Physiologically Based Pharmacokinetic/Pharmacodynamic Modeling to Predict the Impact of CYP2C9 Genetic Polymorphisms, Co-Medication and Formulation on the Pharmacokinetics and Pharmacodynamics of Flurbiprofen

 ,
, 
 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. In Vitro Solubility Experiments
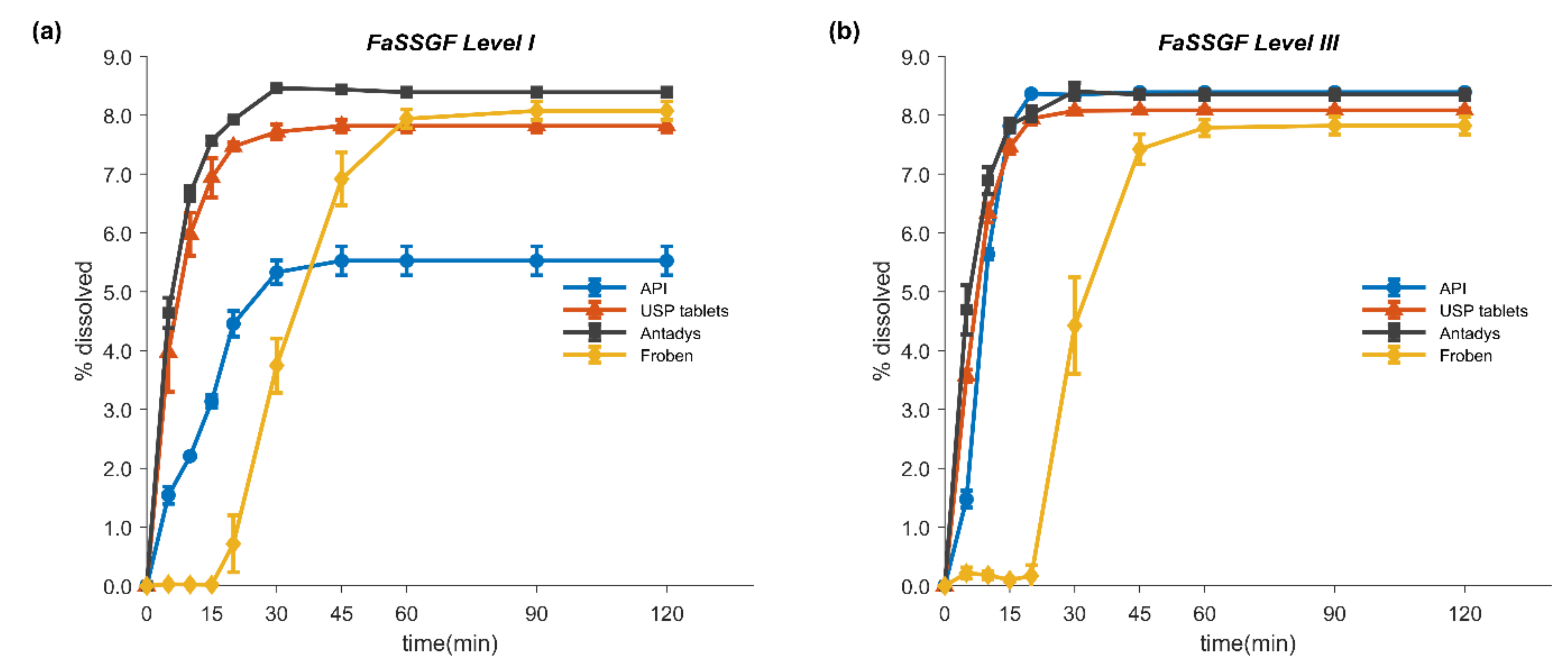
2.3. In Vitro Dissolution Tests
2.4. Two-Stage Dissolution Test
2.5. Quantitative Analysis of Samples
2.6. Model-Based Analysis of In Vitro Solubility Data
2.7. Model-Based Analysis of In Vitro Dissolution Data
2.8. Clinical Studies
2.8.1. PBPK Development and Evaluation Studies
2.8.2. Drug–Drug Interaction (DDI) Studies
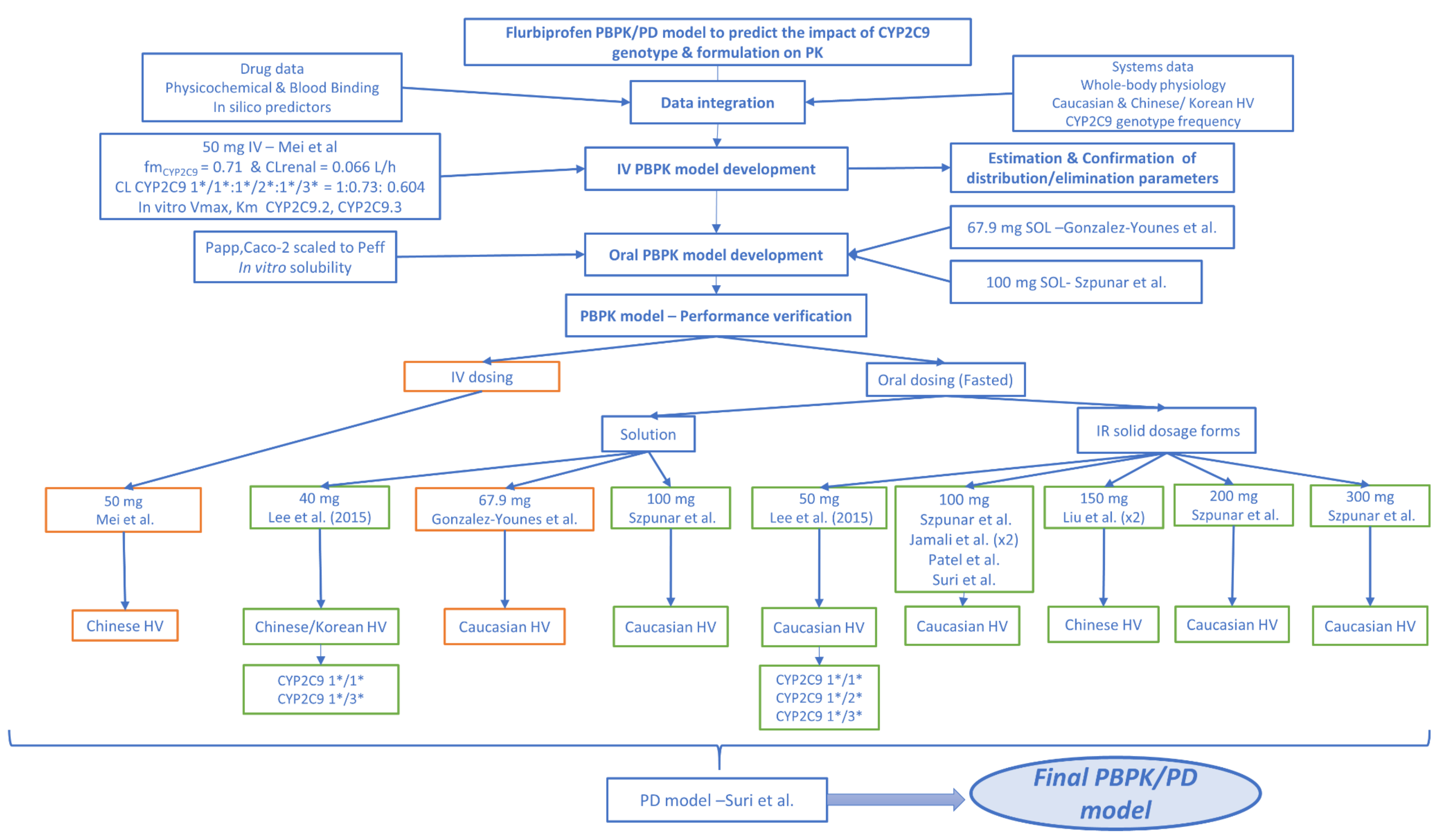
2.9. PBPK Model Development and Verification
2.9.1. Software
2.9.2. PBPK/PD Model Development
Absorption
Distribution
Metabolism and Excretion
Pharmacodynamics
Model Optimization
2.9.3. PBPK/PD Model Validation and Evaluation of Predictive Performance
2.9.4. PBPK DDI Modeling
PBPK Models of Perpetrator Drugs
PBPK DDI Modeling Evaluation
2.9.5. Virtual Populations
2.10. Data Analysis and Model Diagnostics
3. Results
3.1. In Vitro Solubility
3.2. Model-Based Analysis of In Vitro Solubility Data
3.3. In Vitro Dissolution Tests
3.4. Modeling of In Vitro Dissolution
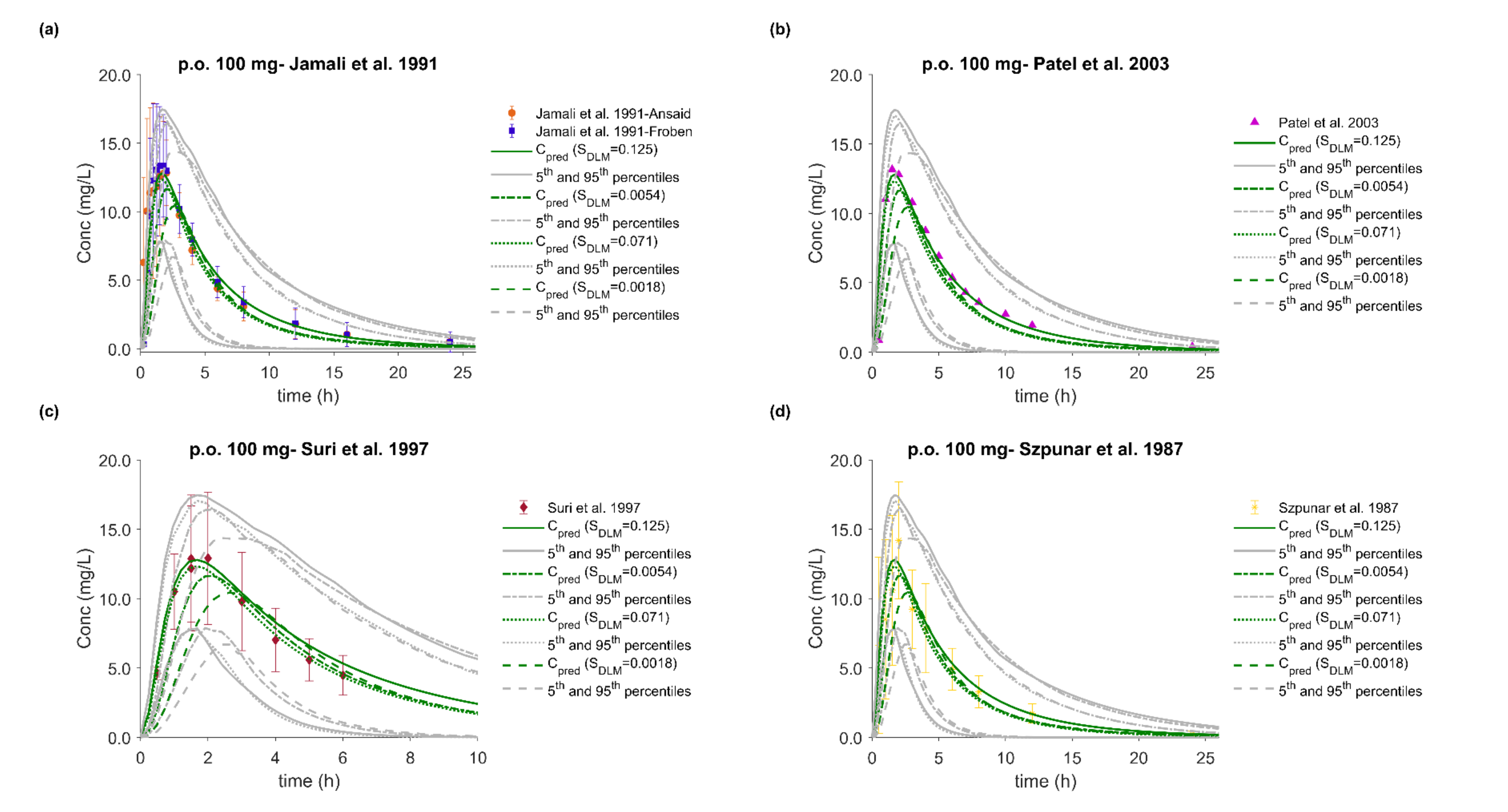
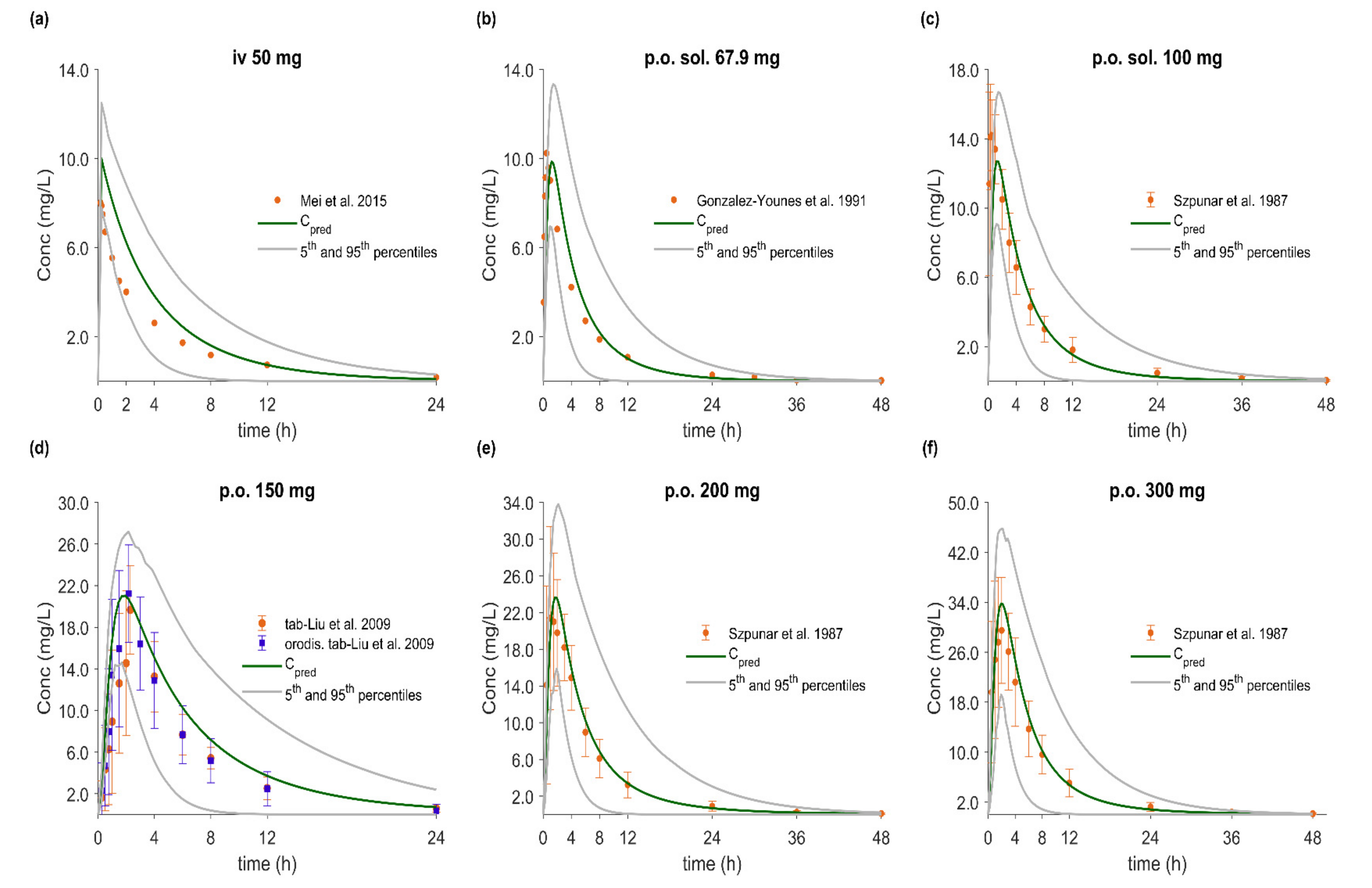
3.5. PBPK/PD Model Development and Evaluation
3.6. Effect of Dissolution Rate
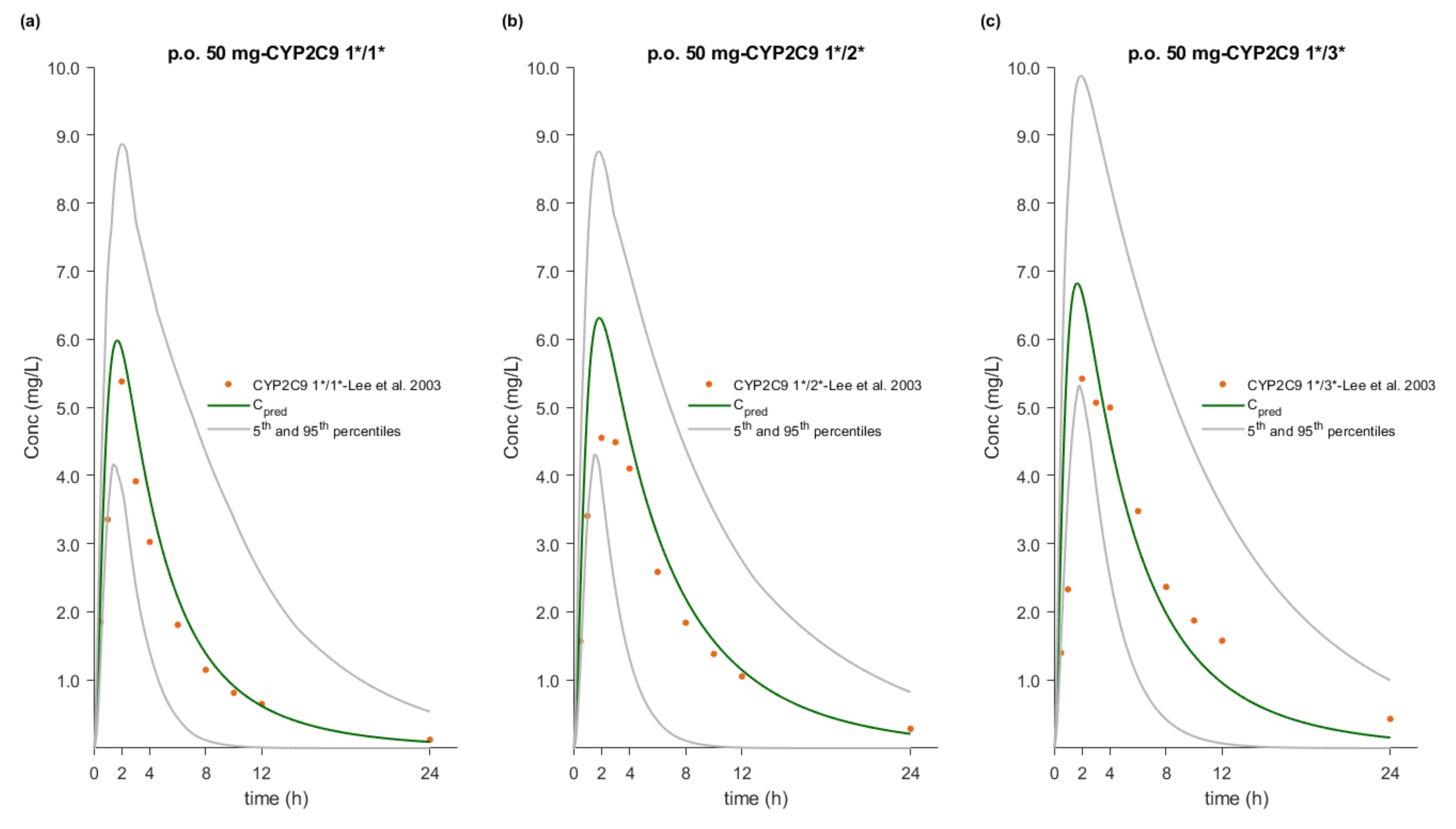
3.7. Effect of CYP2C9 Genetic Polymorphism
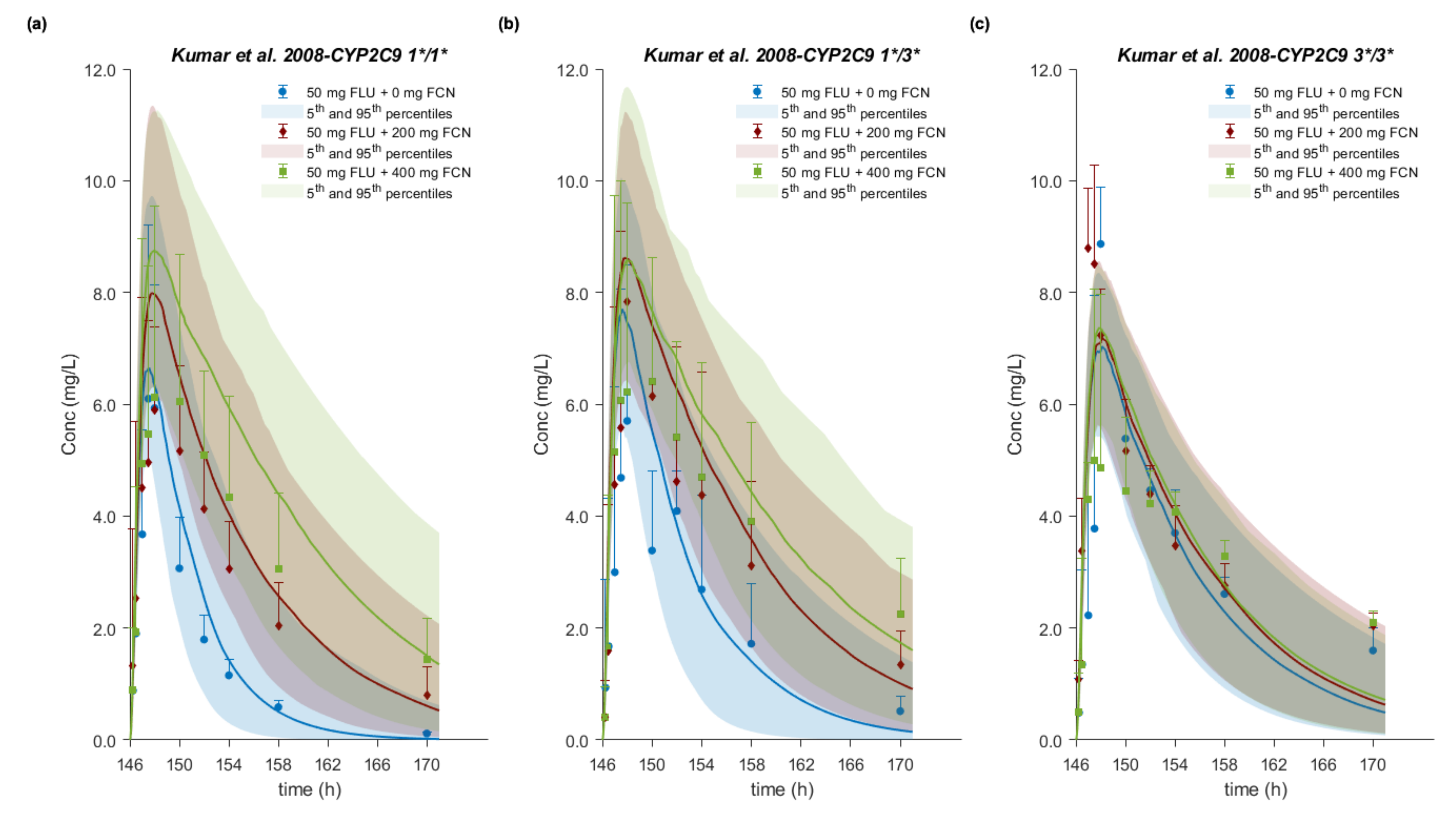
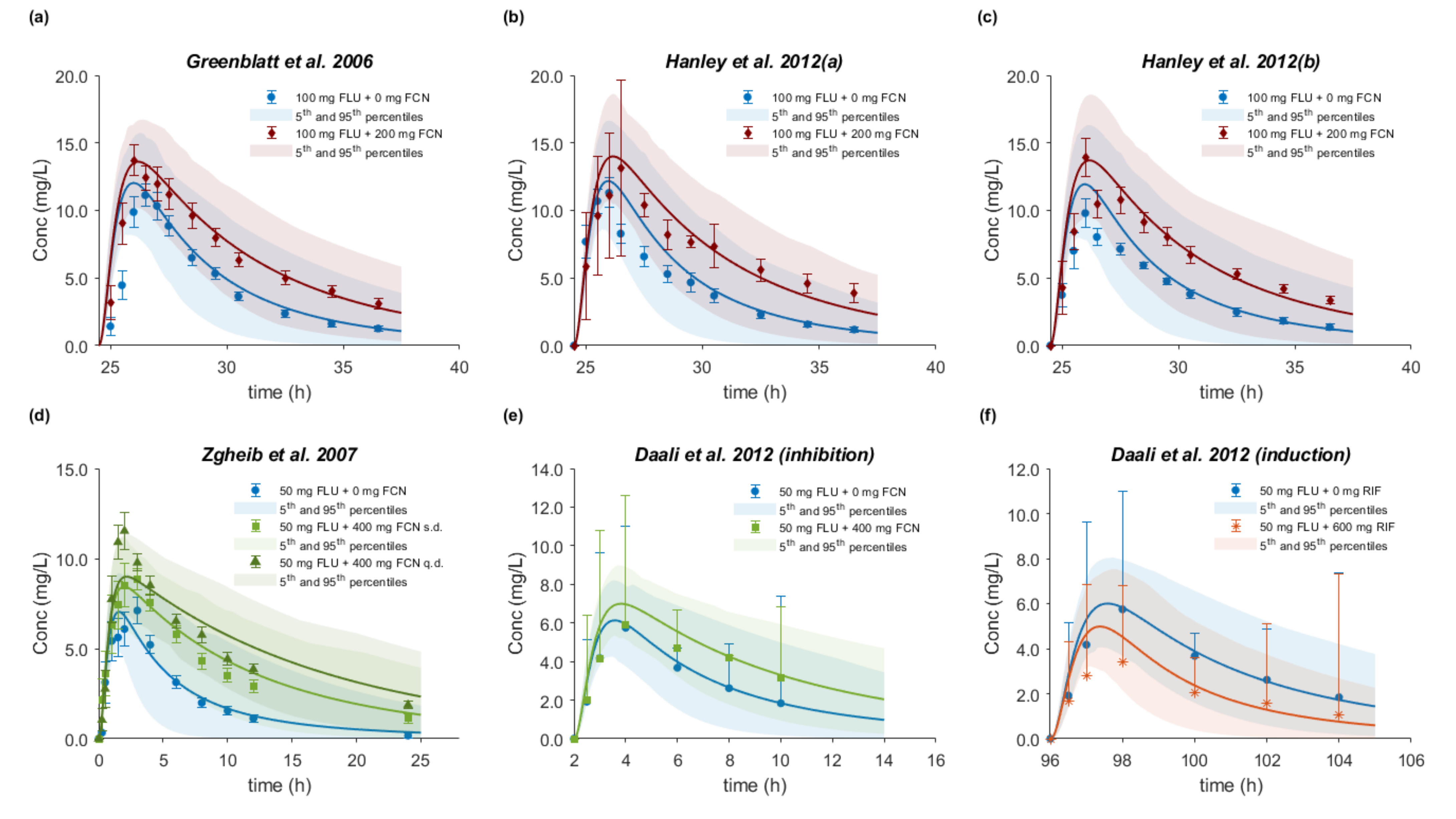
3.8. Drug–Drug–Gene Interactions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Davies, N.M. Clinical pharmacokinetics of flurbiprofen and its enantiomers. Clin. Pharm. 1995, 28, 100–114. [Google Scholar] [CrossRef] [PubMed]
- Dressman, J.B.; Berardi, R.R.; Elta, G.H.; Gray, T.M.; Montgomery, P.A.; Lau, H.S.; Pelekoudas, K.L.; Szpunar, G.J.; Wagner, J.G. Absorption of flurbiprofen in the fed and fasted states. Pharm. Res. 1992, 9, 901–907. [Google Scholar] [CrossRef] [PubMed]
- Ozbay, L.; Unal, D.O.; Cakici, I.; Fenercioglu, A.; Erol, D. Clinical study on the bioequivalence of two tablet formulations of flurbiprofen. Eur. J. Drug Metab. Pharm. 2009, 34, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.K.; Jackson, S.H.D.; Swift, C.G.; Hutt, A.J.; Jackson, S.H.D.; Swift, C.G.; Disposition, A.J.H. Disposition of flurbiprofen in man: Influence of stereochemistry and age. Xenobiotica 2003, 33, 1043–1057. [Google Scholar] [CrossRef]
- Szpunar, G.J.; Albert, K.S.; Wagner, J.G. Pharmacokinetics of flurbiprofen in man. II. Plasma protein binding. Res. Commun. Chem. Pathol. Pharmacol. 1989, 64, 17–30. [Google Scholar]
- Human Cytochrome P450 (CYP) Allele Nomenclature Committee. CYP2C9 Allele Nomenclature. Available online: https://www.pharmvar.org/htdocs/archive/cyp2c9.htm (accessed on 9 May 2020).
- Yamazaki, H.; Inoue, K.; Chiba, K.; Ozawa, N.; Kawai, T.; Suzuki, Y.; Goldstein, J.A.; Guengerich, F.P.; Shimada, T. Comparative Studies on the Catalytic Roles of Cytochrome P450 2C9 and Its Cys-and Leu-Variants in the Oxidation of Warfarin, Flurbiprofen, and Diclofenac by Human Liver Microsomes. Biochem. Pharmacol. 1998, 56, 243–251. [Google Scholar] [CrossRef]
- Van Booven, D.; Marsh, S.; McLeod, H.; Carrillo, M.W.; Sangkuhl, K.; Klein, T.E.; Altman, R.B. Cytochrome P450 2C9-CYP2C9. Pharmacogenet. Genom. 2010, 20, 277–281. [Google Scholar] [CrossRef]
- Scott, S.A.; Khasawneh, R.; Peter, I.; Kornreich, R.; Desnick, R.J. Combined CYP2C9, VKORC1 and CYP4F2 frequencies among racial and ethnic groups. Pharmacogenomics 2010, 11, 781–791. [Google Scholar] [CrossRef]
- Kirchheiner, J.; Brockmöller, J. Clinical consequences of cytochrome P450 2C9 polymorphisms. Clin. Pharmacol. Ther. 2005, 77, 1–16. [Google Scholar] [CrossRef]
- Lee, C.R.; Goldstein, J.A.; Pieper, J.A. Cytochrome P450 2C9 polymorphisms: A comprehensive review of the in-vitro and human data. Pharmacogenetics 2002, 12, 251–263. [Google Scholar] [CrossRef]
- Vieira, M.D.L.T.; Kim, M.J.; Apparaju, S.; Sinha, V.; Zineh, I.; Huang, S.M.; Zhao, P. PBPK model describes the effects of comedication and genetic polymorphism on systemic exposure of drugs that undergo multiple clearance pathways. Clin. Pharmacol. Ther. 2014, 95, 550–557. [Google Scholar] [CrossRef]
- Jin, Y.; Borell, H.; Gardin, A.; Ufer, M.; Huth, F.; Camenisch, G. In vitro studies and in silico predictions of fluconazole and CYP2C9 genetic polymorphism impact on siponimod metabolism and pharmacokinetics. Eur. J. Clin. Pharmacol. 2018, 74, 455–464. [Google Scholar] [CrossRef]
- Abend, A.; Heimbach, T.; Cohen, M.; Kesisoglou, F.; Pepin, X.; Suarez-Sharp, S. Dissolution and Translational Modeling Strategies Enabling Patient-Centric Drug Product Development: The M-CERSI Workshop Summary Report. AAPS J. 2018, 20, 60. [Google Scholar] [CrossRef] [PubMed]
- Sager, J.E.; Yu, J.; Ragueneau-Majlessi, I.; Isoherranen, N. Minireview Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulation Approaches: A Systematic Review of Published Models, Applications, and Model Verifications. DRUG Metab. Dispos. Drug Metab. Dispos. 2015, 43, 1823–1837. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Rowland, M.; Huang, S.-M. Best practice in the use of physiologically based pharmacokinetic modeling and simulation to address clinical pharmacology regulatory questions. Clin. Pharmacol. Ther. 2012, 92, 17–20. [Google Scholar] [CrossRef]
- Djebli, N.; Fabre, D.; Boulenc, X.; Fabre, G.; Sultan, E.; Hurbin, F. Physiologically Based Pharmacokinetic Modeling for Sequential Metabolism: Effect of CYP2C19 Genetic Polymorphism on Clopidogrel and Clopidogrel Active Metabolite Pharmacokinetics. DRUG Metab. Dispos. Drug Metab. Dispos. 2015, 43, 510–522. [Google Scholar] [CrossRef]
- Storelli, F.; Desmeules, J.; Daali, Y. Physiologically-Based Pharmacokinetic Modeling for the Prediction of CYP2D6-Mediated Gene–Drug–Drug Interactions. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 567–576. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, L.; Nguyen, K.; Fretland, A.J. Utility of Intersystem Extrapolation Factors in Early Reaction Phenotyping and the Quantitative Extrapolation of Human Liver Microsomal Intrinsic Clearance Using Recombinant Cytochromes P450. Drug Metab. Dispos. 2011, 39, 373–382. [Google Scholar] [CrossRef]
- Alqahtani, S.; Kaddoumi, A. Development of physiologically based pharmacokinetic/Pharmacodynamic model for Indomethacin disposition in pregnancy. PLoS ONE 2015, 10, e0139762. [Google Scholar] [CrossRef]
- Riedmaier, A.E.; Lindley, D.J.; Hall, J.A.; Castleberry, S.; Slade, R.T.; Stuart, P.; Carr, R.A.; Borchardt, T.B.; Bow, D.A.J.; Nijsen, M. Mechanistic Physiologically Based Pharmacokinetic Modeling of the Dissolution and Food Effect of a Biopharmaceutics Classification System IV Compound-The Venetoclax Story. J. Pharm. Sci. 2018, 107, 495–502. [Google Scholar] [CrossRef]
- Türk, D.; Hanke, N.; Wolf, S.; Frechen, S.; Eissing, T.; Wendl, T.; Schwab, M.; Lehr, T. Physiologically Based Pharmacokinetic Models for Prediction of Complex CYP2C8 and OATP1B1 (SLCO1B1) Drug-Drug-Gene Interactions: A Modeling Network of Gemfibrozil, Repaglinide, Pioglitazone, Rifampicin, Clarithromycin and Itraconazole. Clin. Pharm. 2019, 58, 1595–1607. [Google Scholar] [CrossRef]
- Markopoulos, C.; Andreas, C.J.; Vertzoni, M.; Dressman, J.; Reppas, C. In-vitro simulation of luminal conditions for evaluation of performance of oral drug products: Choosing the appropriate test media. Eur. J. Pharm. Biopharm. 2015, 93, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, A.; Leigh, M.; Kloefer, B.; Dressman, J.B. Advances in the design of fasted state simulating intestinal fluids: FaSSIF-V3. Eur. J. Pharm. Biopharm. 2015, 94, 229–240. [Google Scholar] [CrossRef]
- Loisios-Konstantinidis, I.; Cristofoletti, R.; Fotaki, N.; Turner, D.B.; Dressman, J. Establishing virtual bioequivalence and clinically relevant specifications using in vitro biorelevant dissolution testing and physiologically-based population pharmacokinetic modeling. case example: Naproxen. Eur. J. Pharm. Sci. 2020, 143, 105170. [Google Scholar] [CrossRef]
- Wang, J.; Flanagan, D.R. General solution for diffusion-controlled dissolution of spherical particles. 1. Theory. J. Pharm. Sci. 1999, 88, 731–738. [Google Scholar] [CrossRef]
- Wang, J.; Flanagan, D.R. General solution for diffusion-controlled dissolution of spherical particles. 2. Evaluation of experimental data. J. Pharm. Sci. 2002, 91, 534–542. [Google Scholar] [CrossRef]
- Mooney, K.G.; Mintun, M.A.; Himmelstein, K.J.; Stella, V.J. Dissolution kinetics of carboxylic acids I: Effect of pH under unbuffered conditions. J. Pharm. Sci. 1981, 70, 13–22. [Google Scholar] [CrossRef]
- Mooney, K.G.; Mintun, M.A.; Himmelstein, K.J.; Stella, V.J. Dissolution Kinetics of Carboxylic Acids II: Effect of Buffers. J. Pharm. Sci. 1981, 70, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Mooney, K.G.; Rodriguez-gaxiola, M.; Mintun, M.; Himmelstein, K.J.; Stella, V.J. Dissolution Kinetics of Phenylbutazone. J. Pharm. Sci. 1981, 70, 1358–1365. [Google Scholar] [CrossRef]
- Ozturk, S.S.; Palsson, B.O.; Dressman, J.B. Dissolution of lonizable Drugs in Buffered and Unbuffered Solutions. Pharm. Res. 1988, 5, 272–282. [Google Scholar] [CrossRef]
- Sheng, J.J.; McNamara, D.P.; Amidon, G.L. Toward an In Vivo dissolution methodology: A comparison of phosphate and bicarbonate buffers. Mol. Pharm. 2009, 6, 29–39. [Google Scholar] [CrossRef]
- Serajuddin, A.T.M.; Jarowski, C. Effect of diffusion layer pH and solubility on the dissolution rate of pharmaceutical bases and their hydrochloride salts. I: Phenazopyridine. J. Pharm. Sci. 1985, 74, 142–147. [Google Scholar] [CrossRef]
- Mei, C.; Li, B.; Yin, Q.; Jin, J.; Xiong, T.; He, W.; Gao, X.; Xu, R.; Zhou, P.; Zheng, H.; et al. Liquid chromatography-tandem mass spectrometry for the quantification of flurbiprofen in human plasma and its application in a study of bioequivalence. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2015, 993–994, 69–74. [Google Scholar] [CrossRef]
- Gonzalez-Younes, I.; Wagner, J.G.; Gaines, D.A.; Ferry, J.J.; Hageman, J.M. Absorption of flurbiprofen through human buccal mucosa. J. Pharm. Sci. 1991, 80, 820–823. [Google Scholar] [CrossRef]
- Szpunar, G.J.; Albert, K.S.; Bole, G.G.; Dreyfus, J.N.; Lockwood, G.F.; Wagner, J.G. Pharmacokinetics of flurbiprofen in man. I. Area/dose relationships. Biopharm. Drug Dispos. 1987, 8, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Byeon, J.Y.; Kim, Y.H.; Kim, S.H.; Choi, C.I.; Bae, J.W.; Sohn, U.D.; Jang, C.G.; Lee, J.; Lee, S.Y. Effects of CYP2C9∗1/∗3 genotype on the pharmacokinetics of flurbiprofen in Korean subjects. Arch. Pharm. Res. 2015, 38, 1232–1237. [Google Scholar] [CrossRef]
- Jamali, F.; Collins, D.S.; Berry, B.W.; Molder, S.; Cheung, R.; McColl, K.; Cheung, H. Comparative bioavailability of two flurbiprofen products: Stereospecific versus conventional approach. Biopharm. Drug Dispos. 1991, 12, 435–445. [Google Scholar] [CrossRef]
- Suri, A.; Grundy, B.L.; Derendorf, H. Pharmacokinetics and pharmacodynamics of enantiomers of ibuprofen and flurbiprofen after oral administration. Int. J. Clin. Pharmacol. Ther. 1997, 35, 1–8. [Google Scholar]
- Liu, Y.-M.; Liu, G.-Y.; Liu, Y.; Li, S.-J.; Jia, J.-Y.; Zhang, M.-Q.; Lu, C.; Zhang, Y.-M.; Li, X.-N.; Yu, C. Pharmacokinetic and Bioequivalence Comparison Between Orally Disintegrating and Conventional Tablet Formulations of Flurbiprofen: A Single-Dose, Randomized-Sequence, Open-Label, Two-Period Crossover Study in Healthy Chinese Male Volunteers. Clin. Ther. 2009, 31, 1787–1795. [Google Scholar] [CrossRef]
- Lee, H.-I.; Choi, C.-I.; Byeon, J.-Y.; Lee, J.-E.; Park, S.-Y.; Kim, Y.-H.; Kim, S.-H.; Lee, Y.-J.; Jang, C.-G.; Lee, S.-Y. Simultaneous determination of flurbiprofen and its hydroxy metabolite in human plasma by liquid chromatography-tandem mass spectrometry for clinical application. J. Chromatogr. B 2014, 971, 58–63. [Google Scholar] [CrossRef]
- Lee, C.R.; Pieper, J.A.; Frye, R.F.; Hinderliter, A.L.; Blaisdell, J.A.; Goldstein, J.A. Differences in flurbiprofen pharmacokinetics between CYP2C9*1/*1, *1/*2, and *1/*3 genotypes. Eur. J. Clin. Pharmacol. 2003, 58, 791–794. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Brundage, R.; Oetting, W.S.; Leppik, I.E.; Tracy, T.S. Differential Genotype Dependent Inhibition of CYP2C9 in Humans. Drug Metab. Dispos. 2008, 36, 1242–1248. [Google Scholar] [CrossRef]
- Hanley, M.J.; Masse, G.; Harmatz, J.S.; Court, M.H.; Greenblatt, D.J. Pomegranate juice and pomegranate extract do not impair oral clearance of flurbiprofen in human volunteers: Divergence from in vitro results. Clin. Pharmacol. Ther. 2012, 92, 651–657. [Google Scholar] [CrossRef]
- Hanley, M.J.; Masse, G.; Harmatz, J.S.; Cancalon, P.F.; Dolnikowski, G.G.; Court, M.H.; Greenblatt, D.J. Effect of blueberry juice on clearance of buspirone and flurbiprofen in human volunteers. Br. J. Clin. Pharmacol. 2013, 75, 1041–1052. [Google Scholar] [CrossRef]
- Greenblatt, D.J.; Von Moltke, L.L.; Perloff, E.S.; Luo, Y.; Harmatz, J.S.; Boston, M.A.Z. Interaction of flurbiprofen with cranberry juice, grape juice, tea, and fluconazole: In vitro and clinical studies. Clin. Pharmacol. Ther. 2006, 450, 125–133. [Google Scholar] [CrossRef]
- Zgheib, N.K.; Frye, R.F.; Tracy, T.S.; Romkes, M.; Branch, R.A. Evaluation of flurbiprofen urinary ratios as in vivo indices for CYP2C9 activity. Br. J. Clin. Pharmacol. 2007, 63, 477–487. [Google Scholar] [CrossRef]
- Daali, Y.; Samer, C.; Déglon, J.; Thomas, A.; Chabert, J.; Rebsamen, M.; Staub, C.; Dayer, P.; Desmeules, J. Oral flurbiprofen metabolic ratio assessment using a single-point dried blood spot. Clin. Pharmacol. Ther. 2012, 91, 489–496. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration; A Center for Drug Evaluation and Research (CDER). Physiologically Based Pharmacokinetic Analyses—Format and Content Guidance for Industry; Food and Drug Administration: Silver Spring, MD, USA, 2018. [Google Scholar]
- Kuepfer, L.; Niederalt, C.; Wendl, T.; Schlender, J.-F.; Willmann, S.; Lippert, J.; Block, M.; Eissing, T.; Teutonico, D. Applied Concepts in PBPK Modeling: How to Build a PBPK/PD Model. CPT Pharmacomet. Syst. Pharmacol. 2016, 5, 516–531. [Google Scholar] [CrossRef]
- Ke, A.; Barter, Z.; Rowland-Yeo, K.; Almond, L. Towards a Best Practice Approach in PBPK Modeling: Case Example of Developing a Unified Efavirenz Model Accounting for Induction of CYPs 3A4 and 2B6. CPT Pharmacomet. Syst. Pharmacol. 2016, 5, 367–376. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency (EMA). Committee for Medicinal Products for Human Use (CHMP) Guideline on the Reporting of Physiologically Based Pharmacokinetic (PBPK) Modelling and Simulation; European Medicines Agency: London, UK, 2018. [Google Scholar]
- Shebley, M.; Sandhu, P.; Emami Riedmaier, A.; Jamei, M.; Narayanan, R.; Patel, A.; Peters, S.A.; Reddy, V.P.; Zheng, M.; de Zwart, L.; et al. Physiologically Based Pharmacokinetic Model Qualification and Reporting Procedures for Regulatory Submissions: A Consortium Perspective. Clin. Pharmacol. Ther. 2018, 104, 88–110. [Google Scholar] [CrossRef] [PubMed]
- Avdeef, A. pH-metric Solubility. 1. Solubility-pH Plots. Gibbs Buffer and pK, in Profiles from Bjerrum the Solid State. Pharm. Pharmacol. Commun. Pharm. Pharmacol. Commun. 1998, 4, 165–178. [Google Scholar] [CrossRef]
- Czyrski, A. Determination of the Lipophilicity of Ibuprofen, Naproxen, Ketoprofen, and Flurbiprofen with Thin-Layer Chromatography. J. Chem. 2019, 2019, 3407091. [Google Scholar] [CrossRef]
- Kaiser, D.G.; Brooks, C.D.; Lomen, P.L. Pharmacokinetics of Flurbiprofen. Am. J. Med. 1986, 80, 10–15. [Google Scholar] [CrossRef]
- Risdall, P.C.; Adams, S.S.; Crampton, E.L.; Marchant, B. The Disposition and Metabolism of Flurbiprofen in Several Species Including Man. XENOBIOTICA 1978, 8, 691–704. [Google Scholar] [CrossRef]
- Aarons, L.; Khan, A.Z.; Grennan, D.M.; Alam-Siddiqi, M. The binding of flurbiprofen to plasma proteins. J. Pharm. Pharmacol. 1985, 37, 644–646. [Google Scholar] [CrossRef]
- Lin, J.H.; Cocchetto, D.M.; Duggan, D.E. Protein-binding as a primary determinant of the clinical pharmacokinetic properties of nonsteroidal antiinflammatory drugs. Clin. Pharm. 1987, 12, 402–432. [Google Scholar] [CrossRef]
- Yazdanian, M.; Briggs, K.; Jankovsky, C.; Hawi, A. The “High Solubility” Definition of the Current FDA Guidance on Biopharmaceutical Classification System May Be Too Strict for Acidic Drugs. Pharm. Res. 2004, 21, 293–299. [Google Scholar] [CrossRef]
- Wang, L.; Bao, S.-H.; Pan, P.-P.; Xia, M.; Chen, M.-C.; Liang, B.-Q.; Dai, D.-P.; Cai, J.-P.; Hu, G.-X.; Xia, M.-M. Effect of CYP2C9 genetic polymorphism on the metabolism of flurbiprofen in vitro. Drug Dev. Ind. Pharm. 2015, 41, 1363–1367. [Google Scholar] [CrossRef]
- Wang, H.; Yuan, L.; Zeng, S. Characterizing the effect of UDP-glucuronosyltransferase (UGT) 2B7 and UGT1A9 genetic polymorphisms on enantioselective glucuronidation of flurbiprofen. Biochem. Pharmacol. 2011, 82, 1757–1763. [Google Scholar] [CrossRef]
- Jamei, M.; Turner, D.; Yang, J.; Neuhoff, S.; Polak, S.; Rostami-Hodjegan, A.; Tucker, G. Population-based mechanistic prediction of oral drug absorption. AAPS J. 2009, 11, 225–237. [Google Scholar] [CrossRef]
- Darwich, A.S.; Neuhoff, S.; Jamei, M.; Rostami-Hodjegan, A. Interplay of Metabolism and Transport in Determining Oral Drug Absorption and Gut Wall Metabolism: A Simulation Assessment Using the “Advanced Dissolution, Absorption, Metabolism (ADAM)” Model. Curr. Drug Metab. 2010, 11, 716–729. [Google Scholar] [CrossRef]
- Hens, B.; Brouwers, J.; Anneveld, B.; Corsetti, M.; Symillides, M.; Vertzoni, M.; Reppas, C.; Turner, D.B.; Augustijns, P. Gastrointestinal transfer: In vivo evaluation and implementation in in vitro and in silico predictive tools. Eur. J. Pharm. Sci. 2014, 63, 233–242. [Google Scholar] [CrossRef]
- Psachoulias, D.; Vertzoni, M.; Goumas, K.; Kalioras, V.; Beato, S.; Butler, J.; Reppas, C. Precipitation in and Supersaturation of Contents of the Upper Small Intestine After Administration of Two Weak Bases to Fasted Adults. Pharm. Res. 2011, 28, 3145–3158. [Google Scholar] [CrossRef]
- Cristofoletti, R.; Patel, N.; Dressman, J.B. Differences in Food Effects for 2 Weak Bases With Similar BCS Drug-Related Properties: What Is Happening in the Intestinal Lumen? J. Pharm. Sci. 2016, 105, 2712–2722. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Paixão, P.; Bermejo, M.; Hens, B.; Tsume, Y.; Dickens, J.; Shedden, K.; Salehi, N.; Koenigsknecht, M.J.; Baker, J.R.; Hasler, W.L.; et al. Gastric emptying and intestinal appearance of nonabsorbable drugs phenol red and paromomycin in human subjects: A multi-compartment stomach approach. Eur. J. Pharm. Biopharm. 2018, 129, 162–174. [Google Scholar] [CrossRef]
- Rodgers, T.; Rowland, M. Physiologically based pharmacokinetic modelling 2: Predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J. Pharm. Sci. 2006, 95, 1238–1257. [Google Scholar] [CrossRef] [PubMed]
- Crewe, H.K.; Barter, Z.E.; Rowland Yeo, K.; Rostami-Hodjegan, A. Are there differences in the catalytic activity per unit enzyme of recombinantly expressed and human liver microsomal cytochrome P450 2C9? A systematic investigation into inter-system extrapolation factors. Biopharm. Drug Dispos. 2011, 32, 303–318. [Google Scholar] [CrossRef]
- Kuehl, G.E.; Lampe, J.W.; Potter, J.D.; Bigler, J. Glucuronidation of nonsteroidal intiinflammatory drugs Identifying the enzymes in human liver microsomes. Drug Metab. Dispos. 2005, 33, 1027–1035. [Google Scholar] [CrossRef] [PubMed]
- Mano, Y.; Usui, T.; Kamimura, H. Predominant contribution of UDP-glucuronosyltransferase 2B7 in the glucuronidation of racemic flurbiprofen in the human liver. Drug Metab. Dispos. 2007, 35, 1182–1187. [Google Scholar] [CrossRef]
- Nielsen, L.M.; Sverrisdóttir, E.; Stage, T.B.; Feddersen, S.; Brøsen, K.; Christrup, L.L.; Drewes, A.M.; Olesen, A.E. Lack of genetic association between OCT1, ABCB1, and UGT2B7 variants and morphine pharmacokinetics. Eur. J. Pharm. Sci. 2017, 99, 337–342. [Google Scholar] [CrossRef]
- Ayuso, P.; Neary, M.; Chiong, J.; Owen, A. Meta-analysis of the effect of CYP2B6, CYP2A6, UGT2B7 and CAR polymorphisms on efavirenz plasma concentrations. J. Antimicrob. Chemother. 2019, 74, 3281–3290. [Google Scholar] [CrossRef]
- Edginton, A.N.; Schmitt, W.; Willmann, S. Development and evaluation of a generic physiologically based pharmacokinetic model for children. Clin. Pharm. 2006, 45, 1013–1034. [Google Scholar] [CrossRef]
- Poulin, P.; Theil, F.-P. Development of a novel method for predicting human volume of distribution at steady-state of basic drugs and comparative assessment with existing methods. J. Pharm. Sci. 2009, 98, 4941–4961. [Google Scholar] [CrossRef]
- Obach, R.S.; Baxter, J.G.; Liston, T.E.; Silber, B.M.; Jones, B.C.; MacIntyre, F.; Rance, D.J.; Wastall, P. The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J. Pharmacol. Exp. Ther. 1997, 283, 46–58. [Google Scholar] [PubMed]
- Kim, Y.; Hatley, O.; Rhee, S.J.; Yi, S.; Lee, H.A.; Yoon, S.; Chung, J.Y.; Yu, K.S.; Lee, H. Development of a Korean-specific virtual population for physiologically based pharmacokinetic modelling and simulation. Biopharm. Drug Dispos. 2019, 40, 135–150. [Google Scholar] [CrossRef]
- Myrand, S.P.; Sekiguchi, K.; Man, M.Z.; Lin, X.; Tzeng, R.Y.; Teng, C.H.; Hee, B.; Garrett, M.; Kikkawa, H.; Lin, C.Y.; et al. Pharmacokinetics/genotype associations for major cytochrome P450 enzymes in native and first- and third-generation Japanese populations: Comparison with Korean, Chinese, and Caucasian populations. Clin. Pharmacol. Ther. 2008, 84, 347–361. [Google Scholar] [CrossRef]
- Yoon, Y.-R.; Shon, J.-H.; Kim, M.-K.; Lim, Y.-C.; Lee, H.-R.; Park, J.-Y.; Cha, I.-J.; Shin, J.-G. Frequency of cytochrome P450 2C9 mutant alleles in a Korean population. Br. J. Clin. Pharmacol. 2008, 51, 277–280. [Google Scholar] [CrossRef]
- Avdeef, A.; Berger, C.M. pH-Metric Solubility. 2: Correlation Between the Acid-Base Titration and formulations for use in early animal bioavailability and toxicity studies. Later in development, solubility takes on a broader. Pharm. Res. 2000, 17, 85–89. [Google Scholar] [CrossRef]
- Karow, A.R.; Bahrenburg, S.; Garidel, P. Buffer capacity of biologics-from buffer salts to buffering by antibodies. Biotechnol. Prog. 2013, 29, 480–492. [Google Scholar] [CrossRef]
- Lee, C.B.; Pieper, J.A.; Frye, R.F.; Hinderliter, A.L.; Blaisdell, J.A.; Goldstein, J.A. Tolbutamide, flurbiprofen, and losartan as probes of CYP2C9 activity in humans. J. Clin. Pharmacol. 2003, 43, 84–91. [Google Scholar] [CrossRef]
- Vogl, S.; Lutz, R.W.; Schönfelder, G.; Lutz, W.K. CYP2C9 genotype vs. metabolic phenotype for individual drug dosing-A correlation analysis using flurbiprofen as probe drug. PLoS ONE 2015, 10, e0120403. [Google Scholar] [CrossRef]
- EMA Committee for Proprietary Medicinal Products (CPMP). Note for Guidance on the Investigation of Bioavailability and Bioequivalence; European Medicines Agency: London, UK, 2000. [Google Scholar]
- Dai, D.P.; Xu, R.A.; Hu, L.M.; Wang, S.H.; Geng, P.W.; Yang, J.F.; Yang, L.P.; Qian, J.C.; Wang, Z.S.; Zhu, G.H.; et al. CYP2C9 polymorphism analysis in Han Chinese populations: Building the largest allele frequency database. Pharm. J. 2014, 14, 85–92. [Google Scholar] [CrossRef]
- Scordo, M.G.; Aklillu, E.; Yasar, U.; Dahl, M.-L.; Spina, E.; Ingelman-Sundberg, M. Genetic polymorphism of cytochrome P450 2C9 in a Caucasian and a black African population. Br. J. Clin. Pharmacol. 2001, 52, 447–450. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Administration and Formulation | CYP2C9 Genotype | No. of Subjects | Female Ratio | Ethnicity | Age (years) | BW/BW Range (kg) | BH/BH Range (cm) | Reference |
|---|---|---|---|---|---|---|---|---|
| Intravenous | ||||||||
| 50 mg as 10 mg/mL solution (injection within 2 min) | n.a. | 24 | 0 | Chinese | - | - | - | Mei et al. [34] |
| Oral | ||||||||
| 25 mL of oral solution containing 67.9 mg FLU with 175 mL water; | n.a. | 12 | 0 | Caucasian | 25–31 | - | - | Gonzalez-Younes et al. [35] |
| 40 mL oral solution containing 100 mg FLU with 180 mL water; | n.a. | 15 | 0 | Caucasian | 29 (18–40) | 76.4 (62.3–109.1) | 177 (168–188) | Szpunar et al. [36] |
| Froben solution 40 mg; | 1*/1* | 12 | 0 | Korean | 23.1 (2.4) | 65.1 (7.1) | 174.8 (5.0) | Lee et al. [41] |
| Froben solution 40 mg; | 1*/3* | 8 | 0 | Korean | 22 (2.7) | 64.6 (7.1) | 172.8 (6.4) | Lee et al. [41] |
| USP tablets (Mylan Pharmaceuticals) 50 mg; | 1*/1* | 5 | 0.533 | Caucasian | 24 (5) | 79 (18) | - | Lee et al. [42] |
| USP tablets (Mylan Pharmaceuticals) 50 mg; | 1*/2* | 5 | 0.533 | Caucasian | 24 (5) | 79 (18) | - | Lee et al. [42] |
| USP tablets (Mylan Pharmaceuticals) 50 mg; | 1*/3* | 5 | 0.533 | Caucasian | 24 (5) | 79 (18) | - | Lee et al. [42] |
| Froben 100 mg with 100 mL water; | n.a. | 23 | 0 | Caucasian | 27.2 (18–35) | 71.8 (52.5) | - | Jamali et al. [38] |
| Ansaid 100 mg with 100 mL water; | n.a. | 23 | 0 | Caucasian | 27.2 (18–35) | 71.8 (52.5) | - | Jamali et al. [38] |
| Froben 100 mg with 150 mL water; | n.a. | 4 | 0.5 | Caucasian | 26.8 (2.2) | 67.8 (4.1) | - | Patel et al. [4] |
| 100 mg tablet with 200 mL water; | n.a. | 6 | - | Caucasian | - | - | - | Suri et al. [39] |
| Ansaid 100 mg with 180 mL water; | n.a. | 15 | 0 | Caucasian | 29 (18–40) | 76.4 (62.3–109.1) | 177 (168–188) | Szpunar et al. [36] |
| 3 × 50 mg conventional tablets (reference); | n.a. | 20 | 0 | Chinese | 21.4 (2.5) | 63.2 (5.1) | 174.4 (4.2) | Liu et al. [40] |
| 3 × 50 mg orally disintegrated tablets (test); | n.a. | 20 | 0 | Chinese | 21.4 (2.5) | 63.2 (5.1) | 174.4 (4.2) | Liu et al. [40] |
| 2 × Ansaid 100 mg with 180 mL water; | n.a. | 15 | 0 | Caucasian | 29 (18–40) | 76.4 (62.3–109.1) | 177 (168–188) | Szpunar et al. [36] |
| 3 × Ansaid 100 mg with 180 mL water; | n.a. | 15 | 0 | Caucasian | 29 (18–40) | 76.4 (62.3–109.1) | 177 (168–188) | Szpunaret al. [36] |
| Victim Drug Administration | Perpetrator Drug Administration | Perpetrator in vitro Ki (μΜ) | No. of Doses | Interval (h) | CYP2C9 genotype | No. of Subjects | Female Ratio | Ethnicity | Age (years) | BW/BW Range (kg) | BH/BH Range (cm) | References |
| Flurbiprofen | Fluconazole | |||||||||||
| po 50 mg s.d. | po 200 mg q.d. | 11 | 7 | 2 | 1*/1* | 11 | 0.64 | - | 25 (19–36) | 73.7 (51–108) | 166 (154–193) | Kumar et al. [43] |
| po 50 mg s.d. | po 400 mg q.d. | 11 | 7 | 2 | 1*/1* | 11 | 0.64 | - | 25 (19–36) | 73.7 (51–108) | 166 (154–193) | Kumar et al. [43] |
| po 50 mg s.d. | po 200 mg q.d. | 17 | 7 | 2 | 1*/3* | 8 | 0.63 | - | 23 (19–28) | 66.9 (49–84) | 167 (160–189) | Kumar et al. [43] |
| po 50 mg s.d. | po 400 mg q.d. | 17 | 7 | 2 | 1*/3* | 8 | 0.63 | - | 23 (19–28) | 66.9 (49–84) | 167 (160–189) | Kumar et al. [43] |
| po 50 mg s.d. | po 200 mg q.d. | 23 | 7 | 2 | 3*/3* | 2 | 0.0 | - | (25, 29) | (77, 85) | (177, 179) | Kumar et al. [43] |
| po 50 mg s.d. | po 400 mg q.d. | 23 | 7 | 2 | 3*/3* | 2 | 0.0 | - | (25, 29) | (77, 85) | (177, 179) | Kumar et al. [43] |
| po 100 mg s.d. | po 200 mg b.i.d. | 14.3/20.3 | 2 | 0.5 | - | 12 | 0.25 | Caucasian (n = 8), other (n = 4) a | 19–54 | - | - | Hanley et al. [45] |
| po 100 mg s.d. | po 200 mg b.i.d. | 29.9 | 2 | 0.5 | - | 14 | 0.21 | - | 29 ± 8 | 81 ± 14 | - | Greenblatt et al. [46] |
| po 100 mg s.d. | po 200 mg b.i.d. | 14.3/20.3 | 2 | 0.5 | - | 12 | 0.17 | - | 24–55 | - | - | Hanley et al. [44] |
| Victim Drug Administration | Perpetrator Drug Administration | Perpetrator in vitro Ki (μΜ) | No. of Doses | Interval (h) | CYP2C9 genotype | No. of Subjects | Female Ratio | Ethnicity | Age (years) | BW/BW Range (kg) | BH/BH Range (cm) | References |
| Flurbiprofen | Fluconazole | |||||||||||
| po 50 mg s.d. | po 400 mg s.d | 10 | 1 | 2 | - | 12 | 0.58 | Caucasian (n = 10), other (n = 2) b | 37 ± 3.1 | - | - | Zgheib et al. [47] |
| po 50 mg s.d. | po 400 mg q.d | 10 | 7 | 2 | - | 12 | 0.58 | Caucasian (n = 10), other (n = 2) b | 37 ± 3.1 | - | - | Zgheib et al. [47] |
| po 50 mg s.d. | po 400 mg s.d | 10 | 1 | 2 | - | 10 | 0.0 | Caucasian (n = 9), African (n = 1) | 27 (23–39) | - | - | Daali et al. [48] |
| Rifampicin | ||||||||||||
| po 50 mg s.d. | po 600 mg q.d. | n.a.c | 5 | 0 | - | 10 | 0.0 | Caucasian (n = 9), African (n = 1) | 27 (23–39) | - | - | Daali et al. [48] |
| Parameters | Value | Reference/Comments |
|---|---|---|
| Physicochemical and Blood Binding |  | |
| Chemical Structure | ||
| MW (g/mol) | 244.3 | |
| logPo:w | 3.99 | [54,55] |
| pKa | 4.05 | Updated from in vitro solubility data (see Table 4 and Section 3.2) |
| Blood/plasma ratio | 0.55 | [56] |
| Fraction unbound in plasma | 0.01 | [5,56,57,58,59] |
| Absorption | ||
| Model | ADAM | |
| Papp, Caco-2 (×10−6 cm/s) | 20.1 | Measured value [60] |
| Papp, Caco-2, ref (×10−6 cm/s) | 1.57 | Negative calibrator (Atenolol) value [60] |
| Papp, Caco-2, ref (×10−6 cm/s) | 15.8 | Positive calibrator (Verapamil) value [60] |
| Peff, human (×10−4 cm/s) | 4.83 | Predicted by Simcyp Permeability Calibrator-custom correlation |
| Formulation type | Immediate Release | |
| S0 (mg/mL) | 0.018 | In vitro data (see Table 4 and Section 3.1) |
| logKm:w neutral | 5.37 | Estimated from in vitro data (see Table 5 and Section 2.6 and Section 3.2) |
| logKm:w ion | 2.46 | Estimated from in vitro data (see Table 5 and Section 2.6 and Section 3.2) |
| In vivo dissolution | see Table 6 and Table 7 | Estimated DLM scalars from in vitro data (see Section 2.7) |
| Distribution | ||
| Model | Full PBPK | |
| Vss (L/kg) | 0.074 | Predicted by Method 2 |
| Kp scalar | 0.7 | Optimized on the basis of IV data–PE module |
| Elimination | ||
| Fox | 0.71 | [4] |
| Model | Allelic-specific enzyme kinetics | |
| CYP2C9 1*/1*-Vmax (pmol/min/pmol CYP) | 15.79 | Recombinant CYP (fu,mic = 1) [61] |
| CYP2C9 1*/1*-Km (μM) | 8.756 | Recombinant CYP (fu,mic = 1) [61] |
| CYP2C9 1*/2*-Vmax (pmol/min/pmol CYP) | 11.53 | Scaled for CLCYP2C9 1*/1*/CLCYP2C9 1*/3* = 0.73 [42] |
| CYP2C9 1*/2*-Km (μM) | 8.756 | Recombinant CYP (fu,mic = 1) [61] |
| CYP2C9 1*/3*-Vmax (pmol/min/pmol CYP) | 9.55 | Scaled for CLCYP2C9 1*/1*/CLCYP2C9 1*/3* = 0.605 [42] |
| CYP2C9 1*/3*-Km (μM) | 8.756 | Recombinant CYP (fu,mic = 1) [61] |
| CYP2C9 2*/2*-Vmax (pmol/min/pmol CYP) | 10.04 | Recombinant CYP (fu,mic = 1) [61] |
| CYP2C9 2*/2*-Km (μM) | 10.39 | Recombinant CYP (fu,mic = 1) [61] |
| CYP2C9 3*/3*-Vmax (pmol/min/pmol CYP) | 8.901 | Recombinant CYP (fu,mic = 1) [61] |
| CYP2C9 3*/3*-Km (μM) | 23.25 | Recombinant CYP (fu,mic = 1) [61] |
| CYP2C9-ISEF | 0.3 | Optimized on the basis of IV data–PE module |
| UGT2B7-Vmax (pmol/min/mg protein) | 119.7 | Recombinant UGT [62] |
| UGT2B7-Km (μM) | 50.21 | Recombinant UGT [62] |
| UGT1A9-Vmax (pmol/min/mg protein) | 3.286 | Recombinant UGT [62] |
| UGT1A9-Km (μM) | 182.2 | Recombinant UGT [62] |
| Additional HLM liver CLint (μL/min/mg proein) | 7.88 | Retrograde model for a target fmCYP2C9 = 0.71 |
| Clrenal (L/h) | 0.066 | [4] |
| Pharmacodynamics | ||
| Model | Effect compartment linked to inhibitory Emax model | [39] |
| keo (h−1) (%CV) | 0.56 (43) | PD endpoint: Evoked Potentials |
| IC50 (mg/L) | 25.8 (21) | |
| keo (h−1) (%CV) | 0.89 (24) | PD endpoint: Pain rating score |
| IC50 (mg/L) | 27.6 (10) |
| Medium | Flurbiprofen | |
|---|---|---|
| pHfinal | Solubility (μg/mL) | |
| Aqueous buffers | ||
| FaSSGF Level I (pH = 1.6) | 1.6 | 18.1 (0.17) |
| Acetate buffer (pH = 4.5) | 4.7 | 101.1 (7.06) |
| FeSSIF V1 Level I (pH = 5.0) | 5.1 | 225.4 (5.6) |
| Phosphate buffer (pH = 6.5) | 6.1 | 2024.4 (128.2) |
| Phosphate buffer (pH = 6.8) | 6.3 | 3127.1 (194.9) |
| Fasted state biorelevant media | ||
| Level III FaSSGF (pH = 1.6) | 1.6 | 18.5 (1.6) |
| Level II FaSSIF V1 (pH = 6.5) | 6.0 | 1954.9 (3.9) |
| Level II FaSSIF V3 (pH = 6.7) | 5.9 | 1585.4 (172.1) |
| pKa | logKm:w Neutral | logKm:w Ion | |
|---|---|---|---|
| Estimate (95% CI) | 4.05 (4.42–4.44) | 5.36 (4.61–6.11) | 2.56 (1.38–5.02) |
| R2 | 0.9990 | 0.9999 | |
| Dissolution Medium | Formulation | ||
|---|---|---|---|
| API Powder | USP Tablets | Antadys | |
| FaSSGF Level III | |||
| SDLM (95% CI) | 0.0218 (0.0161–0.0274) | 0.0929 (0.0731–0.113) | 0.107 (0.087–0.127) |
| R2 | 0.944 | 0.973 | 0.982 |
| FaSSIF V3 Level I | |||
| SDLM (95% CI) | 0.00185 (0.001–0.00312) | 0.0791 (0.0589–0.993) | 0.120 (0.0979–0.142) |
| R2 | 0.974 | 0.986 | 0.995 |
| FaSSIF V3 Level II | |||
| SDLM (95% CI) | 0.0965 (0.0544–0.139) | 0.0622 (0.0398–0.0847) | 0.125 (0.106–0.143) |
| R2 | 0.971 | 0.976 | 0.996 |
| Ph. Eur. Phosphate Buffer | |||
| SDLM (95% CI) | 0.00542 (0.00468–0.00617) | 0.0150 (0.0110–0.0189) | 0.0449 (0.0448–0.0450) |
| R2 | 0.986 | 0.983 | 0.999 |
| Dissolution Model/Media | Formulation |
|---|---|
| Froben | |
| First order disintegration/all intestinal media | |
| kd (h−1) (95% CI) | 0.127 (0.00844–0.0253) |
| Tlag (min) (95% CI) | 14.6 (8.91–20.1) |
| R2 | 0.941 |
| Serial Dilution/Two-stage (FaSSGF Level III + FaSSIF V3 Level II) | |
| SDLM, Gastric (95% CI) | 0.001 (0.001–0.0244) |
| SDLM, Intestinal (95% CI) | 0.0712 (0.0576–0.0849) |
| R2 | 0.991 |
| Route of Administration | Dose (mg) | Flurbiprofen MRD | Reference |
|---|---|---|---|
| iv (s.d.) | 50 | 1.41 | [34] |
| po (sol, s.d.) | 67.9 | 1.85 | [35] |
| po (sol, s.d.) | 100 | 1.60 | [36] |
| po (sol, s.d., CYP2C9 1*/1*) | 40 | 1.52 | [37] |
| po (sol, s.d., CYP2C9 1*/3*) | 40 | 1.30 | [37] |
| po (tab, s.d., CYP2C9 1*/1*) | 50 | 1.24 | [42] |
| po (tab, s.d., CYP2C9 1*/2*) | 50 | 1.30 | [42] |
| po (tab, s.d., CYP2C9 1*/3*) | 50 | 1.62 | [42] |
| po (tab, s.d., CYP2C9 1*/1*) | 50 | 1.26 | [43] |
| po (tab, s.d., CYP2C9 1*/1*) | 50 | 1.25 | [43] |
| po (tab, s.d., CYP2C9 1*/3*) | 50 | 1.28 | [43] |
| po (tab, s.d., CYP2C9 1*/3*) | 50 | 1.25 | [43] |
| po (tab, s.d., CYP2C9 3*/3*) | 50 | 1.25 | [43] |
| po (tab, s.d., CYP2C9 3*/3*) | 50 | 1.20 | [43] |
| po (tab (Ansaid), s.d.) | 100 | 1.31–2.38 | [38] |
| SDLM_SI = 0.125 | 1.31 | ||
| SDLM_SI = 0.0054 | 1.72 | ||
| kd = 0.127 h−1 and Tlag = 14.6 min | 1.64 | ||
| SDLM_SI = 0.0712 | 1.65 | ||
| SDLM_SI = 0.0018 | 2.38 | ||
| po (tab (Froben), s.d.) | 100 | 1.70–2.43 | [38] |
| SDLM_SI = 0.125 | 1.70 | ||
| SDLM_SI = 0.0054 | 1.88 | ||
| kd = 0.127 h−1 and Tlag = 14.6 min | 1.80 | ||
| SDLM_SI = 0.0712 | 1.83 | ||
| SDLM_SI = 0.0018 | 2.43 | ||
| po (tab (Froben), s.d.) | 100 | 1.69–1.92 | [4] |
| SDLM_SI = 0.125 | 1.69 | ||
| SDLM_SI = 0.0054 | 1.78 | ||
| kd = 0.127 h−1 and Tlag = 14.6 min | 1.92 | ||
| SDLM_SI = 0.0712 | 1.90 | ||
| SDLM_SI = 0.0018 | 1.79 | ||
| po (tab, s.d.) | 100 | 1.04–1.74 | [39] |
| SDLM_SI = 0.125 | 1.11 | ||
| SDLM_SI = 0.0054 | 1.20 | ||
| kd = 0.127 h−1 and Tlag = 14.6 min | 1.12 | ||
| SDLM_SI = 0.0712 | 1.04 | ||
| SDLM_SI = 0.0018 | 1.74 | ||
| po (tab, s.d) | 150 | 1.51 | [40] |
| po (orod, s.d.) | 150 | 1.42 | [40] |
| po (tab (Ansaid), s.d.) | 200 | 1.20 | [36] |
| po (tab (Ansaid), s.d.) | 300 | 1.34 | [36] |
| MRD (range) | 1.54 (1.04–2.43) | ||
| MRD≤ 1.25 | 9/38 | ||
| MRD≤ 2 | 36/38 |
| AUCinf (mg/L·h) | Cmax (mg/L) | CL/F (L/h) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Route of Administration | Dose (mg) | obs | pred | Rpred/obs | obs | pred | Rpred/obs | obs | pred | Rpred/obs | Reference |
| iv (s.d.) | 50 | 35.2 | 43.7 | 1.24 | - | - | - | 1.50 | 1.36 | 0.91 | [34] |
| po (sol, s.d.) | 67.9 | 55.1 | 56.1 | 1.01 | 10.8 | 9.99 | 0.92 | - | - | - | [35] |
| po (sol, s.d.) | 100 | 82.7 | 78.0 | 0.94 | 14.2 | 12.9 | 0.91 | 1.28 | 1.50 | 1.17 | [36] |
| po (sol, s.d., CYP2C9 1*/1*) | 40 | 29.3 | 29.1 | 0.99 | 5.54 | 5.86 | 1.06 | 1.39 | 1.16 | 0.83 | [37] |
| po (sol, s.d., CYP2C9 1*/3*) | 40 | 47.6 | 44.2 | 0.93 | 6.93 | 6.22 | 0.90 | 0.88 | 0.67 | 0.76 | [37] |
| po (tab, s.d., CYP2C9 1*/1*) | 50 | 29.4 | 28.6 | 0.97 | 5.38 | 5.84 | 1.09 | 1.77 | 1.67 | 0.83 | [42] |
| po (tab, s.d., CYP2C9 1*/2*) | 50 | 40.7 | 45.6 | 1.12 | 4.55 | 6.34 | 1.39 | 1.30 | 1.20 | 0.92 | [42] |
| po (tab, s.d., CYP2C9 1*/3*) | 50 | 51.1 | 46.4 | 0.91 | 5.42 | 6.68 | 1.23 | 1.00 | 1.03 | 1.03 | [42] |
| po (tab, s.d., CYP2C9 1*/1*) | 50 | 30.8a | 35.8 a | 1.16 | 6.1 a | 6.91 a | 1.13 | 1.6 a | 1.4 a | 0.88 | [43] |
| po (tab, s.d., CYP2C9 1*/1*) | 50 | 30.8 a | 36 a | 1.17 | 6.1 a | 6.8 a | 1.11 | 1.6 a | 1.43 a | 0.89 | [43] |
| po (tab, s.d., CYP2C9 1*/3*) | 50 | 53.7 a | 54.6 a | 1.02 | 8.9 a | 7.7 a | 0.87 | 0.9 a | 0.98 a | 1.09 | [43] |
| po (tab, s.d., CYP2C9 1*/3*) | 50 | 53.7 a | 53.1 a | 0.99 | 8.9 a | 7.44 a | 0.84 | 0.9 a | 0.96 a | 1.07 | [43] |
| po (tab, s.d., CYP2C9 3*/3*) | 50 | (85.8, 119) b | 76.1 | (0.89, 0.64) | (8, 9.4) b | 6.99 | (0.87, 0.74) | (0.6, 0.4) b | 0.64 | (1.07, 1.6) | [43] |
| po (tab, s.d., CYP2C9 3*/3*) | 50 | (85.8, 119) b | 77.7 | (0.91, 0.65) | (8, 9.4) b | 7.1 | (0.89, 0.76) | (0.6, 0.4) b | 0.68 | (1.13, 1.7) | [43] |
| po (tab (Ansaid), s.d.) | 100 | [38] | |||||||||
| SDLM_SI = 0.125 | 80.5 | 81.1 | 1.01 | 12.8 | 12.8 | 1.00 | - | - | - | ||
| SDLM_SI = 0.0054 | 66.5 | 0.83 | 11.6 | 0.90 | |||||||
| kd = 0.127 h−1 and Tlag = 14.6 min | 68.8 | 0.85 | 11.8 | 0.92 | |||||||
| SDLM_SI = 0.0712 | 67.2 | 0.83 | 12.3 | 0.96 | |||||||
| SDLM_SI = 0.0018 | 62.7 | 0.78 | 10.4 | 0.81 | |||||||
| po (tab (Froben), s.d.) | 100 | [38] | |||||||||
| SDLM_SI = 0.125 | 82.3 | 81.1 | 0.99 | 13.3 | 12.8 | 0.96 | - | - | - | ||
| SDLM_SI = 0.0054 | 66.5 | 0.81 | 11.6 | 0.87 | |||||||
| kd = 0.127 h−1 and Tlag = 14.6 min | 68.8 | 0.84 | 11.8 | 0.88 | |||||||
| SDLM_SI = 0.0712 | 67.2 | 0.82 | 12.3 | 0.92 | |||||||
| SDLM_SI = 0.0018 | 62.7 | 0.76 | 10.4 | 0.78 | |||||||
| po (tab (Froben), s.d.) | 100 | [4] | |||||||||
| SDLM_SI = 0.125 | 87.8 | 81.1 | 0.92 | 13.2 | 12.7 | 0.96 | 1.27 | 1.42 | 1.12 | ||
| SDLM_SI = 0.0054 | 66.5 | 0.76 | 11.6 | 0.88 | 1.68 | 1.32 | |||||
| kd = 0.127 h−1 and Tlag = 14.6 min | 68.8 | 0.78 | 11.8 | 0.89 | 1.66 | 1.31 | |||||
| SDLM_SI = 0.0712 | 67.2 | 0.77 | 12.2 | 0.92 | 1.66 | 1.31 | |||||
| SDLM_SI = 0.0018 | 62.7 | 0.71 | 11.0 | 0.84 | 1.77 | 1.39 | |||||
| po (tab, s.d.) | 100 | [39] | |||||||||
| SDLM_SI = 0.125 | 67.7 | 81.1 | 1.20 | 12.9 | 12.7 | 0.98 | 1.52 | 1.42 | 0.93 | ||
| SDLM_SI = 0.0054 | 66.5 | 0.98 | 11.6 | 0.90 | 1.68 | 1.11 | |||||
| kd = 0.127 h−1 and Tlag = 14.6 min | 68.8 | 1.02 | 11.8 | 0.91 | 1.66 | 1.09 | |||||
| SDLM_SI = 0.0712 | 67.2 | 0.99 | 12.2 | 0.95 | 1.66 | 1.09 | |||||
| SDLM_SI = 0.0018 | 62.7 | 0.93 | 11.0 | 0.86 | 1.77 | 1.16 | |||||
| po (tab, s.d) | 150 | 124.3 | 154.4 | 1.24 | 15.2 | 20.7 | 1.36 | - | - | - | [40] |
| po (orod, s.d.) | 150 | 129.8 | 154.4 | 1.19 | 16.8 | 20.7 | 1.23 | - | - | - | [40] |
| po (tab (Ansaid), s.d.) | 200 | 161.3 | 159.9 | 0.99 | 21.4 | 23.6 | 1.10 | 1.32 | 1.50 | 1.14 | [36] |
| po (tab (Ansaid), s.d.) | 300 | 233.9 | 228.9 | 0.98 | 29.5 | 33.7 | 1.14 | 1.36 | 1.55 | 1.14 | [36] |
| GMFE (range) | 1.15 (1.01–1,56) | 1.14 (1.00–1.39) | 1.18 (1.03–1.7) | ||||||||
| GMFE≤ 1.25 | 30/38 | 31/37 | 18/25 | ||||||||
| GMFE≤ 2 | 38/38 | 37/37 | 25/25 | ||||||||
| DDI AUC Ratio | DDI Cmax Ratio | DDI CL/F Ratio | Reference | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Victim Drug Administration | Perpetrator Drug Administration | No. of Doses | Interval (h) | CYP2C9 Genotype | obs | pred | Rpred/obs | obs | pred | Rpred/obs | obs | pred | Rpred/obs | |
| Flurbiprofen | Fluconazole | |||||||||||||
| po 50 mg s.d. | po 200 mg q.d. | 7 | 2 | 1*/1* | 2.02 a | 1.94 | 0.96 | 1.03 a | 1.18 | 1.15 | 0.5 a | 0.51 | 1.02 | [43] |
| po 50 mg s.d. | po 400 mg q.d. | 7 | 2 | 1*/1* | 3.03 | 2.36 | 0.78 | 0.99 | 1.23 | 1.24 | 0.31 | 0.42 | 1.35 | [43] |
| po 50 mg s.d. | po 200 mg q.d. | 7 | 2 | 1*/3* | 1.8 | 1.58 | 0.88 | 0.87 | 1.11 | 1.28 | 0.56 | 0.63 | 1.13 | [43] |
| po 50 mg s.d. | po 400 mg q.d. | 7 | 2 | 1*/3* | 2.48 | 1.84 | 0.74 | 0.94 | 1.14 | 1.21 | 0.44 | 0.54 | 1.23 | [43] |
| po 50 mg s.d. | po 200 mg q.d. | 7 | 2 | 3*/3* | (1.58, 1.28)# | 1.09 | 0.76 | (1.08, 0.91) # | 1.02 | 1.02 | (0.75, 0.66) # | 0.92 | 1.30 | [43] |
| po 50 mg s.d. | po 400 mg q.d. | 7 | 2 | 3*/3* | (1.39, 1.12)# | 1.16 | 0.92 | (0.54, 0.90) # | 1.03 | 1.43 | (1.00, 0.66) # | 0.86 | 1.04 | [43] |
| po 100 mg s.d. | po 200 mg b.i.d. | 2 | 0.5 | n.a. | 1.71 b | 1.65 | 0.97 | 1.16 b | 1.15 | 0.99 | 0.57 | 0.61 | 1.07 | [45] |
| po 100 mg s.d. | po 200 mg b.i.d. | 2 | 0.5 | n.a. | 1.81 | 1.51 | 0.83 | 1.23 | 1.13 | 0.92 | 0.55 | 0.68 | 1.24 | [46] |
| po 100 mg s.d. | po 200 mg b.i.d. | 2 | 0.5 | n.a. | 1.97 b | 1.62 | 0.82 | 1.47 b | 1.15 | 0.78 | 0.5 | 0.62 | 1.24 | [44] |
| po 50 mg s.d. | po 400 mg s.d. | 1 | 2 | n.a. | 2.16 | 2.23 | 1.03 | 1.24 | 1.2 | 0.97 | 0.46 | 0.48 | 1.04 | [47] |
| po 50 mg s.d. | po 400 mg q.d. | 7 | 2 | n.a. | 2.81 | 2.87 | 1.02 | 1.37 | 1.25 | 0.91 | 0.35 | 0.39 | 1.11 | [47] |
| po 50 mg s.d. | po 400 mg s.d. | 1 | 2 | n.a. | 1.21 | 1.53 | 1.26 | 1.14 | 1.14 | 1.00 | 0.67 | 0.67 | 1.00 | [48] |
| Rifampicin | ||||||||||||||
| po 50 mg s.d. | po 600 mg q.d. | 5 | 0 | n.a. | 0.56 | 0.63 | 1.13 | 0.71 | 0.83 | 1.17 | 1.85 | 1.73 | 0.94 | Daali et al. |
| GMFE (range) | 1.17 (1.02–1.35) | 1.16 (1.00–1.43) | 1.15 (1.00–1.35) | |||||||||||
| GMFE≤ 1.25 | 8/12 | 9/12 | 10/12 | |||||||||||
| GMFE≤ 2 | 12/12 | 12/12 | 12/12 | |||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loisios-Konstantinidis, I.; Cristofoletti, R.; Jamei, M.; Turner, D.; Dressman, J. Physiologically Based Pharmacokinetic/Pharmacodynamic Modeling to Predict the Impact of CYP2C9 Genetic Polymorphisms, Co-Medication and Formulation on the Pharmacokinetics and Pharmacodynamics of Flurbiprofen. Pharmaceutics 2020, 12, 1049. https://doi.org/10.3390/pharmaceutics12111049
Loisios-Konstantinidis I, Cristofoletti R, Jamei M, Turner D, Dressman J. Physiologically Based Pharmacokinetic/Pharmacodynamic Modeling to Predict the Impact of CYP2C9 Genetic Polymorphisms, Co-Medication and Formulation on the Pharmacokinetics and Pharmacodynamics of Flurbiprofen. Pharmaceutics. 2020; 12(11):1049. https://doi.org/10.3390/pharmaceutics12111049
Chicago/Turabian StyleLoisios-Konstantinidis, Ioannis, Rodrigo Cristofoletti, Masoud Jamei, David Turner, and Jennifer Dressman. 2020. "Physiologically Based Pharmacokinetic/Pharmacodynamic Modeling to Predict the Impact of CYP2C9 Genetic Polymorphisms, Co-Medication and Formulation on the Pharmacokinetics and Pharmacodynamics of Flurbiprofen" Pharmaceutics 12, no. 11: 1049. https://doi.org/10.3390/pharmaceutics12111049
APA StyleLoisios-Konstantinidis, I., Cristofoletti, R., Jamei, M., Turner, D., & Dressman, J. (2020). Physiologically Based Pharmacokinetic/Pharmacodynamic Modeling to Predict the Impact of CYP2C9 Genetic Polymorphisms, Co-Medication and Formulation on the Pharmacokinetics and Pharmacodynamics of Flurbiprofen. Pharmaceutics, 12(11), 1049. https://doi.org/10.3390/pharmaceutics12111049