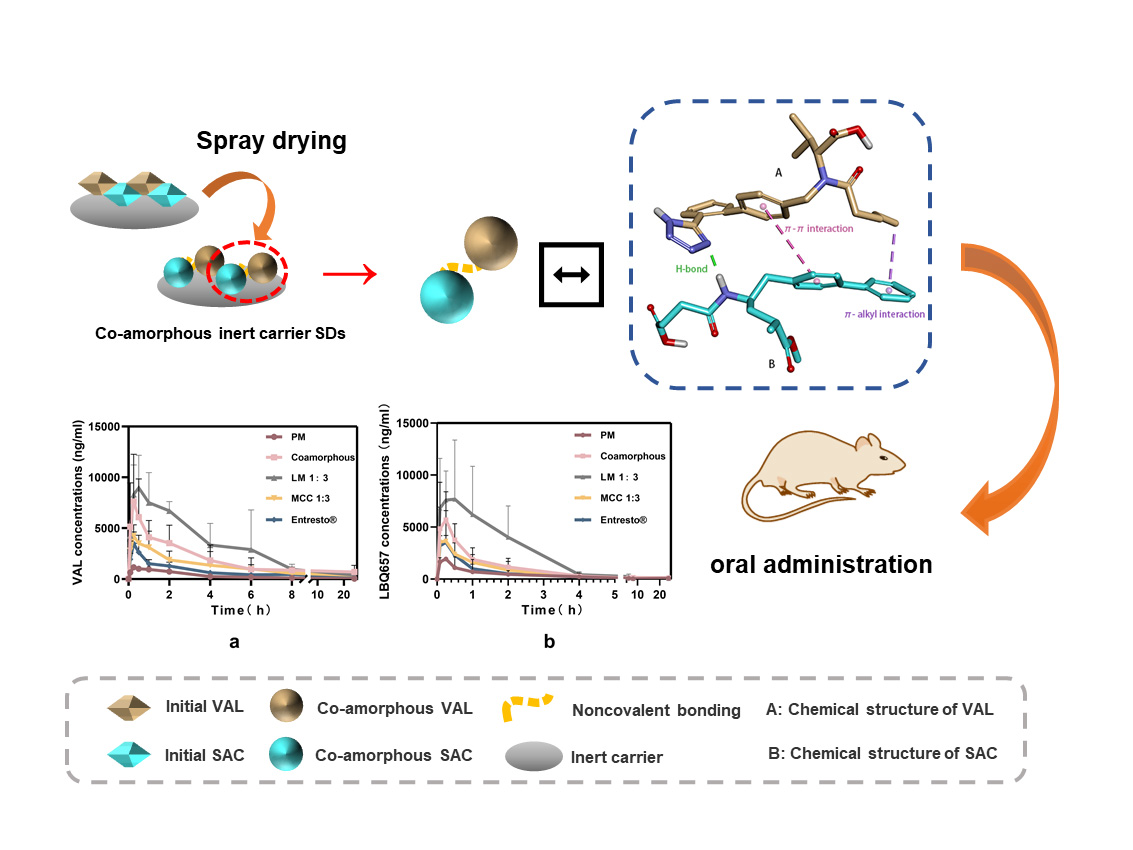
2.2. Preparation of Co-Amorphous SDs
2.3. Characterization of Co-Amorphous SDs
2.3.1. Powder X-ray Diffraction (PXRD) and Fourier Transform Infrared (FT-IR)
2.3.2. Differential Scanning Calorimetry (DSC)
2.4. Particle Size Distribution and Morphology Observations
2.5. Apparent Equilibrium Solubility Study
2.6. Dissolution and Permeation Study
2.6.1. No-Sink Dissolution/Permeation (D/P) Study
2.6.2. Sink Condition Dissolubility Study
2.7. Compactibility TEST
2.8. Stability Study
2.9. Pharmacokinetic Study
2.9.1. Animal Experiments
2.9.2. Bioanalytical Method
2.10. Statistical Analyses
3. Results
3.1. Solid State Characterization
3.2. Particle Size and Morphology
3.3. Apparent Equilibrium Solubility
3.4. Dissolution and Permeation Behavior
3.5. Compactibility Test
3.6. Stability Study
3.7. Pharmacokinetic Studies
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Horter, D.; Dressman, J.B. Influence of physicochemical properties on dissolution of drugs in the gastrointestinal tract. Adv. Drug Deliv. Rev. 2001, 46, 75–87. [Google Scholar] [CrossRef]
- Saal, C.; Becker, A. Pharmaceutical salts: A summary on doses of salt formers from the orange book. Eur. J. Pharm. Sci. 2013, 49, 614–623. [Google Scholar] [CrossRef] [PubMed]
- Schultheiss, N.; Newman, A. Pharmaceutical cocrystals and their physicochemical properties. Cryst. Growth Des. 2009, 9, 2950–2967. [Google Scholar] [CrossRef] [PubMed]
- Rautio, J.; Kumpulainen, H.; Heimbach, T.; Oliyai, R.; Oh, D.; Jarvinen, T.; Savolainen, J. Prodrugs: Design and clinical applications. Nat. Rev. Drug Discov. 2008, 7, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Rabinow, B.E. Nanosuspensions in drug delivery. Nat. Rev. Drug Discov. 2004, 3, 785–796. [Google Scholar] [CrossRef]
- Dengale, S.J.; Grohganz, H.; Rades, T.; Lobmann, K. Recent advances in co-amorphous drug formulations. Adv. Drug Deliv. Rev. 2016, 100, 116–125. [Google Scholar] [CrossRef]
- Karagianni, A.; Kachrimanis, K.; Nikolakakis, I. Co-amorphous solid dispersions for solubility and absorption improvement of drugs: Composition, preparation, characterization and formulations for oral delivery. Pharmaceutics 2018, 10, 98. [Google Scholar] [CrossRef]
- Knapik, J.; Wojnarowska, Z.; Grzybowska, K.; Jurkiewicz, K.; Tajber, L.; Paluch, M. Molecular dynamics and physical stability of coamorphous ezetimib and indapamide mixtures. Mol. Pharm. 2015, 12, 3610–3619. [Google Scholar] [CrossRef] [PubMed]
- Aaltonen, J.; Rades, T. Towards physico-relevant dissolution testing: The importance of solid-state analysis in dissolution. Dissolut. Technol. 2009, 16, 47–54. [Google Scholar] [CrossRef]
- Engers, D.; Teng, J.; Jimenez-Novoa, J.; Gent, P.; Hossack, S.; Campbell, C.; Thomson, J.; Ivanisevic, I.; Templeton, A.; Byrn, S.; et al. A solid-state approach to enable early development compounds: Selection and animal bioavailability studies of an itraconazole amorphous solid dispersion. J. Pharm. Sci. 2010, 99, 3901–3922. [Google Scholar] [CrossRef]
- Brouwers, J.; Brewster, M.E.; Augustijns, P. Supersaturating drug delivery systems: The answer to solubility-limited oral bioavailability? J. Pharm. Sci. 2009, 98, 2549–2572. [Google Scholar] [CrossRef]
- Vasconcelos, T.; Marques, S.; das Neves, J.; Sarmento, B. Amorphous solid dispersions: Rational selection of a manufacturing process. Adv. Drug Deliv. Rev. 2016, 100, 85–101. [Google Scholar] [CrossRef]
- Jensen, K.T.; Larsen, F.H.; Cornett, C.; Lobmann, K.; Grohganz, H.; Rades, T. Formation mechanism of coamorphous drug-amino acid mixtures. Mol. Pharm. 2015, 12, 2484–2492. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, T.; Sarmento, B.; Costa, P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov. Today 2007, 12, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Noe, A.; Chandra, P.; Al-Fayoumi, S.; Ligueros-Saylan, M.; Sarangapani, R.; Maahs, S.; Ksander, G.; Rigel, D.F.; Jeng, A.Y. Pharmacokinetics and pharmacodynamics of lcz696, a novel dual-acting angiotensin receptor—Neprilysin inhibitor (arni). J. Clin. Pharmacol. 2010, 50, 401–414. [Google Scholar] [CrossRef]
- Dhumal, R.; Biradar, S.; Yamamura, S.; Paradkar, A.; York, P. Preparation of amorphous cefuroxime axetil nanoparticles by sonoprecipitation for enhancement of bioavailability. Eur. J. Pharm. Biopharm. 2008, 70, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.; Serrano, D.R.; Worku, Z.A.; Norris, B.A.; Healy, A.M. Production of cocrystals in an excipient matrix by spray drying. Int. J. Pharm. 2018, 536, 467–477. [Google Scholar] [CrossRef]
- Ahmed, M. Stability indicating rp-hplc method for simultaneous estimation of sacubitril and valsartan in bulk and combined pharmaceutical dosage form. World J. Pharm. Pharm. Sci. 2017, 6, 1714–1728. [Google Scholar] [CrossRef]
- Guo, M.; Wang, K.; Qiao, N.; Yardley, V.; Li, M. Investigating permeation behavior of flufenamic acid cocrystals using a dissolution and permeation system. Mol. Pharm. 2018, 15, 4257–4272. [Google Scholar] [CrossRef]
- Guo, M.; Wang, K.; Qiao, N.; Fabian, L.; Sadiq, G.; Li, M. Insight into flufenamic acid cocrystal dissolution in the presence of a polymer in solution: From single crystal to powder dissolution. Mol. Pharm. 2017, 14, 4583–4596. [Google Scholar] [CrossRef]
- Metre, S.; Mukesh, S.; Samal, S.K.; Chand, M.; Sangamwar, A.T. Enhanced biopharmaceutical performance of rivaroxaban through polymeric amorphous solid dispersion. Mol. Pharm. 2018, 15, 652–668. [Google Scholar] [CrossRef]
- Rubtsov, I.V.; Wang, J.; Hochstrasser, R.M. Dual frequency 2d-ir of peptide amide-a and amide-i modes. J. Chem. Phys. 2003, 118, 7733–7736. [Google Scholar] [CrossRef]
- Ardiana, F.; Indrayanto, G. Valsartan. In Profiles of Drug Substances, Excipients and Related Methodology; Elsevier: Amsterdam, The Netherlands, 2015; Volume 40, pp. 431–493. [Google Scholar]
- Zhao, J.; Wang, J. Direct anionic effect on water structure and indirect anionic effect on peptide backbone hydration state revealed by thin-layer infrared spectroscopy. J. Phys. Chem. B 2018, 122, 68–76. [Google Scholar] [CrossRef]
- Lu, Y.Q.; Miller, J.D. Carboxyl stretching vibrations of spontaneously adsorbed and lb-transferred calcium carboxylates as determined by ftir internal reflection spectroscopy. J. Colloid Interface Sci. 2002, 256, 41–52. [Google Scholar] [CrossRef]
- Loebmann, K.; Strachan, C.; Grohganz, H.; Rades, T.; Korhonen, O.; Laitinen, R. Co-amorphous simvastatin and glipizide combinations show improved physical stability without evidence of intermolecular interactions. Eur. J. Pharm. Biopharm. 2012, 81, 159–169. [Google Scholar] [CrossRef]
- Shakiba, S.; Mansouri, S.; Selomulya, C.; Woo, M.W. The role of the intermediate stage of drying on particle in-situ crystallization in spray dryers. Powder Technol. 2018, 323, 357–366. [Google Scholar] [CrossRef]
- Kian, L.K.; Jawaid, M.; Ariffin, H.; Alothman, O.Y. Isolation and characterization of microcrystalline cellulose from roselle fibers. Int. J. Biol. Macromol. 2017, 103, 931–940. [Google Scholar] [CrossRef]
- Skotnicki, M.; Gawel, A.; Cebe, P.; Pyda, M. Thermal behavior and phase identification of valsartan by standard and temperature-modulated differential scanning calorimetry. Drug Dev. Ind. Pharm. 2013, 39, 1508–1514. [Google Scholar] [CrossRef]
- Guo, Y.; Shalaev, E.; Smith, S. Physical stability of pharmaceutical formulations: Solid-state characterization of amorphous dispersions. Trac Trends Anal. Chem. 2013, 49, 137–144. [Google Scholar] [CrossRef]
- Gamble, J.F.; Terada, M.; Holzner, C.; Lavery, L.; Nicholson, S.J.; Timmins, P.; Tobyn, M. Application of x-ray microtomography for the characterisation of hollow polymer-stabilised spray dried amorphous dispersion particles. Int. J. Pharm. 2016, 510, 1–8. [Google Scholar] [CrossRef]
- Amidon, G.L.; Lennernäs, H.; Shah, V.P.; Crison, J.R. A theoretical basis for a biopharmaceutic drug classification: The correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef]
- Guo, M.; Wang, K.; Hamill, N.; Lorimer, K.; Li, M. Investigating the influence of polymers on supersaturated flufenamic acid cocrystal solutions. Mol. Pharm. 2016, 13, 3292–3307. [Google Scholar] [CrossRef]
- Heng, W.; Wei, Y.; Xue, Y.; Cheng, H.; Zhang, L.; Zhang, J.; Gao, Y.; Qian, S. Gel formation induced slow dissolution of amorphous indomethacin. Pharm. Res. 2019, 36, 159. [Google Scholar] [CrossRef]
- Paudel, A.; Worku, Z.A.; Meeus, J.; Guns, S.; Van den Mooter, G. Manufacturing of solid dispersions of poorly water soluble drugs by spray drying: Formulation and process considerations. Int. J. Pharm. 2013, 453, 253–284. [Google Scholar] [CrossRef]
- Kim, J.-S.; Kim, M.-S.; Park, H.J.; Jin, S.-J.; Lee, S.; Hwang, S.-J. Physicochemical properties and oral bioavailability of amorphous atorvastatin hemi-calcium using spray-drying and sas process. Int. J. Pharm. 2008, 359, 211–219. [Google Scholar] [CrossRef]
- Craye, G.; Lobmann, K.; Grohganz, H.; Rades, T.; Laitinen, R. Characterization of amorphous and co-amorphous simvastatin formulations prepared by spray drying. Molecules 2015, 20, 21532–21548. [Google Scholar] [CrossRef]
- Vehring, R. Pharmaceutical particle engineering via spray drying. Pharm. Res. 2008, 25, 999–1022. [Google Scholar] [CrossRef]
- Satoh, T.; Hidaka, F.; Miyake, K.; Yoshiyama, N.; Takeda, K.; Matsuura, T.; Imanaka, H.; Ishida, N.; Imamura, K. Surfactant-free solid dispersion of fat-soluble flavour in an amorphous sugar matrix. Food Chem. 2016, 197, 1136–1142. [Google Scholar] [CrossRef]
- Tu Van, D.; Van den Mooter, G. The role of the carrier in the formulation of pharmaceutical solid dispersions. Part ii: Amorphous carriers. Expert Opin. Drug Deliv. 2016, 13, 1681–1694. [Google Scholar]
- Craig, D.Q.M. The mechanisms of drug release from solid dispersions in water-soluble polymers. Int. J. Pharm. 2002, 231, 131–144. [Google Scholar] [CrossRef]
- Kirubakaran, P.; Wang, K.; Rosbottom, I.; Cross, R.B.M.; Li, M. Understanding the effects of a polymer on the surface dissolution of pharmaceutical cocrystals using combined experimental and molecular dynamics simulation approaches. Mol. Pharm. 2020, 17, 517–529. [Google Scholar] [CrossRef]
- Shah, V.P.; Amidon, G.L. Gl amidon, h. Lennernas, vp shah, and jr crison. A theoretical basis for a biopharmaceutic drug classification: The correlation of in vitro drug product dissolution and in vivo bioavailability. AAPS J. 2014, 16, 894–898. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Yamazoe, C.; Yasuda, Y.; Higashi, K.; Kawakami, K.; Moribe, K. Mechanism of enhanced nifedipine dissolution by polymer-blended solid dispersion through molecular-level characterization. Mol. Pharm. 2018, 15, 4099–4109. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | SAC (g) | VAL (g) | LM (g) | Formulation | SAC (g) | VAL (g) | MCC (g) |
|---|---|---|---|---|---|---|---|
| Co-amorphous | 1.33 | 1.34 | - | ||||
| Co-amorphous LM 1:1 | 1.33 | 1.34 | 2.67 | Co-amorphous MCC 1:1 | 1.33 | 1.34 | 2.67 |
| Co-amorphous LM 1:2 | 1.33 | 1.34 | 5.34 | Co-amorphous MCC 1:2 | 1.33 | 1.34 | 5.34 |
| Co-amorphous LM 1:3 | 1.33 | 1.34 | 8.01 | Co-amorphous MCC 1:3 | 1.33 | 1.34 | 8.01 |
| Co-amorphous LM 1:4 | 1.33 | 1.34 | 10.68 | Co-amorphous MCC 1:4 | 1.33 | 1.34 | 10.68 |
| Parameters | VAL | ||||
| PM | Co-amorphous | LM 1:3 | MCC 1:3 | Entresto | |
| AUC(0-t)(ng h/mL) | 3574.74 ± 933.15 | 57,022.08 ± 833.40 | 63,013.11 ± 23,471.31 | 48,748.19 ± 53,022.52 | 40,916.24 ± 19,884.30 |
| Cmax (ng/mL) | 1175.18 ± 220.28 | 11,352.98± 4598.69 | 7699.03 ± 3200.25 | 6163.12 ± 1599.95 | 5289.11 ± 1851.86 |
| Relative bioavailability % | 10.63 | 139.36 | 154.01 | 119.14 | - |
| Parameters | LBQ657 | ||||
| PM | Co-amorphous | LM 1:3 | MCC 1:3 | Entresto | |
| AUC(0-t)(ng h/mL) | 3625.72 ± 1716.67 | 13,258.26 ± 1325.30 | 20,454.46 ± 7323.42 | 7159.44 ± 18,014.82 | 5741.35 ± 1137.78 |
| Cmax (ng/mL) | 3074.73 ± 949.17 | 6363.13 ± 2706.14 | 11,243.79 ± 3936.34 | 4134.42 ± 624.32 | 3841.42 ± 3473.21 |
| Relative bioavailability % | 63.15 | 230.93 | 356.27 | 124.70 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Gao, Y.; Du, X.; Guan, R.; He, Z.; Liu, H. Combining Co-Amorphous-Based Spray Drying with Inert Carriers to Achieve Improved Bioavailability and Excellent Downstream Manufacturability. Pharmaceutics 2020, 12, 1063. https://doi.org/10.3390/pharmaceutics12111063
Zhang Y, Gao Y, Du X, Guan R, He Z, Liu H. Combining Co-Amorphous-Based Spray Drying with Inert Carriers to Achieve Improved Bioavailability and Excellent Downstream Manufacturability. Pharmaceutics. 2020; 12(11):1063. https://doi.org/10.3390/pharmaceutics12111063
Chicago/Turabian StyleZhang, Yingxi, Yuan Gao, Xiaoxiao Du, Rou Guan, Zhonggui He, and Hongzhuo Liu. 2020. "Combining Co-Amorphous-Based Spray Drying with Inert Carriers to Achieve Improved Bioavailability and Excellent Downstream Manufacturability" Pharmaceutics 12, no. 11: 1063. https://doi.org/10.3390/pharmaceutics12111063
APA StyleZhang, Y., Gao, Y., Du, X., Guan, R., He, Z., & Liu, H. (2020). Combining Co-Amorphous-Based Spray Drying with Inert Carriers to Achieve Improved Bioavailability and Excellent Downstream Manufacturability. Pharmaceutics, 12(11), 1063. https://doi.org/10.3390/pharmaceutics12111063