Bioavailability Improvement of Carbamazepine via Oral Administration of Modified-Release Amorphous Solid Dispersions in Rats



Abstract
1. Introduction
2. Materials and Methods
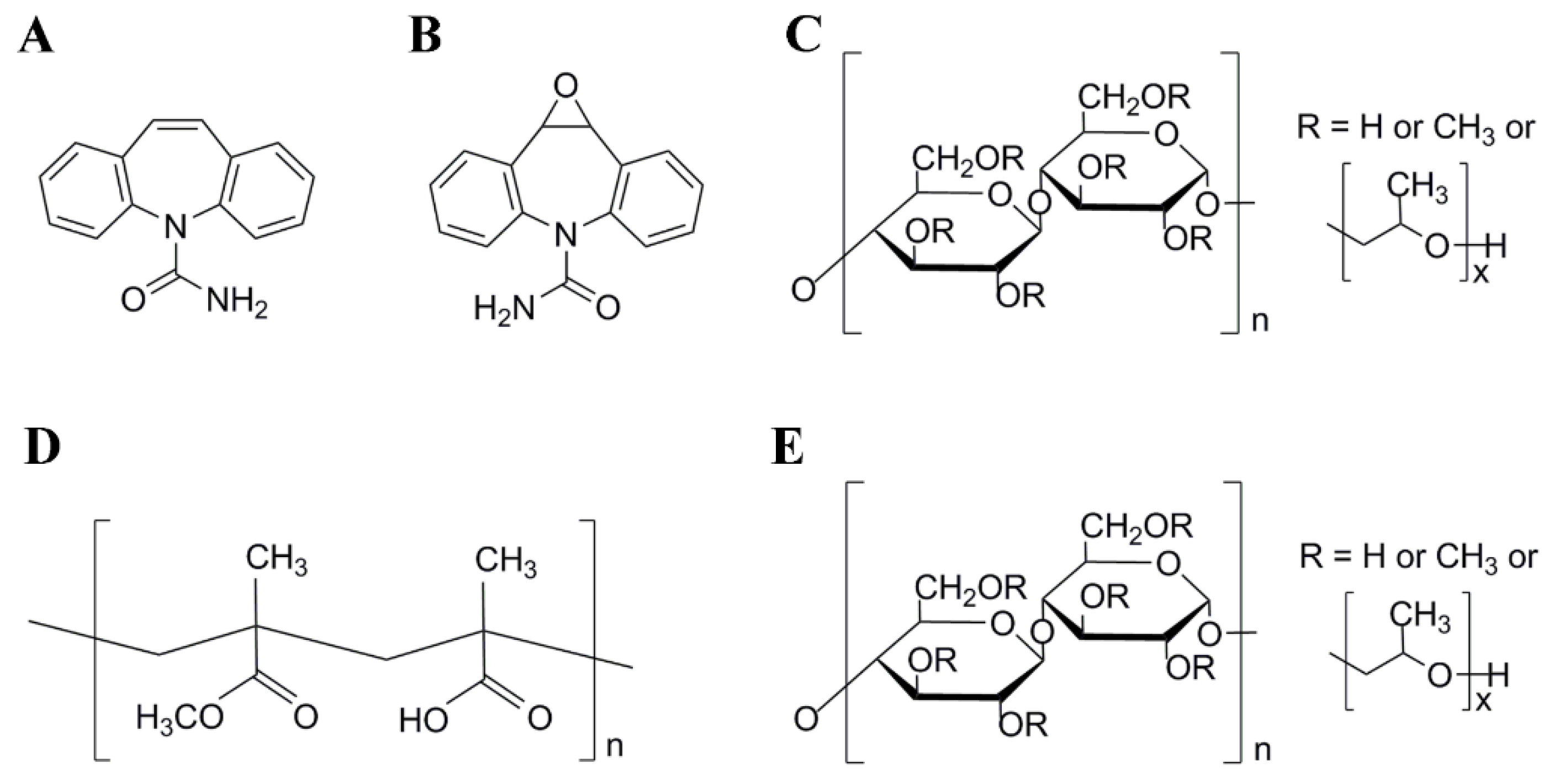
2.1. Materials
2.2. Preparation of CBZ-Mr-ASD
2.3. Preparation of CBZ-Mr-ASD Capsule Formulations
2.4. Characterization of Amorphous Solid Dispersions
2.4.1. Scanning Electron Microscopy (SEM)
2.4.2. Thermal Analysis
2.4.3. X-Ray Powder Diffraction (XRPD)
2.4.4. Hygroscopic Property
Moisture Property
Water Content Measurements
2.4.5. Compressibility and Fluidity Analysis
2.4.6. Drug Content Determination
2.4.7. In Vitro Dissolution Testing
2.5. In Vivo Pharmacokinetic Studies
2.5.1. Pharmacokinetic Studies in Rats after a Single Oral Dose
2.5.2. Quantification of CBZ and CBZ-E Concentrations in Plasma
3. Results and Discussion
3.1. Preparation of CBZ-Mr-ASD and Related Capsule Formulations
3.2. Physicochemical Properties of CBZ-Mr-ASD and Related Capsule Formulations
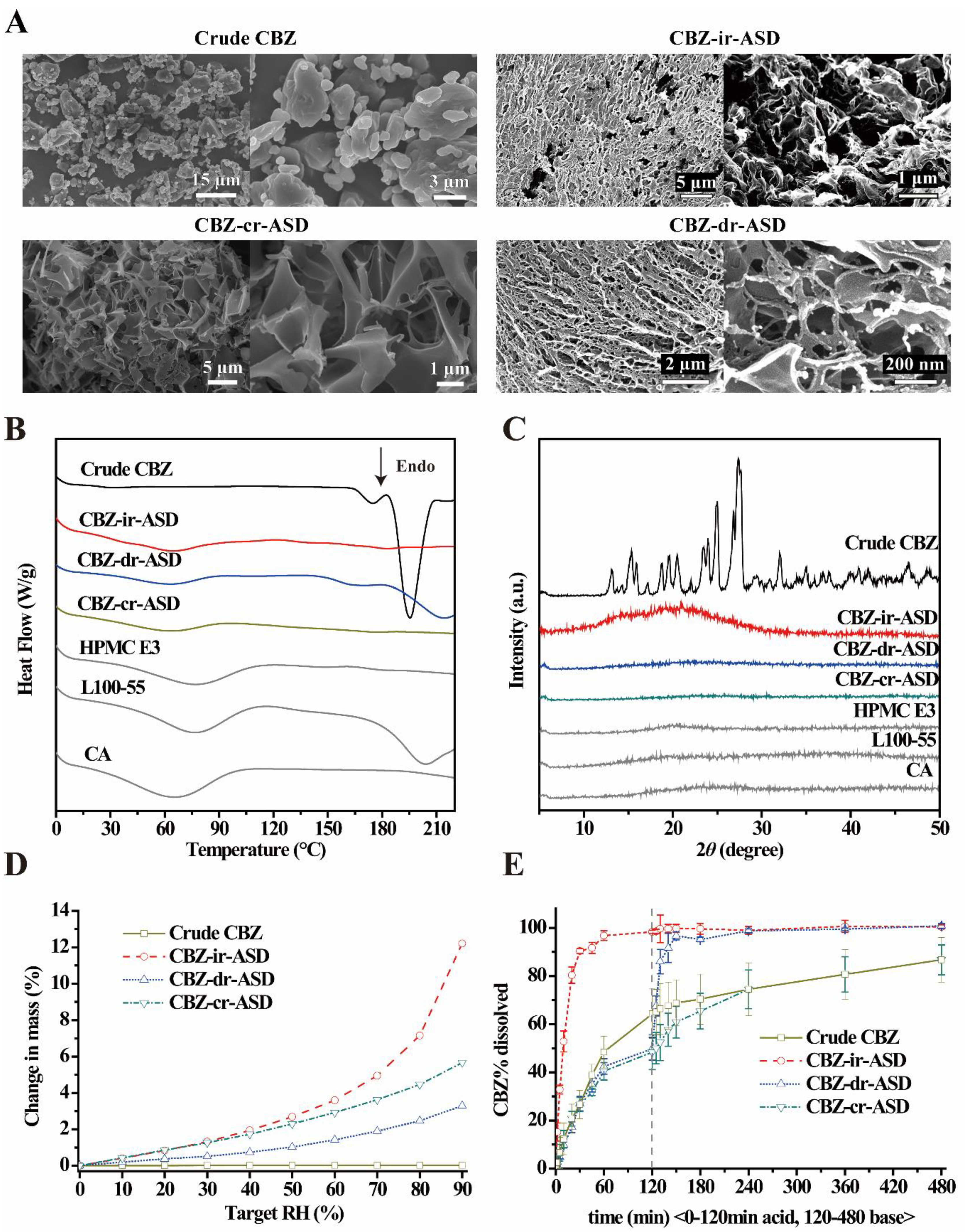
3.2.1. Scanning Electron Microscopy (SEM) of CBZ-Mr-ASD
3.2.2. Amorphous State of CBZ-Mr-ASD
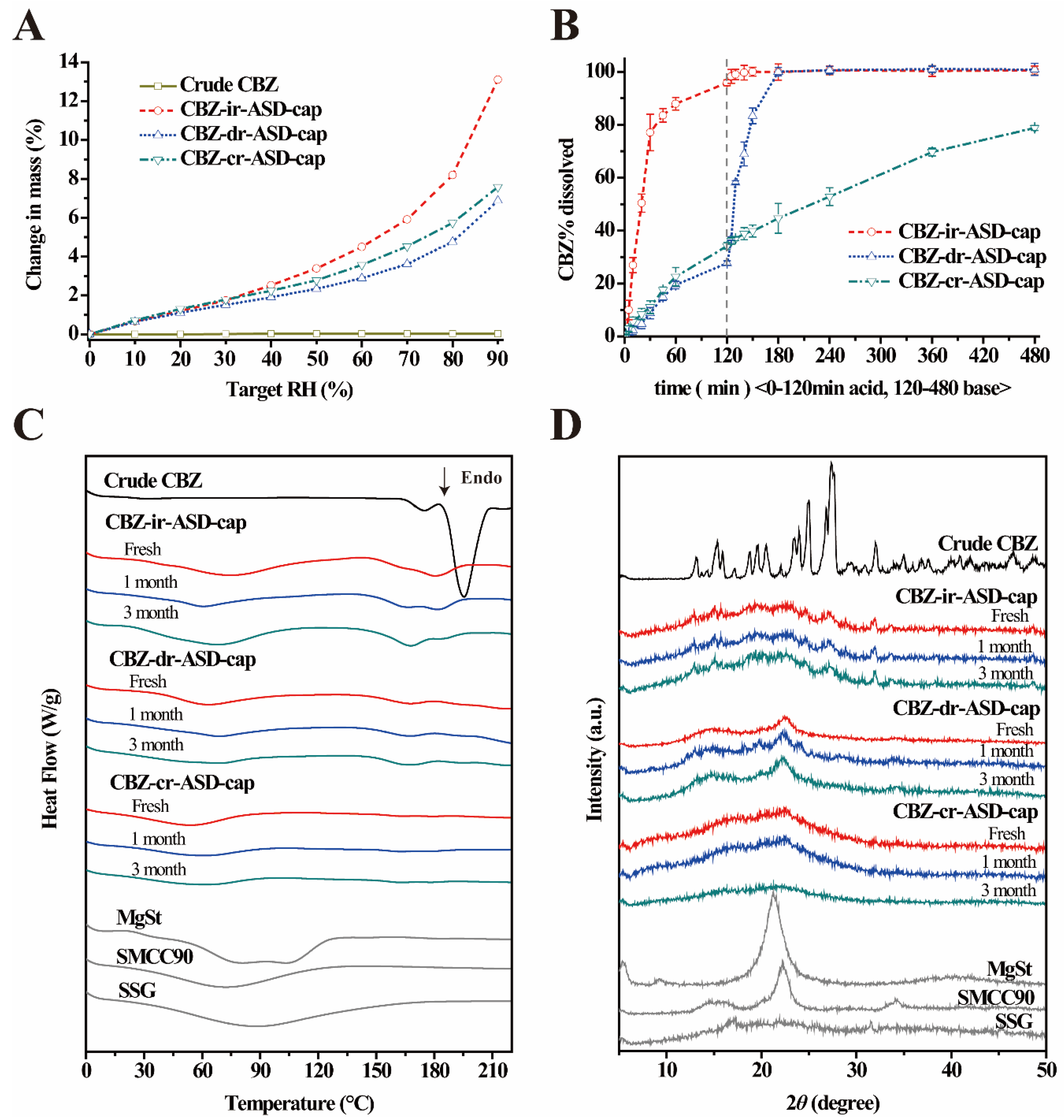
3.2.3. Hygroscopic Property
3.2.4. Drug Content and Flowability
3.2.5. Dissolution Testing Under Sink Conditions
3.2.6. Physical Stability Studies
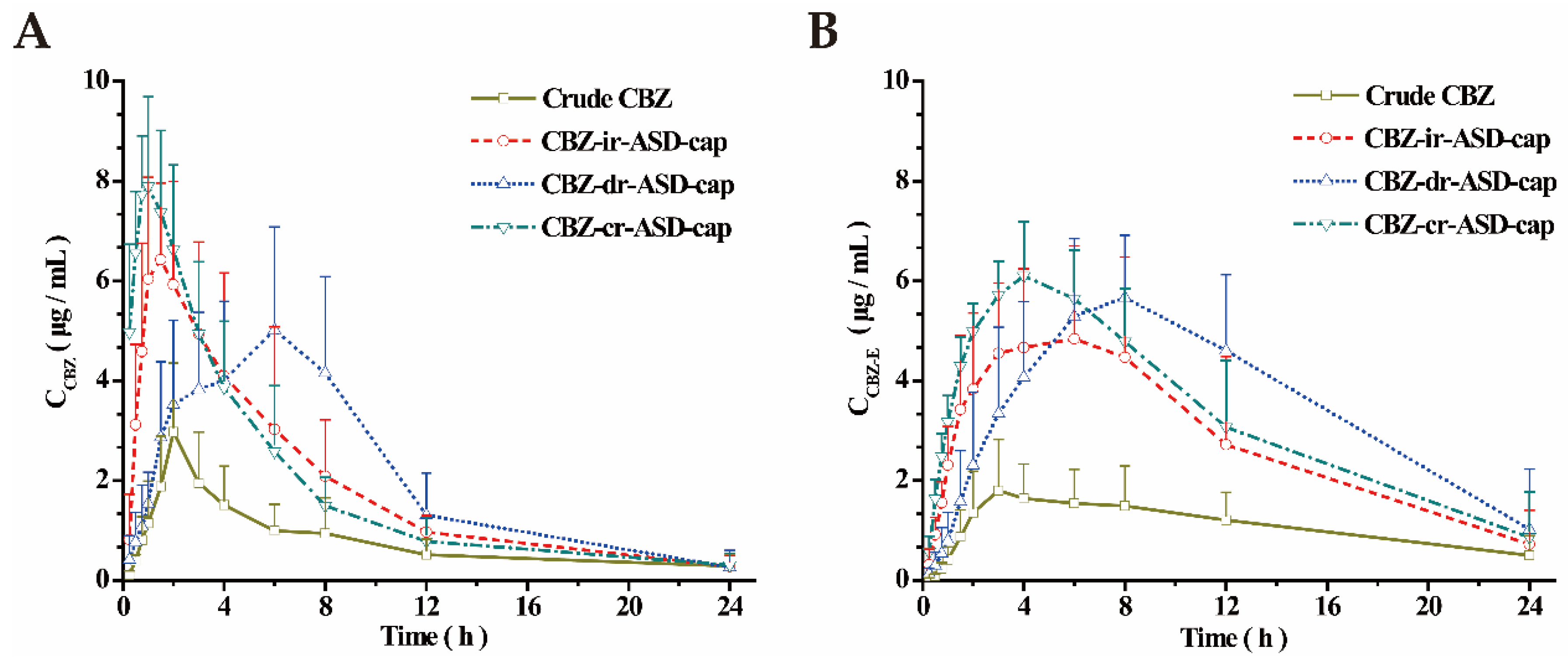
3.3. Pharmacokinetic Studies in Rats
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hens, B.; Corsetti, M.; Bermejo, M.; Löbenberg, R.; González, P.M.; Mitra, A.; Desai, D.; Chilukuri, D.M.; Aceituno, A. “Development of Fixed Dose Combination Products” Workshop Report: Considerations of Gastrointestinal Physiology and Overall Development Strategy. Aaps. J. 2019, 21, 75. [Google Scholar] [CrossRef] [PubMed]
- Siew, A. Solving poor solubility to unlock a drug’s potential. Pharm. Tech. 2015, 27, 20–25. [Google Scholar]
- Taylor, L.S.; Zhang, G.G.Z. Physical chemistry of supersaturated solutions and implications for oral absorption. Adv. Drug. Deliv. Rev. 2016, 101, 122–142. [Google Scholar] [CrossRef] [PubMed]
- Amidon, G.L.; Lennernas, H.; Shah, V.P.; Crison, J.R. A Theoretical Basis for a Biopharmaceutic Drug Classification—The Correlation of in-vitro Drug Product Dissolution and in-vivo Bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef]
- Kawakami, K. Crystallization Tendency of Pharmaceutical Glasses: Relevance to Compound Properties, Impact of Formulation Process, and Implications for Design of Amorphous Solid Dispersions. Pharmaceutics 2019, 11, 202. [Google Scholar] [CrossRef]
- Medarevic, D.; Kachrimanis, K.; Djuric, Z.; Ibric, S. Influence of hydrophilic polymers on the complexation of carbamazepine with hydroxypropyl-beta-cyclodextrin. Eur. J. Pharm. Sci. 2015, 78, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.S.; Date, A.A.; Pissurlenkar, R.R.S.; Coutinho, E.C.; Nagarsenker, M.S. Sulfobutyl Ether(7) beta-Cyclodextrin (SBE7 beta-CD) Carbamazepine Complex: Preparation, Characterization, Molecular Modeling, and Evaluation of In Vivo Anti-epileptic Activity. AAPS Pharmscitech 2011, 12, 1163–1175. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, H.; Sun, C.C. Expedited Tablet Formulation Development of a Highly Soluble Carbamazepine Cocrystal Enabled by Precipitation Inhibition in Diffusion Layer. Pharm. Res. 2019, 36, 90. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, H.; Sun, C.C. Improving Dissolution Rate of Carbamazepine-Glutaric Acid Cocrystal Through Solubilization by Excess Coformer. Pharm. Res. 2018, 35, 4. [Google Scholar] [CrossRef]
- Li, Z.; Matzger, A.J. Influence of Coformer Stoichiometric Ratio on Pharmaceutical Cocrystal Dissolution: Three Cocrystals of Carbamazepine/4-Aminobenzoic Acid. Mol. Pharm. 2016, 13, 990–995. [Google Scholar] [CrossRef]
- Wu, W.; Wang, Y.; Lobmann, K.; Grohganz, H.; Rades, T. Transformations between Co-Amorphous and Co-Crystal Systems and Their Influence on the Formation and Physical Stability of Co-Amorphous Systems. Mol. Pharm. 2019, 16, 1294–1304. [Google Scholar] [CrossRef] [PubMed]
- Kuminek, G.; Kratz, J.M.; Ribeiro, R.; Kelmann, R.G.; de Araujo, B.V.; Teixeira, H.F.; Simoes, C.M.O.; Koester, L.S. Pharmacokinetic study of a carbamazepine nanoemulsion in beagle dogs. Int. J. Pharm. 2009, 378, 146–148. [Google Scholar] [CrossRef] [PubMed]
- Rathod, S.; Tripathi, R.; Verma, G.; Aswal, V.K.; Bahadur, P.; Tiwari, S. Bioadhesive polymeric film-based integrative platform for the unidirectional carbamazepine release from a volatile microemulsion. Colloid. Surf. B 2018, 170, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Douroumis, D.; Fahr, A. Stable carbamazepine colloidal systems using the cosolvent technique. Eur. J. Pharm. Sci. 2007, 30, 367–374. [Google Scholar] [CrossRef]
- Jain, S.; Pandey, S.; Sola, P.; Pathan, H.; Patil, R.; Ray, D.; Aswal, V.K.; Bahadur, P.; Tiwari, S. Solubilization of Carbamazepine in TPGS Micelles: Effect of Temperature and Electrolyte Addition. Aaps Pharmscitech 2019, 20, 203. [Google Scholar] [CrossRef]
- Ana, R.; Mendes, M.; Sousa, J.; Pais, A.; Falcao, A.; Fortuna, A.; Vitorino, C. Rethinking carbamazepine oral delivery using polymer-lipid hybrid nanoparticles. Int. J. Pharm. 2019, 554, 352–365. [Google Scholar] [CrossRef]
- Elmowafy, M.; Shalaby, K.; Badran, M.M.; Ali, H.M.; Abdel-Bakky, M.S.; Ibrahim, H.M. Multifunctional carbamazepine loaded nanostructured lipid carrier (NLC) formulation. Int. J. Pharm. 2018, 550, 359–371. [Google Scholar] [CrossRef]
- Gandhi, A.V.; Thipsay, P.; Kirthivasan, B.; Squillante, E. Adsorption onto Mesoporous Silica Using Supercritical Fluid Technology Improves Dissolution Rate of Carbamazepine-a Poorly Soluble Compound. Aaps Pharmscitech 2017, 18, 3140–3150. [Google Scholar] [CrossRef]
- Ueda, H.; Wu, W.; Lobmann, K.; Grohganz, H.; Mullertz, A.; Rades, T. Application of a Salt Coformer in a Co-Amorphous Drug System Dramatically Enhances the Glass Transition Temperature: A Case Study of the Ternary System Carbamazepine, Citric Acid, and L-Arginine. Mol. Pharm. 2018, 15, 2036–2044. [Google Scholar] [CrossRef]
- Warnken, Z.; Puppolo, M.; Hughey, J.; Duarte, I.; Jansen-Varnum, S. In Vitro-In Vivo Correlations of Carbamazepine Nanodispersions for Application in Formulation Development. J. Pharm. Sci. 2018, 107, 453–465. [Google Scholar] [CrossRef]
- Ma, X.; Müller, F.; Huang, S.; Lowinger, M.; Liu, X.; Schooler, R.; Williams, R.O., III. Influence of Carbamazepine Dihydrate on the Preparation of Amorphous Solid Dispersions by Hot Melt Extrusion. Pharmaceutics 2020, 12, 379. [Google Scholar] [CrossRef] [PubMed]
- Fael, H.; Demirel, A.L. Tannic acid as a co-former in co-amorphous systems: Enhancing their physical stability, solubility and dissolution behavior. Int. J. Pharm. 2020, 581, 119284. [Google Scholar] [CrossRef] [PubMed]
- Janssens, S.; Van den Mooter, G. Review: Physical chemistry of solid dispersions. J. Pharm. Pharm. 2009, 61, 1571–1586. [Google Scholar] [CrossRef]
- Hancock, B.C.; Shamblin, S.L.; Zografi, G. Molecular Mobility of Amorphous Pharmaceutical Solids below Their Glass-Transition Temperatures. Pharm. Res. 1995, 12, 799–806. [Google Scholar] [CrossRef]
- Brouwers, J.; Brewster, M.E.; Augustijns, P. Supersaturating Drug Delivery Systems: The Answer to Solubility-Limited Oral Bioavailability? J. Pharm. Sci. 2009, 98, 2549–2572. [Google Scholar] [CrossRef]
- Miller, J.M.; Beig, A.; Carr, R.A.; Spence, J.K.; Dahan, A. A Win-Win Solution in Oral Delivery of Lipophilic Drugs: Supersaturation via Amorphous Solid Dispersions Increases Apparent Solubility without Sacrifice of Intestinal Membrane Permeability. Mol. Pharm. 2012, 9, 2009–2016. [Google Scholar] [CrossRef]
- Mendonsa, N.; Almutairy, B.; Kallakunta, V.R.; Sarabu, S.; Thipsay, P.; Bandari, S.; Repka, M.A. Manufacturing strategies to develop amorphous solid dispersions: An overview. J. Drug. Deliv. Sci. Technol. 2020, 55, 101459. [Google Scholar] [CrossRef]
- Miller, D.A. Editorial for Theme Issue: Applications of KinetiSol Dispersing for Advanced Amorphous Solid Dispersions. Aaps Pharmscitech 2018, 19, 1931–1932. [Google Scholar] [CrossRef] [PubMed]
- Ellenberger, D.J.; Miller, D.A.; Williams, R.O., III. Expanding the Application and Formulation Space of Amorphous Solid Dispersions with KinetiSolA (R): A Review. Aaps Pharmscitech 2018, 19, 1933–1956. [Google Scholar] [CrossRef]
- Ren, Y.; Mei, L.; Zhou, L.; Guo, G. Recent Perspectives in Hot Melt Extrusion-Based Polymeric Formulations for Drug Delivery: Applications and Innovations. Aaps Pharmscitech 2019, 20, 92. [Google Scholar] [CrossRef]
- Moon, C.; Watts, A.B.; Lu, X.; Su, Y.; Williams, R.O., III. Enhanced Aerosolization of High Potency Nanoaggregates of Voriconazole by Dry Powder Inhalation. Mol. Pharm. 2019, 16, 1799–1812. [Google Scholar] [CrossRef]
- Vasconcelos, T.; Marques, S.; das Neves, J.; Sarmento, B. Amorphous solid dispersions: Rational selection of a manufacturing process. Adv. Drug Del. Rev. 2016, 100, 85–101. [Google Scholar] [CrossRef]
- Overhoff, K.A.; Engstrom, J.D.; Chen, B.; Scherzer, B.D.; Milner, T.E.; Johnston, K.P.; Williams, R.O., III. Novel ultra-rapid freezing particle engineering process for enhancement of dissolution rates of poorly water-soluble drugs. Eur. J. Pharm. Biopharm. 2007, 65, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Lang, B.; McGinity, J.W.; Williams, R.O., III. Dissolution enhancement of itraconazole by hot-melt extrusion alone and the combination of hot-melt extrusion and rapid freezing--effect of formulation and processing variables. Mol. Pharm. 2014, 11, 186–196. [Google Scholar] [CrossRef]
- Maincent, J.; Williams, R.O., III. Sustained-release amorphous solid dispersions. Drug. Deliv. Transl. Res. 2018, 8, 1714–1725. [Google Scholar] [CrossRef] [PubMed]
- LaFountaine, J.S.; Prasad, L.K.; Miller, D.A.; McGinity, J.W.; Williams, R.O., III. Mucoadhesive amorphous solid dispersions for sustained release of poorly water soluble drugs. Eur. J. Pharm. Biopharm. 2017, 113, 157–167. [Google Scholar] [CrossRef]
- Tres, F.; Treacher, K.; Booth, J.; Hughes, L.P.; Wren, S.A.C.; Aylott, J.W.; Burley, J.C. Indomethacin-Kollidon VA64 Extrudates: A Mechanistic Study of pH-Dependent Controlled Release. Mol. Pharm. 2016, 13, 1166–1175. [Google Scholar] [CrossRef]
- Sun, D.D.; Lee, P.I. Evolution of Supersaturation of Amorphous Pharmaceuticals: The Effect of Rate of Supersaturation Generation. Mol. Pharm. 2013, 10, 4330–4346. [Google Scholar] [CrossRef]
- Landmark, C.J.; Johannessen, S.I.; Tomson, T. Host factors affecting antiepileptic drug delivery-Pharmacokinetic variability. Adv. Drug Del. Rev. 2012, 64, 896–910. [Google Scholar] [CrossRef]
- Deng, J.; Staufenbiel, S.; Bodmeier, R. Evaluation of a biphasic in vitro dissolution test for estimating the bioavailability of carbamazepine polymorphic forms. Eur. J. Pharm. Sci. 2017, 105, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Hiemke, C.; Bergemann, N.; Clement, H.W.; Conca, A.; Deckert, J.; Domschke, K.; Eckermann, G.; Egberts, K.; Gerlach, M.; Greiner, C.; et al. Consensus Guidelines for Therapeutic Drug Monitoring in Neuropsychopharmacology: Update 2017. Pharmacopsychiatry 2018, 51, 9–62. [Google Scholar] [PubMed]
- Zhang, M.; Li, H.; Lang, B.; O’Donnell, K.; Zhang, H.; Wang, Z.; Dong, Y.; Wu, C.; Williams, R.O., III. Formulation and delivery of improved amorphous fenofibrate solid dispersions prepared by thin film freezing. Eur. J. Pharm. Biopharm. 2012, 82, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Thakkar, S.G.; Ruwona, T.B.; Williams, R.O., III; Cui, Z. A method of lyophilizing vaccines containing aluminum salts into a dry powder without causing particle aggregation or decreasing the immunogenicity following reconstitution. J. Control. Release 2015, 204, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Watts, A.B.; Du, J.; Bui, A.; Hengsawas, S.; Peters, J.I.; Williams, R.O., III. Formulation of a novel fixed dose combination of salmeterol xinafoate and mometasone furoate for inhaled drug delivery. Eur. J. Pharm. Biopharm. 2015, 96, 132–142. [Google Scholar] [CrossRef]
- Wang, Y.-B.; Watts, A.B.; Peters, J.I.; Liu, S.; Batra, A.; Williams, R.O., III. In Vitro and In Vivo Performance of Dry Powder Inhalation Formulations: Comparison of Particles Prepared by Thin Film Freezing and Micronization. Aaps Pharmscitech 2014, 15, 981–993. [Google Scholar] [CrossRef]
- Yeom, D.W.; Son, H.Y.; Kim, J.H.; Kim, S.R.; Lee, S.G.; Song, S.H.; Chae, B.R.; Choi, Y.W. Development of a solidified self-microemulsifying drug delivery system (S-SMEDDS) for atorvastatin calcium with improved dissolution and bioavailability. Int. J. Pharm. 2016, 506, 302–311. [Google Scholar] [CrossRef]
- Eichelbaum, M.; Tomson, T.; Tybring, G.; Bertilsson, L. Carbamazepine Metabolism in Man Induction and Pharmacogenetic Aspects. Clin. Pharm. 1985, 10, 80–90. [Google Scholar] [CrossRef]
- Patsalos, P.N.; Spencer, E.P.; Berry, D.J. Therapeutic Drug Monitoring of Antiepileptic Drugs in Epilepsy: A 2018 Update. Drug. Monit. 2018, 40, 526–548. [Google Scholar] [CrossRef]
- Kacirova, I.; Grundmann, M.; Brozmanova, H. Concentrations of carbamazepine and carbamazepine-10,11-epoxide in maternal and umbilical cord blood at birth: Influence of co-administration of valproic acid or enzyme-inducing antiepileptic drugs. Epilepsy. Res. 2016, 122, 84–90. [Google Scholar] [CrossRef]
- Overhoff, K.A.; Johnston, K.P.; Tam, J.; Engstrom, J.; Williams, R.O., III. Use of thin film freezing to enable drug delivery: A review. J. Drug. Deliv. Sci. Technol. 2009, 19, 89–98. [Google Scholar] [CrossRef]
- Mugheirbi, N.A.; O’Connell, P.; Serrano, D.R.; Healy, A.M.; Taylor, L.S.; Tajber, L. A Comparative Study on the Performance of Inert and Functionalized Spheres Coated with Solid Dispersions Made of Two Structurally Related Antifungal Drugs. Mol. Pharm. 2017, 14, 3718–3728. [Google Scholar] [CrossRef]
- Decroix, C.; Chalamet, Y.; Sudre, G.; Caroll, V. Thermo-mechanical properties and blend behaviour of cellulose acetate/lactates and acid systems: Natural-based plasticizers. Carbohydr. Polym. 2020, 237, 116072. [Google Scholar] [CrossRef]
- Chen, X.; Partheniadis, I.; Nikolakakis, I.; Al-Obaidi, H. Solubility Improvement of Progesterone from Solid Dispersions Prepared by Solvent Evaporation and Co-milling. Polymers 2020, 12, 854. [Google Scholar] [CrossRef]
- Zhou, Q.; Loh, Z.H.; Yu, J.; Sun, S.-P.; Gengenbach, T.; Denman, J.A.; Li, J.; Chan, H.-K. How Much Surface Coating of Hydrophobic Azithromycin Is Sufficient to Prevent Moisture-Induced Decrease in Aerosolisation of Hygroscopic Amorphous Colistin Powder? AAPS J. 2016, 18, 1213–1224. [Google Scholar] [CrossRef]
- Vasanthavada, M.; Tong, W.Q.; Joshi, Y.; Kislalioglu, M.S. Phase Behavior of amorphous molecular dispersions I: Determination of the degree and mechanism of solid solubility. Pharm. Res. 2004, 21, 1598–1606. [Google Scholar] [CrossRef]
- Wang, X.; Michoel, A.; Van den Mooter, G. Solid state characteristics of ternary solid dispersions composed of PVPVA64, Myrj 52 and itraconazole. Int. J. Pharm. 2005, 303, 54–61. [Google Scholar] [CrossRef]
- Szabo, E.; Demuth, B.; Galata, D.L.; Vass, P.; Hirsch, E.; Csontos, I.; Marosi, G.; Nagy, Z.K. Continuous Formulation Approaches of Amorphous Solid Dispersions: Significance of Powder Flow Properties and Feeding Performance. Pharmaceutics 2019, 11, 654. [Google Scholar] [CrossRef]
- Guerin, E.; Tchoreloff, P.; Leclerc, B.; Tanguy, D.; Deleuil, M.; Couarraze, G. Rheological characterization of pharmaceutical powders using tap testing, shear cell and mercury porosimeter. Int. J. Pharm. 1999, 189, 91–103. [Google Scholar] [CrossRef]
- Dokoumetzidis, A.; Macheras, P. A century of dissolution research: From Noyes and Whitney to the Biopharmaceutics Classification System. Int. J. Pharm. 2006, 321, 1–11. [Google Scholar] [CrossRef]
- Sun, D.D.; Lee, P.I. Probing the mechanisms of drug release from amorphous solid dispersions in medium-soluble and medium-insoluble carriers. J. Control. Release 2015, 211, 85–93. [Google Scholar] [CrossRef]
- Leite, C.E.; Petersen, G.O.; Lunardelli, A.; Thiesen, F.V. A high-performance liquid chromatography method for the determination of carbamazepine and carbamazepine-10,11-epoxide and its comparison with chemiluminescent immunoassay. Clin. Chem. Lab. Med. 2009, 47, 458–463. [Google Scholar] [CrossRef]
- Yoshida, T.; Imai, K.; Motohashi, S.; Hamano, S.-I.; Sato, M. Simultaneous determination of zonisamide, carbamazepine and carbamazepine-10,11-epoxide in infant serum by high-performance liquid chromatography. J. Pharm. Biomed. Anal. 2006, 41, 1386–1390. [Google Scholar] [CrossRef]
- Thakore, S.D.; Thakur, P.S.; Shete, G.; Gangwal, R.; Narang, A.S.; Sangamwar, A.T.; Bansal, A.K. Assessment of Biopharmaceutical Performance of Supersaturating Formulations of Carbamazepine in Rats Using Physiologically Based Pharmacokinetic Modeling. AAPS Pharmscitech 2019, 20, 179. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ingredients | Function | Types of CBZ-mr-ASD | ||
|---|---|---|---|---|
| CBZ-ir-ASD | CBZ-dr-ASD | CBZ-cr-ASD | ||
| CBZ-mr-ASD | Excipient | HPMC E3 | L100-55 | CA |
| Ratio of CBZ/Excipient (w/w) | 1:1 | 2:1 | 1:2 | |
| Solvent | 1,4-Dioxane/water (8:2, v/v) | 1,4-Dioxane | 1,4-Dioxane | |
| Solids in solution (w/v, %) | 2.0 | 2.0 | 2.0 | |
| Active and carrier | 900 mg | 750 mg | 900 mg | |
| SMCC90 | Diluent | 80 mg | 200 mg | 80 mg |
| SSG | Disintegrant | 18 mg | 45 mg | 18 mg |
| MgSt | Lubricant | 2 mg | 5 mg | 2 mg |
| Size | 3 (60 mg) | 3 (60 mg) | 1 (100 mg) | |
| Formulation | Drug Content (%) | Residual Water Content (%) | Carr Index | Flowability * | |
|---|---|---|---|---|---|
| CBZ-mr-ASD | CBZ-ir-ASD | 49.71 ± 0.68 | 0.782 ± 0.188 | 19.02 ± 0.49 | Fair |
| CBZ-dr-ASD | 65.62 ± 0.70 | 0.493 ± 0.059 | 16.40 ± 0.91 | Fair | |
| CBZ-cr-ASD | 33.07 ± 0.32 | 0.186 ± 0.018 | 24.67 ± 0.58 | Passable | |
| CBZ-ir-ASD-cap | 44.51 ± 0.20 | 0.574 ± 0.158 | 10.26 ± 1.47 | Good | |
| CBZ-mr-ASD-cap | CBZ-dr-ASD-cap | 48.25 ± 0.32 | 0.492 ± 0.059 | 12.42 ± 0.55 | Good |
| CBZ-cr-ASD-cap | 29.56 ± 0.29 | 0.553 ± 0.120 | 15.97 ± 0.25 | Good | |
| Parameters | Tmax (h) | Cmax (μg/mL) | AUC(0–t) (μg/mL × h) | |
|---|---|---|---|---|
| CBZ | Crude drug | 2.0 ± 0.55 | 3.41 ± 0.89 | 18.85 ± 5.68 |
| CBZ-ir-ASD-cap | 1.67 ± 0.75 | 7.38 ± 1.36 | 44.87 ± 16.06 | |
| CBZ-dr-ASD-cap | 5.17 ± 2.22 | 6.38 ± 1.01 | 49.63 ± 9.93 | |
| CBZ-cr-ASD-cap | 1.08 ± 0.47 | 8.25 ± 1.50 | 44.95 ± 12.22 | |
| CBZ-E | Crude drug | 4.67 ± 2.66 | 2.24 ± 0.84 | 24.52 ± 10.51 |
| CBZ-ir-ASD-cap | 5.33 ± 1.97 | 5.26 ± 1.76 | 66.88 ± 25.01 | |
| CBZ-dr-ASD-cap | 7.67 ± 0.82 | 6.01 ± 1.48 | 83.01 ± 10.68 | |
| CBZ-cr-ASD-cap | 4.17 ± 0.98 | 6.14 ± 1.04 | 78.47 ± 20.27 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Zhang, M.; Xiong, L.; Feng, W.; Williams, R.O., III. Bioavailability Improvement of Carbamazepine via Oral Administration of Modified-Release Amorphous Solid Dispersions in Rats. Pharmaceutics 2020, 12, 1023. https://doi.org/10.3390/pharmaceutics12111023
Li H, Zhang M, Xiong L, Feng W, Williams RO III. Bioavailability Improvement of Carbamazepine via Oral Administration of Modified-Release Amorphous Solid Dispersions in Rats. Pharmaceutics. 2020; 12(11):1023. https://doi.org/10.3390/pharmaceutics12111023
Chicago/Turabian StyleLi, Houli, Meimei Zhang, Lilong Xiong, Weiyi Feng, and Robert O. Williams, III. 2020. "Bioavailability Improvement of Carbamazepine via Oral Administration of Modified-Release Amorphous Solid Dispersions in Rats" Pharmaceutics 12, no. 11: 1023. https://doi.org/10.3390/pharmaceutics12111023
APA StyleLi, H., Zhang, M., Xiong, L., Feng, W., & Williams, R. O., III. (2020). Bioavailability Improvement of Carbamazepine via Oral Administration of Modified-Release Amorphous Solid Dispersions in Rats. Pharmaceutics, 12(11), 1023. https://doi.org/10.3390/pharmaceutics12111023