Combining Surface Templating and Confinement for Controlling Pharmaceutical Crystallization


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
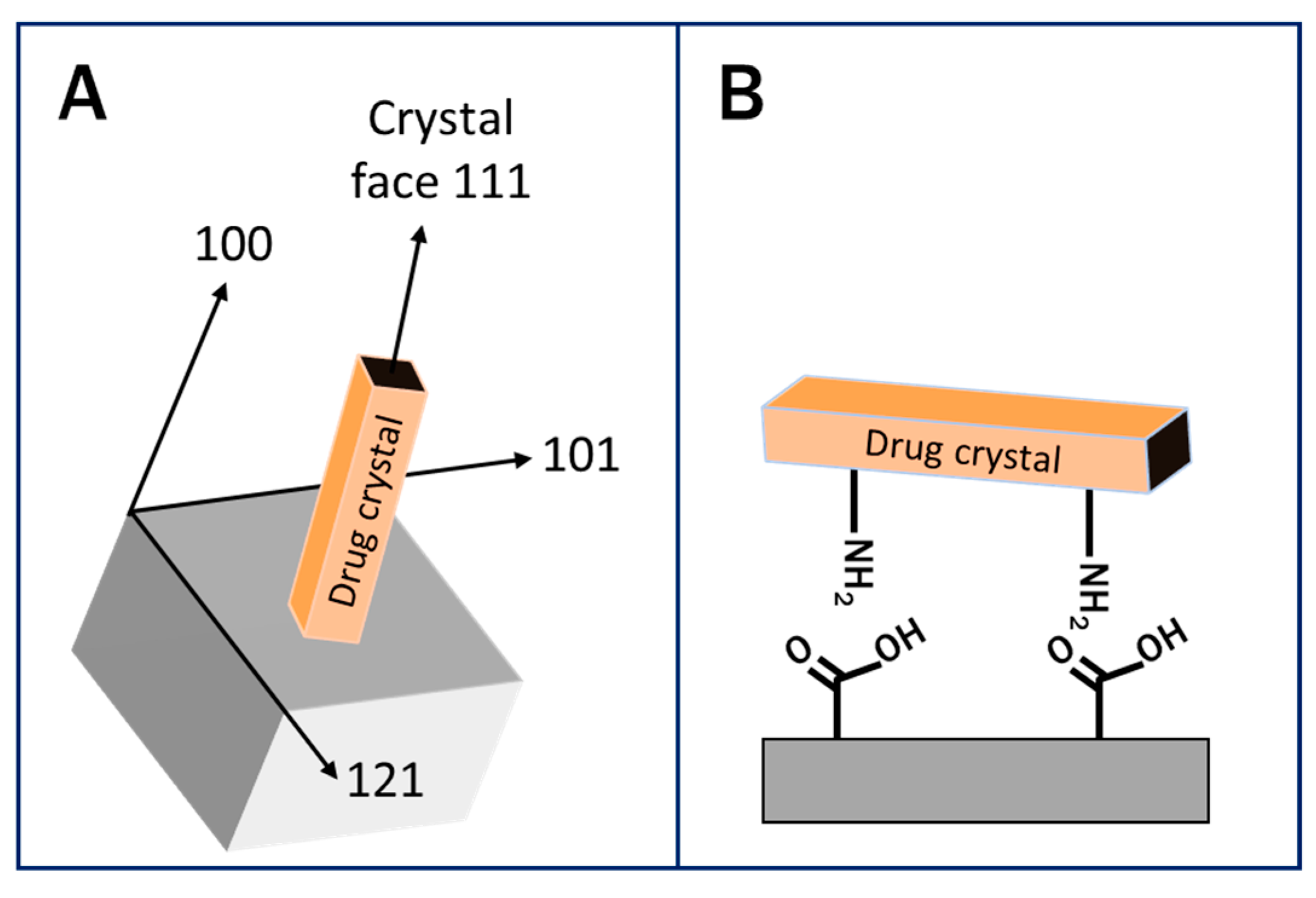{kind=link}
{kind=link}
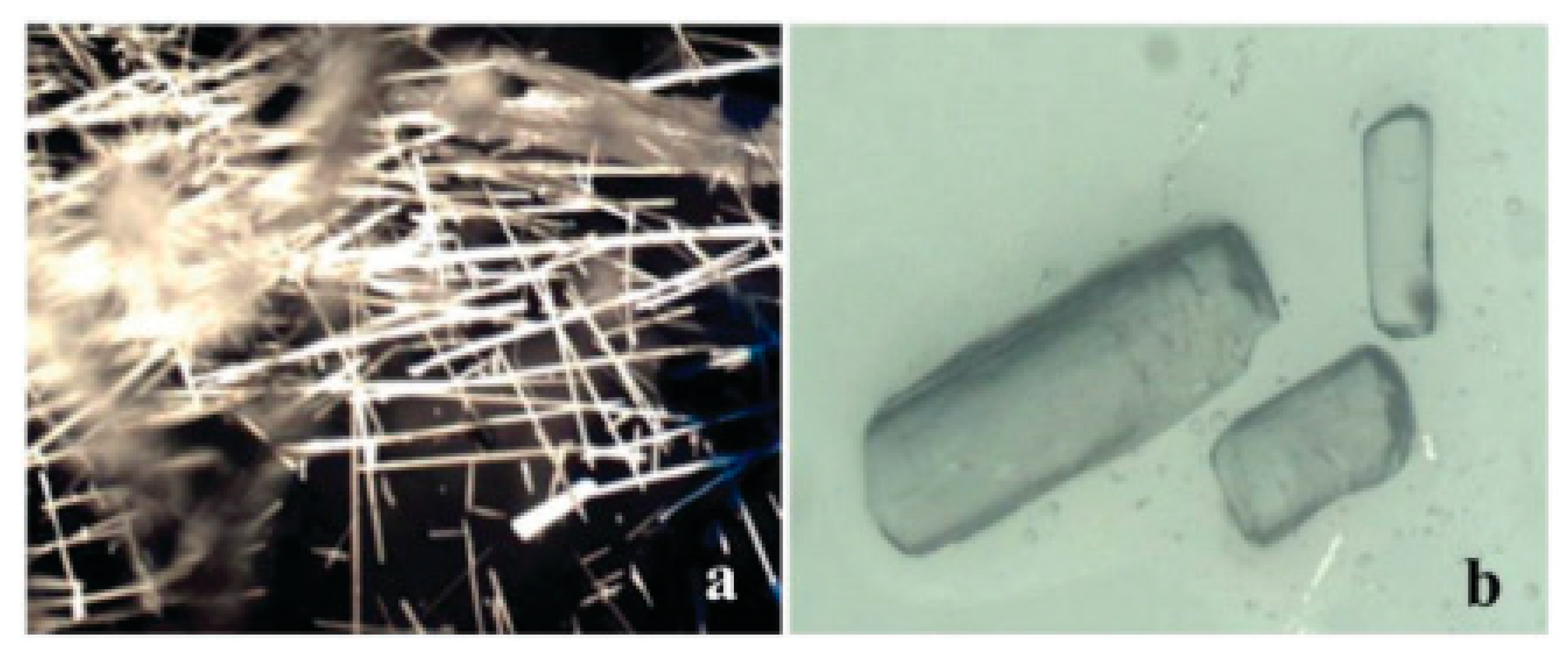{kind=link}
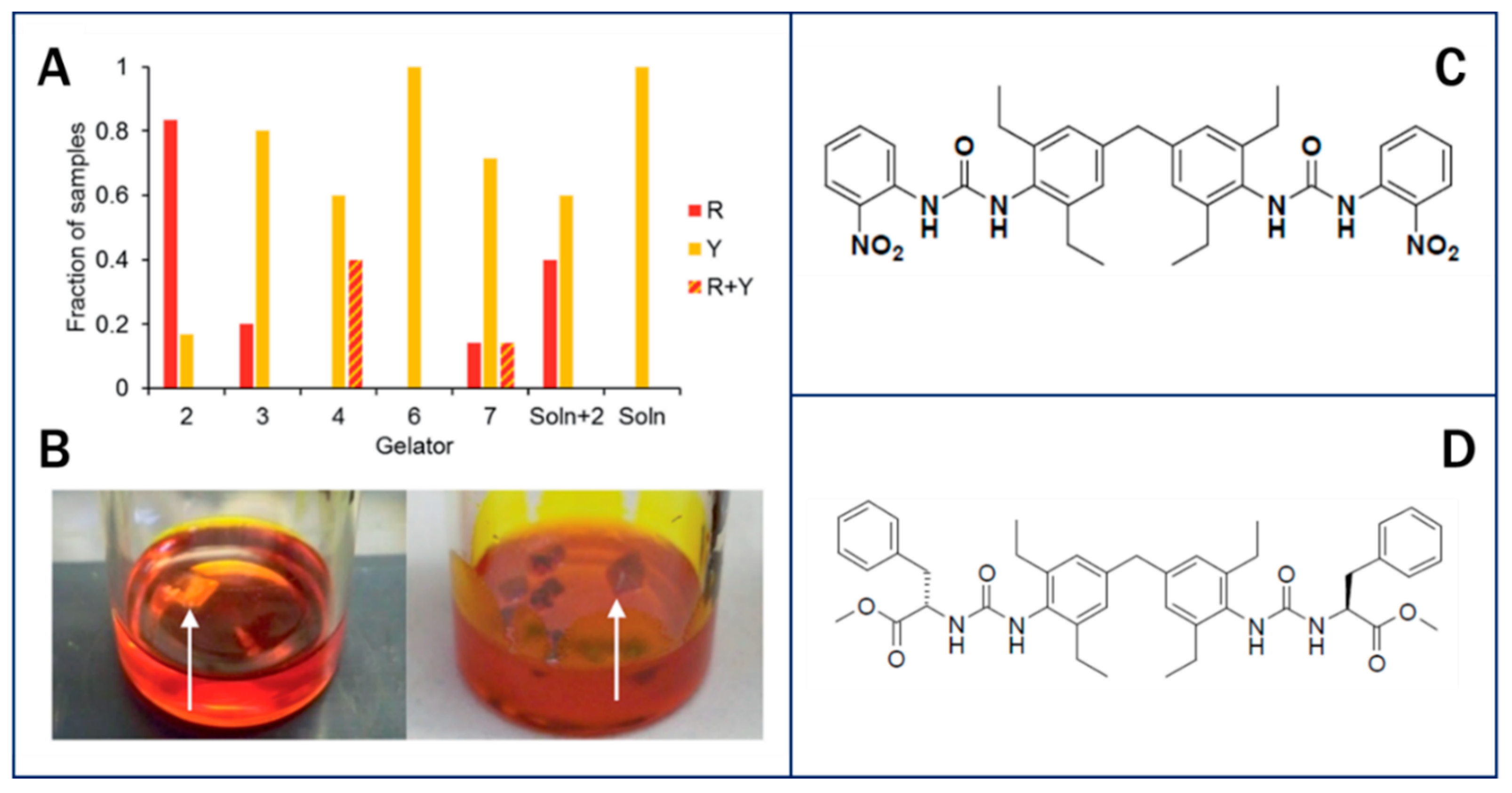{kind=link}
1. Introduction
2. Polymorphism in Pharmaceuticals
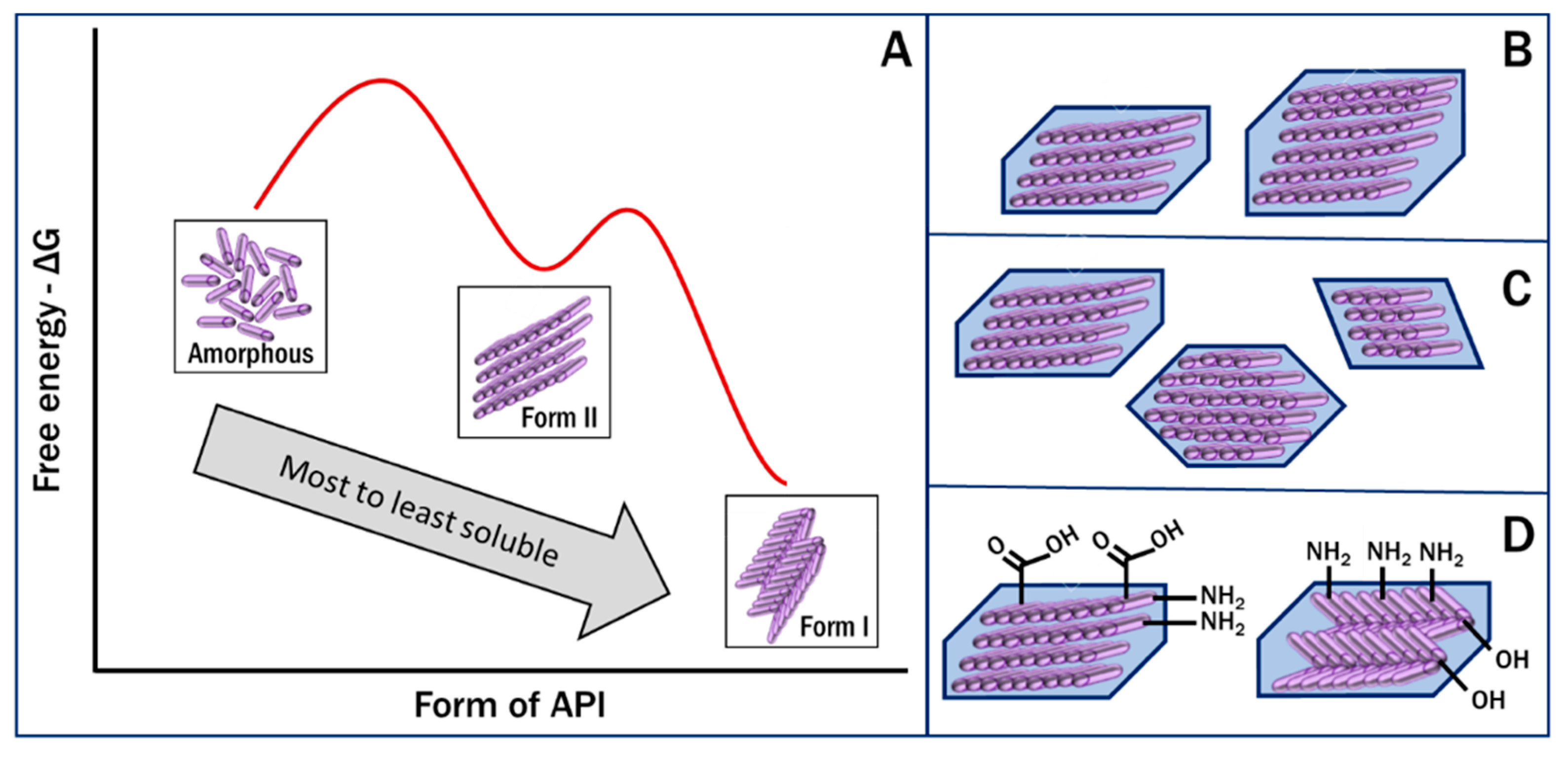
2.1. The Amorphous Form
2.2. Polymorph Stability and Metastable Forms
3. Individual Approaches to Polymorph Control
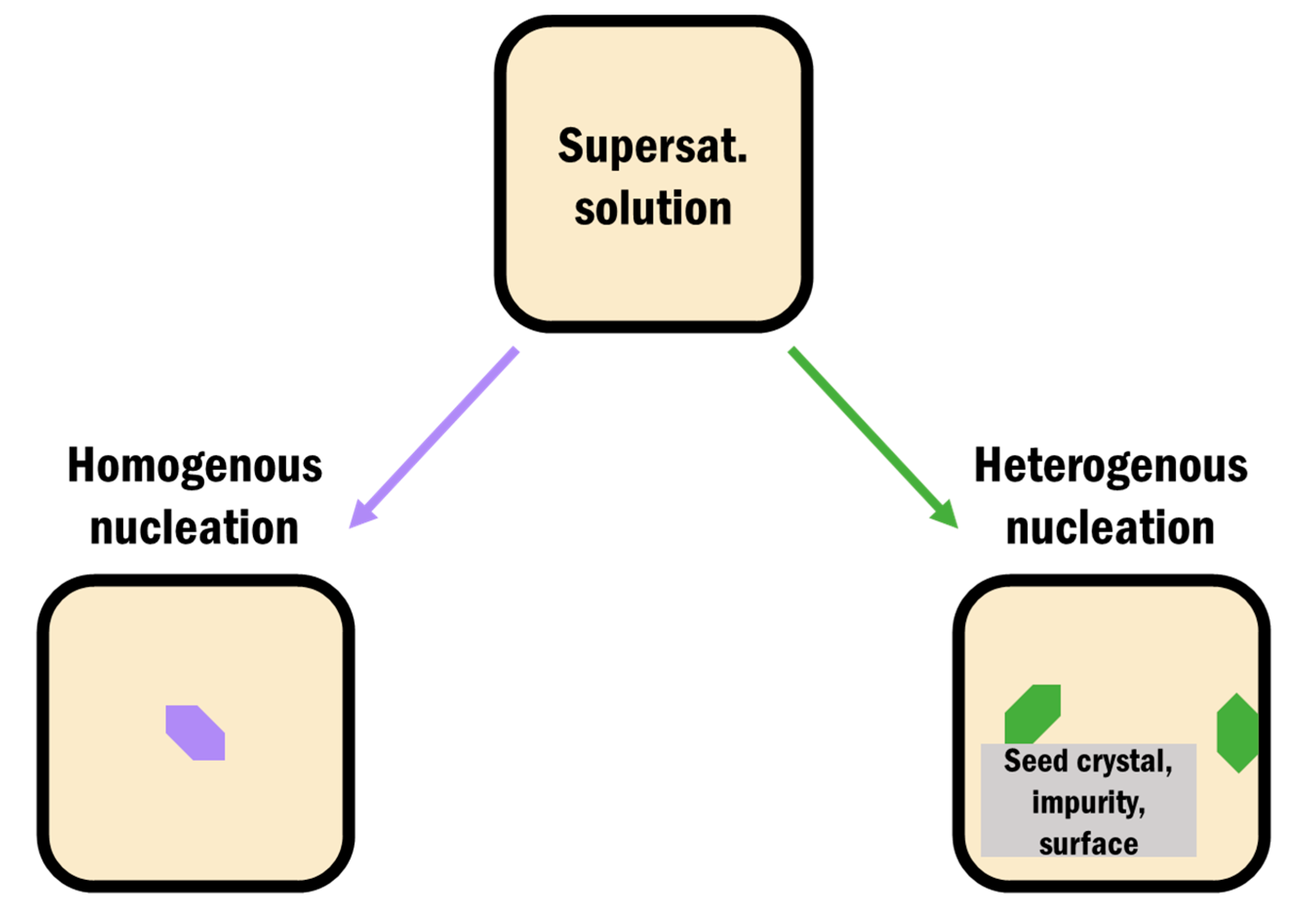
3.1. Heterogeneous Crystallization on Surfaces
3.2. Confinement in Pores
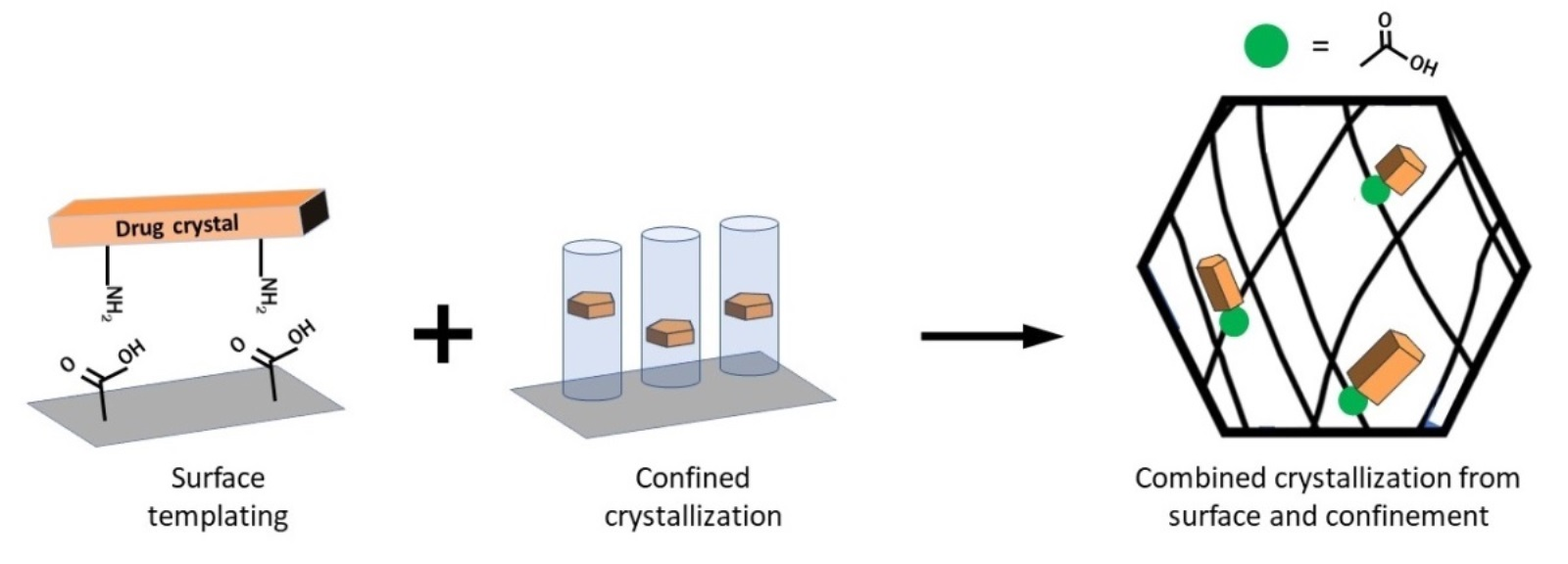
4. Combining Surface Chemistry and Confinement for Pharmaceutical Crystallization
4.1. Crystallization in Cross-Linked Polymer Networks
4.2. Crystallization in Physical Gels
4.3. Porous Particles of Organic Small Molecules
5. Use of Surface Chemistry and Confinement Outside of Pharmaceuticals
6. Summary and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sedo, K. 2018 Global Drug Delivery & Formulation Report: Part. 1, a Global Review; Drug Development and Delivery: Montville, NJ, USA, 2019. [Google Scholar]
- Sinha, B.; Müller, R.H.; Möschwitzer, J.P. Bottom-up approaches for preparing drug nanocrystals: Formulations and factors affecting particle size. Int. J. Pharm. 2013, 453, 126–141. [Google Scholar] [CrossRef] [PubMed]
- Censi, R.; di Martino, P. Polymorph impact on the bioavailability and stability of poorly soluble drugs. Molecules 2015, 20, 18759–18776. [Google Scholar] [CrossRef] [PubMed]
- Shekunov, B.Y.; York, P. Crystallization processes in pharmaceutical technology and drug delivery design. J. Cryst. Growth 2000, 211, 122–136. [Google Scholar] [CrossRef]
- FDA. Bioavailability and Bioequivalence Studies Submitted in NDAs or INDs—General Considerations; Food and Drug Administration Guidance for Industry; Food and Drug Administration: Rockville, MD, USA, 2014.
- Kalepu, S.; Nekkanti, V. Insoluble drug delivery strategies: Review of recent advances and business prospects. Acta Pharm. Sin. B 2015, 5, 442–453. [Google Scholar] [CrossRef]
- Williams, H.D.; Trevaskis, N.L.; Charman, S.A.; Shanker, R.M.; Charman, W.N.; Pouton, C.W.; Porter, C.J.H. Strategies to Address Low Drug Solubility in Discovery and Development. Pharmacol. Rev. 2013, 65, 315–499. [Google Scholar] [CrossRef]
- Lee, E.H. A practical guide to pharmaceutical polymorph screening & selection. Asian J. Pharm. Sci. 2014, 9, 163–175. [Google Scholar] [CrossRef]
- Bernstein, J. Polymorphism in Molecular Crystals; Oxford University Press, Inc.: Oxford, UK, 2010; ISBN 9780199236565. [Google Scholar]
- Saifee, M.; Inamdar, N.; Dhamecha, D.L.; Rathi, A.A. Drug polymorphism: A review. Int. J. Health Res. 2009, 2, 291–306. [Google Scholar] [CrossRef]
- Singhal, D.; Curatolo, W. Drug polymorphism and dosage form design: A practical perspective. Adv. Drug Deliv. Rev. 2004, 56, 335–347. [Google Scholar] [CrossRef]
- Behera, L.; Sahoo, S.; Patil, S. Enhancement of Solubility: A pharmaceutical Overview. Der Pharm. Lett. 2010, 2, 310–318. [Google Scholar]
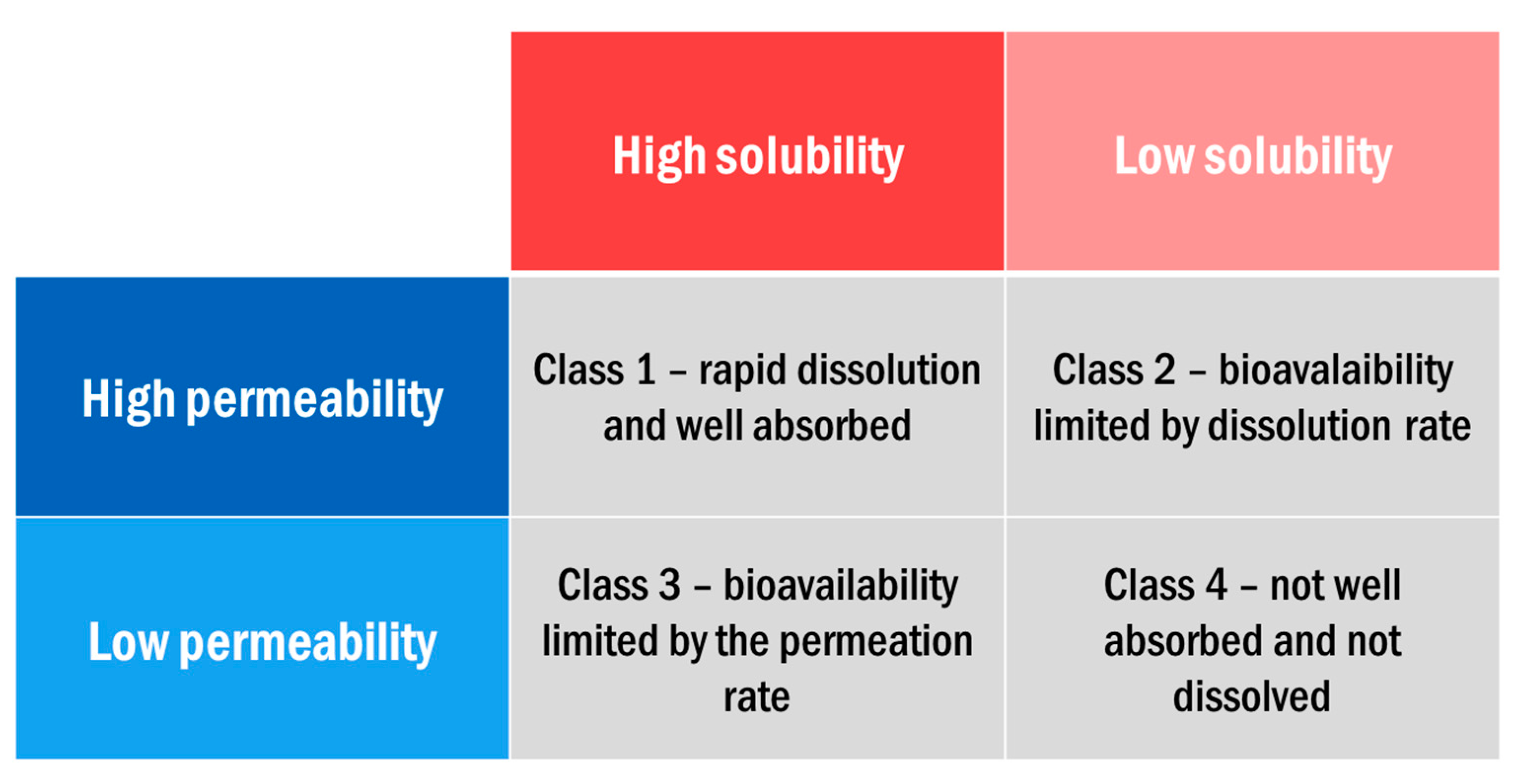
- Amidon, G.L.; Lennernas, H.; Shah, V.P.; Crison, J.R. A Theoretical Basis for a Biopharmaceutic Drug Classification: The Correlation of in Vitro Drug Product Dissolution and in Vivo Bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef]
- Benet, L.Z. The Role of BCS (Biopharmaceutics Classification System) and BDDCS (Biopharmaceutics Drug Disposition Classification System) in Drug Development. J. Pharm. Sci. 2013, 102, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Ting, J.M.; Porter, W.W.; Mecca, J.M.; Bates, F.S.; Reineke, T.M. Advances in Polymer Design for Enhancing Oral Drug Solubility and Delivery. Bioconjug. Chem. 2018, 29, 939–952. [Google Scholar] [CrossRef] [PubMed]
- Babu, N.J.; Nangia, A. Solubility Advantage of Amorphous Drugs and Pharmaceutical Cocrystals. Cryst. Growth Des. 2011, 11, 2662–2679. [Google Scholar] [CrossRef]
- Jamzad, S.; Fassihi, R. Role of surfactant and pH on dissolution properties of fenofibrate and glipizide—A technical note. AAPS PharmSciTech 2006, 7, E17–E22. [Google Scholar] [CrossRef]
- The U.S. Department of Health and Human Services. Waiver of In Vivo Immediate-Release Solid Oral Bioequivalence Studies for Bioavailability and Biopharmaceutics Classification Dosage Forms Based on a Biopharmaceutics Classification System; Food and Drug Administration Guidance for Industry; Food and Drug Administration: Rockville, MD, USA, 2017.
- Elder, D.P.; Holm, R.; de Diego, H.L. Use of pharmaceutical salts and cocrystals to address the issue of poor solubility. Int. J. Pharm. 2013, 453, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Schultheiss, N.; Newman, A. Pharmaceutical Cocrystals and Their Physicochemical Properties. Cryst. Growth Des. 2009, 9, 2950–2967. [Google Scholar] [CrossRef]
- Yadav, A.; Shete, A.; Dabke, A.; Kulkarni, P.; Sakhare, S. Co-crystals: A novel approach to modify physicochemical properties of active pharmaceutical ingredients. Indian J. Pharm. Sci. 2009, 71, 359. [Google Scholar] [CrossRef]
- Aitipamula, S.; Banerjee, R.; Bansal, A.K.; Biradha, K.; Cheney, M.L.; Choudhury, A.R.; Desiraju, G.R.; Dikundwar, A.G.; Dubey, R.; Duggirala, N.; et al. Polymorphs, Salts, and Cocrystals: What’s in a Name? Cryst. Growth Des. 2012, 12, 2147–2152. [Google Scholar] [CrossRef]
- Serajuddin, A.T.M. Salt formation to improve drug solubility. Adv. Drug Deliv. Rev. 2007, 59, 603–616. [Google Scholar] [CrossRef]
- Gupta, D.; Bhatia, D.; Dave, V.; Sutariya, V.; Varghese Gupta, S. Salts of Therapeutic Agents: Chemical, Physicochemical, and Biological Considerations. Molecules 2018, 23, 1719. [Google Scholar] [CrossRef]
- Karimi-Jafari, M.; Padrela, L.; Walker, G.M.; Croker, D.M. Creating Cocrystals: A Review of Pharmaceutical Cocrystal Preparation Routes and Applications. Cryst. Growth Des. 2018, 18, 6370–6387. [Google Scholar] [CrossRef]
- Sun, C.C. Cocrystallization for successful drug delivery. Expert Opin. Drug Deliv. 2013, 10, 201–213. [Google Scholar] [CrossRef]
- Artusio, F.; Pisano, R. Surface-induced crystallization of pharmaceuticals and biopharmaceuticals: A review. Int. J. Pharm. 2018, 547, 190–208. [Google Scholar] [CrossRef] [PubMed]
- Thakore, S.D.; Sood, A.; Bansal, A.K. Emerging role of primary heterogeneous nucleation in pharmaceutical crystallization. Drug Dev. Res. 2020, 81, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, B.D.; Ha, J.-M.; Hillmyer, M.A.; Ward, M.D. Manipulating Crystal Growth and Polymorphism by Confinement in Nanoscale Crystallization Chambers. Acc. Chem. Res. 2012, 45, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.C.L.; Bimbo, L.M. Crystallisation behaviour of pharmaceutical compounds confined within mesoporous silicon. Pharmaceutics 2020, 12, 214. [Google Scholar] [CrossRef]
- Jiang, Q.; Ward, M.D. Crystallization under nanoscale confinement. Chem. Soc. Rev. 2014, 43, 2066–2079. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Y.; Lee, I.S.; Dette, S.S.; Boerner, J.; Myerson, A.S. Crystallization on Confined Engineered Surfaces: A Method to Control Crystal Size and Generate Different Polymorphs. J. Am. Chem. Soc. 2005, 127, 14982–14983. [Google Scholar] [CrossRef] [PubMed]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug Solubility: Importance and Enhancement Techniques. ISRN Pharm. 2012, 2012, 195727. [Google Scholar] [CrossRef]
- Blagden, N.; de Matas, M.; Gavan, P.T.; York, P. Crystal engineering of active pharmaceutical ingredients to improve solubility and dissolution rates. Adv. Drug Deliv. Rev. 2007, 59, 617–630. [Google Scholar] [CrossRef]
- Munroe, Á.; Rasmuson, Å.C.; Hodnett, B.K.; Croker, D.M. Relative Stabilities of the Five Polymorphs of Sulfathiazole. Cryst. Growth Des. 2012, 12, 2825–2835. [Google Scholar] [CrossRef]
- Miller, J.; Collman, B.; Greene, L.; Grant, D.; Blackburn, A. Identifying the Stable Polymorph Early in the Drug Discovery-Development Process. Pharm. Dev. Technol. 2005, 10, 291–297. [Google Scholar] [CrossRef] [PubMed]
- de Villiers, M.M.; van der Watt, J.G.; Lötter, A.P. The interconversion of the polymorphic forms of chloramphenicol palmitate (CAP) as a function of environmental temperature. Drug Dev. Ind. Pharm. 1991, 17, 1295–1303. [Google Scholar] [CrossRef]
- Csakurda-Harmathy, Z.; Thege, I.K. Transformation of chloramphenicol palmitate from therapeutically inactive polymorph A to active polymorph B. J. Therm. Anal. 1997, 50, 867–871. [Google Scholar] [CrossRef]
- Aguiar, A.J.; Krc, J.; Kinkel, A.W.; Samyn, J.C. Effect of polymorphism on the absorption of chloramphenicol from chloramphenicol palmitate. J. Pharm. Sci. 1967, 56, 847–853. [Google Scholar] [CrossRef]
- Hancock, B.C.; Zografi, G. Characteristics and Significance of the Amorphous State in Pharmaceutical Systems. J. Pharm. Sci. 1997, 86, 1–12. [Google Scholar] [CrossRef]
- Hancock, B.C.; Parks, M. What is the true solubility advantage for amorphous pharmaceuticals? Pharm. Res. 2000, 17, 397–404. [Google Scholar] [CrossRef]
- Yu, L. Amorphous pharmaceutical solids: Preparation, characterization and stabilization. Adv. Drug Deliv. Rev. 2001, 48, 27–42. [Google Scholar] [CrossRef]
- Aucamp, M.; Odendaal, R.; Liebenberg, W.; Hamman, J. Amorphous azithromycin with improved aqueous solubility and intestinal membrane permeability. Drug Dev. Ind. Pharm. 2015, 41, 1100–1108. [Google Scholar] [CrossRef]
- Dhumal, R.; Biradar, S.; Yamamura, S.; Paradkar, A.; York, P. Preparation of amorphous cefuroxime axetil nanoparticles by sonoprecipitation for enhancement of bioavailability. Eur. J. Pharm. Biopharm. 2008, 70, 109–115. [Google Scholar] [CrossRef]
- Watanabe, T.; Wakiyama, N.; Usui, F.; Ikeda, M.; Isobe, T.; Senna, M. Stability of amorphous indomethacin compounded with silica. Int. J. Pharm. 2001, 226, 81–91. [Google Scholar] [CrossRef]
- Rajaram, S.; Sivaranjani, S.; Soundammal, S.; Sowmitha, S.; Sowmiya, B. Enhancement of solubility and dissolution rate of poorly water-soluble etravirine by solid dispersion technique for antiretroviral therapy. Int. J. Res. Pharm. Sci. 2019, 10, 1–7. [Google Scholar] [CrossRef]
- Yuan, X.; Sperger, D.; Munson, E.J. Investigating Miscibility and Molecular Mobility of Nifedipine-PVP Amorphous Solid Dispersions Using Solid-State NMR Spectroscopy. Mol. Pharm. 2014, 11, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Hailu, S.A.; Bogner, R.H. Effect of the pH Grade of Silicates on Chemical Stability of Coground Amorphous Quinapril Hydrochloride and its Stabilization Using pH-Modifiers. J. Pharm. Sci. 2009, 98, 3358–3372. [Google Scholar] [CrossRef]
- Brettmann, B.; Bell, E.; Myerson, A.; Trout, B. Solid-State NMR Characterization of High-Loading Solid Solutions of API and Excipients Formed by Electrospinning. J. Pharm. Sci. 2012, 101, 1538–1545. [Google Scholar] [CrossRef]
- Brettmann, B.K.; Myerson, A.S.; Trout, B.L. Solid-state nuclear magnetic resonance study of the physical stability of electrospun drug and polymer solid solutions. J. Pharm. Sci. 2012. [Google Scholar] [CrossRef]
- Baghel, S.; Cathcart, H.; O’Reilly, N.J. Polymeric Amorphous Solid Dispersions: A Review of Amorphization, Crystallization, Stabilization, Solid-State Characterization, and Aqueous Solubilization of Biopharmaceutical Classification System Class II Drugs. J. Pharm. Sci. 2016, 105, 2527–2544. [Google Scholar] [CrossRef]
- Telford, R.; Seaton, C.C.; Clout, A.; Buanz, A.; Gaisford, S.; Williams, G.R.; Prior, T.J.; Okoye, C.H.; Munshi, T.; Scowen, I.J. Stabilisation of metastable polymorphs: The case of paracetamol form III. Chem. Commun. 2016, 52, 12028–12031. [Google Scholar] [CrossRef]
- Beiner, M.; Rengarajan, G.T.; Pankaj, S.; Enke, D.; Steinhart, M. Manipulating the crystalline state of pharmaceuticals by nanoconfinement. Nano Lett. 2007, 7, 1381–1385. [Google Scholar] [CrossRef]
- Matsuda, Y.; Akazawa, R.; Teraoka, R.; Otsuka, M. Pharmaceutical Evaluation of Carbamazepine Modifications: Comparative Study for Photostability of Carbamazepine Polymorphs by using Fourier-transformed Reflection-absorption Infrared Spectroscopy and Colorimetric Measurement. J. Pharm. Pharmacol. 1994, 46, 162–167. [Google Scholar] [CrossRef]
- Kaneniwa, N.; Yamaguchi, T.; Watari, N.; Otsuka, M. Hygroscopicity of carbamazepine crystalline powders. Yakugaku Zasshi 1984, 104, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Callister Jr, W.D.; Rethwisch, D.G. Materials Science and Engineering: An Introduction, 8th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2009; ISBN 978-0470419977. [Google Scholar]
- Dubbini, A.; Censi, R.; Martena, V.; Hoti, E.; Ricciutelli, M.; Malaj, L.; di Martino, P. Influence of pH and method of crystallization on the solid physical form of indomethacin. Int. J. Pharm. 2014, 473, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Arribas Bueno, R.; Crowley, C.M.; Hodnett, B.K.; Hudson, S.; Davern, P. Influence of Process Parameters on the Heterogeneous Nucleation of Active Pharmaceutical Ingredients onto Excipients. Org. Process Res. Dev. 2017, 21, 559–570. [Google Scholar] [CrossRef]
- Sun, J.; Wang, F.; Sui, Y.; She, Z.; Zhai, W.; Wang, C.; Deng, Y. Effect of particle size on solubility, dissolution rate, and oral bioavailability: Evaluation using coenzyme Q10 as naked nanocrystals. Int. J. Nanomed. 2012, 7, 5733–5744. [Google Scholar] [CrossRef]
- Bernstein, J. Polymorphism in Molecular Crystals, 1st ed.; International Union of Crystallography: Chester, UK, 2007; ISBN 9780191707940. [Google Scholar]
- Modi, S.R.; Dantuluri, A.K.R.; Perumalla, S.R.; Sun, C.C.; Bansal, A.K. Effect of Crystal Habit on Intrinsic Dissolution Behavior of Celecoxib Due to Differential Wettability. Cryst. Growth Des. 2014, 14, 5283–5292. [Google Scholar] [CrossRef]
- Chadha, R.; Gupta, S.; Shukla, G. Crystal habit, characterization and pharmacological activity of various crystal forms of arteether. Acta Pharm. Sin. B 2011, 1, 129–135. [Google Scholar] [CrossRef][Green Version]
- Verma, V.; Zeglinski, J.; Hudson, S.; Davern, P.; Hodnett, B.K. Dependence of Heterogeneous Nucleation on Hydrogen Bonding Lifetime and Complementarity. Cryst. Growth Des. 2018, 18, 7158–7172. [Google Scholar] [CrossRef]
- Louhi-Kultanen, M.; Karjalainen, M.; Rantanen, J.; Huhtanen, M.; Kallas, J. Crystallization of glycine with ultrasound. Int. J. Pharm. 2006, 320, 23–29. [Google Scholar] [CrossRef]
- Kim, A.I.; Akers, M.J.; Nail, S.L. The Physical State of Mannitol after Freeze-Drying: Effects of Mannitol Concentration, Freezing Rate, and a Noncrystallizing Cosolute. J. Pharm. Sci. 1998, 87, 931–935. [Google Scholar] [CrossRef]
- Gao, Z.; Rohani, S.; Gong, J.; Wang, J. Recent Developments in the Crystallization Process: Toward the Pharmaceutical Industry. Engineering 2017, 3, 343–353. [Google Scholar] [CrossRef]
- Yazdanpanah, N.; Testa, C.J.; Perala, S.R.K.; Jensen, K.D.; Braatz, R.D.; Myerson, A.S.; Trout, B.L. Continuous Heterogeneous Crystallization on Excipient Surfaces. Cryst. Growth Des. 2017, 17, 3321–3330. [Google Scholar] [CrossRef]
- Myerson, A.S. (Ed.) Handbook of Industrial Crystallization; Butterworth-Heinemann: Oxford, UK, 2002; ISBN 978-0-7506-7012-8. [Google Scholar]
- Hiremath, R.; Basile, J.A.; Varney, S.W.; Swift, J.A. Controlling Molecular Crystal Polymorphism with Self-Assembled Monolayer Templates. J. Am. Chem. Soc. 2005, 127, 18321–18327. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.A.; Kaplan, K.; Yuk, S.A.; Saboo, S.; Melkey, K.; Chadwick, K. Utilization of Surface Equilibria for Controlling Heterogeneous Nucleation: Making the “Disappeared” Polymorph of 3-Aminobenzensulfonic Acid “Reappear”. Cryst. Growth Des. 2016, 16, 6933–6940. [Google Scholar] [CrossRef]
- Pfund, L.Y.; Price, C.P.; Frick, J.J.; Matzger, A.J. Controlling pharmaceutical crystallization with designed polymeric heteronuclei. J. Am. Chem. Soc. 2015, 137, 871–875. [Google Scholar] [CrossRef] [PubMed]
- Quon, J.L.; Chadwick, K.; Wood, G.P.F.; Sheu, I.; Brettmann, B.K.; Myerson, A.S.; Trout, B.L. Templated nucleation of acetaminophen on spherical excipient agglomerates. Langmuir 2013, 29, 3292–3300. [Google Scholar] [CrossRef] [PubMed]
- Beckmann, W.; Otto, W.; Budde, U. Crystallisation of the Stable Polymorph of Hydroxytriendione: Seeding Process and Effects of Purity. Org. Process Res. Dev. 2001, 5, 387–392. [Google Scholar] [CrossRef]
- Beckmann, W.; Nickisch, K.; Budde, U. Development of a Seeding Technique for the Crystallization of the Metastable A Modification of Abecarnil. Org. Process Res. Dev. 1998, 2, 298–304. [Google Scholar] [CrossRef]
- Nichols, G.; Frampton, C.S. Physicochemical Characterization of the Orthorhombic Polymorph of Paracetamol Crystallized from Solution. J. Pharm. Sci. 1998, 87, 684–693. [Google Scholar] [CrossRef]
- Bakar, M.R.A.; Nagy, Z.K.; Rielly, C.D. Seeded Batch Cooling Crystallization with Temperature Cycling for the Control of Size Uniformity and Polymorphic Purity of Sulfathiazole Crystals. Org. Process Res. Dev. 2009, 13, 1343–1356. [Google Scholar] [CrossRef]
- Ling, J.; Chadwick, K. Heterogeneous Crystallization inside Microporous Polymer Particles as a Process Intensification Technology for the Manufacture of Drug Formulations. Org. Process Res. Dev. 2017, 21, 827–834. [Google Scholar] [CrossRef]
- Diao, Y.; Myerson, A.S.; Hatton, T.A.; Trout, B.L. Surface design for controlled crystallization: The role of surface chemistry and nanoscale pores in heterogeneous nucleation. Langmuir 2011, 27, 5324–5334. [Google Scholar] [CrossRef] [PubMed]
- López-Mejías, V.; Myerson, A.S.; Trout, B.L. Geometric Design of Heterogeneous Nucleation Sites on Biocompatible Surfaces. Cryst. Growth Des. 2013, 13, 3835–3841. [Google Scholar] [CrossRef]
- Mitchell, C.A.; Yu, L.; Ward, M.D. Selective nucleation and discovery of organic polymorphs through epitaxy with single crystal substrates. J. Am. Chem. Soc. 2001, 123, 10830–10839. [Google Scholar] [CrossRef] [PubMed]
- Chadwick, K.; Chen, J.; Myerson, A.S.; Trout, B.L. Toward the Rational Design of Crystalline Surfaces for Heteroepitaxy: Role of Molecular Functionality. Cryst. Growth Des. 2012, 12, 1159–1166. [Google Scholar] [CrossRef]
- Hiremath, R.; Varney, S.W.; Swift, J.A. Selective growth of a less stable polymorph of 2-iodo-4-nitroaniline on a self-assembled monolayer template. Chem. Commun. 2004, 2676. [Google Scholar] [CrossRef]
- Parambil, J.V.; Poornachary, S.K.; Tan, R.B.H.; Heng, J.Y.Y. Template-induced polymorphic selectivity: The effects of surface chemistry and solute concentration on carbamazepine crystallisation. CrystEngComm 2014, 16, 4927–4930. [Google Scholar] [CrossRef]
- Tan, L.; Davis, R.M.; Myerson, A.S.; Trout, B.L. Control of Heterogeneous Nucleation via Rationally Designed Biocompatible Polymer Surfaces with Nanoscale Features. Cryst. Growth Des. 2015, 15, 2176–2186. [Google Scholar] [CrossRef]
- Yang, H.; Song, C.L.; Lim, Y.X.S.; Chen, W.; Heng, J.Y.Y. Selective crystallisation of carbamazepine polymorphs on surfaces with differing properties. CrystEngComm 2017, 19, 6573–6578. [Google Scholar] [CrossRef]
- Lee, E.H.; Boerrigter, S.X.M.; Byrn, S.R. Epitaxy of a Structurally Related Compound on the (100) Faces of Flufenamic Acid Form I and III Single Crystals. Cryst. Growth Des. 2010, 10, 518–527. [Google Scholar] [CrossRef]
- Bonafede, S.J.; Ward, M.D. Selective Nucleation and Growth of an Organic Polymorph by Ledge-Directed Epitaxy on a Molecular Crystal Substrate. J. Am. Chem. Soc. 1995, 117, 7853–7861. [Google Scholar] [CrossRef]
- Carter, P.W.; Ward, M.D. Directing Polymorph Selectivity during Nucleation of Anthranilic Acid on Molecular Substrates. J. Am. Chem. Soc. 1994, 116, 769–770. [Google Scholar] [CrossRef]
- Raghavan, S.L.; Trividic, A.; Davis, A.F.; Hadgraft, J. Crystallization of hydrocortisone acetate: Influence of polymers. Int. J. Pharm. 2001, 212, 213–221. [Google Scholar] [CrossRef]
- Lang, M.; Grzesiak, A.L.; Matzger, A.J. The use of polymer heteronuclei for crystalline polymorph selection. J. Am. Chem. Soc. 2002, 124, 14834–14835. [Google Scholar] [CrossRef] [PubMed]
- Caridi, A.; Kulkarni, S.A.; di Profio, G.; Curcio, E.; ter Horst, J.H. Template-Induced Nucleation of Isonicotinamide Polymorphs. Cryst. Growth Des. 2014, 14, 1135–1141. [Google Scholar] [CrossRef]
- Moshe, H.; Levi, G.; Mastai, Y. Polymorphism stabilization by crystal adsorption on a self-assembled monolayer. CrystEngComm 2013, 15, 9203. [Google Scholar] [CrossRef]
- Yang, X.; Sarma, B.; Myerson, A.S. Polymorph Control of Micro/Nano-Sized Mefenamic Acid Crystals on Patterned Self-Assembled Monolayer Islands. Cryst. Growth Des. 2012, 12, 5521–5528. [Google Scholar] [CrossRef]
- Singh, A.; Lee, I.S.; Kim, K.; Myerson, A.S. Crystal growth on self-assembled monolayers. CrystEngComm 2011, 13, 24–32. [Google Scholar] [CrossRef]
- Dressler, D.H.; Mastai, Y. Controlling Polymorphism by Crystallization on Self-Assembled Multilayers. Cryst. Growth Des. 2007, 7, 847–850. [Google Scholar] [CrossRef]
- Ha, J.M.; Wolf, J.H.; Hillmyer, M.A.; Ward, M.D. Polymorph Selectivity under Nanoscopic Confinement. J. Am. Chem. Soc. 2004, 126, 3382–3383. [Google Scholar] [CrossRef]
- Dwyer, L.M.; Michaelis, V.K.; O’Mahony, M.; Griffin, R.G.; Myerson, A.S. Confined crystallization of fenofibrate in nanoporous silica. CrystEngComm 2015, 17, 7922–7929. [Google Scholar] [CrossRef]
- Lee, H.; Lee, J. Confined crystallization of drug in directionally freeze-dried water-soluble template. J. Ind. Eng. Chem. 2015, 21, 1183–1190. [Google Scholar] [CrossRef]
- Rengarajan, G.T.; Enke, D.; Steinhart, M.; Beiner, M. Stabilization of the amorphous state of pharmaceuticals in nanopores. J. Mater. Chem. 2008, 18, 2537. [Google Scholar] [CrossRef]
- Yang, X.; Ong, T.-C.; Michaelis, V.K.; Heng, S.; Griffin, R.G.; Myerson, A.S. Formation of organic molecular nanocrystals under soft confinement. CrystEngComm 2015, 17, 6044–6052. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, B.D.; Hillmyer, M.A.; Ward, M.D. Glycine Polymorphism in Nanoscale Crystallization Chambers. Cryst. Growth Des. 2008, 8, 3368–3375. [Google Scholar] [CrossRef]
- Al-Ani, A.J.; Herdes, C.; Wilson, C.C.; Castro-Dominguez, B. Engineering a New Access Route to Metastable Polymorphs with Electrical Confinement. Cryst. Growth Des. 2020, 20, 1451–1457. [Google Scholar] [CrossRef]
- Jackson, C.L.; McKenna, G.B. Vitrification and Crystallization of Organic Liquids Confined to Nanoscale Pores. Chem. Mater. 1996, 8, 2128–2137. [Google Scholar] [CrossRef]
- Pauchet, M.; Morelli, T.; Coste, S.; Malandain, J.-J.J.; Coquerel, G. Crystallization of (±)-Modafinil in Gel: Access to Form I, Form III, and Twins. Cryst. Growth Des. 2006, 6, 1881–1889. [Google Scholar] [CrossRef]
- Eral, H.B.; López-Mejías, V.; O’Mahony, M.; Trout, B.L.; Myerson, A.S.; Doyle, P.S. Biocompatible Alginate Microgel Particles as Heteronucleants and Encapsulating Vehicles for Hydrophilic and Hydrophobic Drugs. Cryst. Growth Des. 2014, 14, 2073–2082. [Google Scholar] [CrossRef]
- Diao, Y.; Helgeson, M.E.; Myerson, A.S.; Hatton, T.A.; Doyle, P.S.; Trout, B.L. Controlled nucleation from solution using polymer microgels. J. Am. Chem. Soc. 2011, 133, 3756–3759. [Google Scholar] [CrossRef]
- Diao, Y.; Whaley, K.E.; Helgeson, M.E.; Woldeyes, M.A.; Doyle, P.S.; Myerson, A.S.; Hatton, T.A.; Trout, B.L. Gel-induced selective crystallization of polymorphs. J. Am. Chem. Soc. 2012, 134, 673–684. [Google Scholar] [CrossRef]
- Esposito, C.L.; Kirilov, P.; Roullin, V.G. Organogels, promising drug delivery systems: An update of state-of-the-art and recent applications. J. Control. Release 2018, 271, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Kalshetti, P.P.; Rajendra, V.B.; Dixit, D.N.; Parekh, P.P. Hydrogels as a drug delivery system and applications: A review. Int. J. Pharm. Pharm. Sci. 2012, 4, 1–7. [Google Scholar]
- Sagiri, S.S.; Singh, V.K.; Kulanthaivel, S.; Banerjee, I.; Basak, P.; Battachrya, M.K.; Pal, K. Stearate organogel–gelatin hydrogel based bigels: Physicochemical, thermal, mechanical characterizations and in vitro drug delivery applications. J. Mech. Behav. Biomed. Mater. 2015, 43, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, L.; Kennedy, S.R.; Jones, C.D.; Steed, J.W. Cavity-containing supramolecular gels as a crystallization tool for hydrophobic pharmaceuticals. Chem. Commun. 2016, 52, 10113–10116. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.A.; Piepenbrock, M.-O.M.; Lloyd, G.O.; Clarke, N.; Howard, J.A.K.K.; Steed, J.W. Anion-switchable supramolecular gels for controlling pharmaceutical crystal growth. Nat. Chem. 2010, 2, 1037–1043. [Google Scholar] [CrossRef]
- Tokuyama, H.; Kato, Y. Preparation of thermosensitive polymeric organogels and their drug release behaviors. Eur. Polym. J. 2010, 46, 277–282. [Google Scholar] [CrossRef]
- Vintiloiu, A.; Leroux, J.C. Organogels and their use in drug delivery—A review. J. Control. Release 2008, 125, 179–192. [Google Scholar] [CrossRef]
- Aparicio, F.; Matesanz, E.; Sánchez, L. Cooperative self-assembly of linear organogelators. Amplification of chirality and crystal growth of pharmaceutical ingredients. Chem. Commun. 2012, 48, 5757–5759. [Google Scholar] [CrossRef]
- Ruiz-Palomero, C.; Kennedy, S.R.; Soriano, M.L.; Jones, C.D.; Valcarcel, M.; Steed, J.W.; Valcárcel, M.; Steed, J.W. Pharmaceutical crystallization with nanocellulose organogels. Chem. Commun. 2016, 52, 7782–7785. [Google Scholar] [CrossRef]
- Banerjee, M.; Saraswatula, S.; Willows, L.G.; Woods, H.; Brettmann, B. Pharmaceutical crystallization in surface-modified nanocellulose organogels. J. Mater. Chem. B 2018, 6, 7317–7328. [Google Scholar] [CrossRef]
- Foster, J.A.; Damodaran, K.K.; Maurin, A.; Day, G.M.; Thompson, H.P.G.; Cameron, G.J.; Bernal, J.C.; Steed, J.W. Pharmaceutical polymorph control in a drug-mimetic supramolecular gel. Chem. Sci. 2017, 8, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Bronstein, H.; Nielsen, C.B.; Schroeder, B.C.; McCulloch, I. The role of chemical design in the performance of organic semiconductors. Nat. Rev. Chem. 2020, 4, 66–77. [Google Scholar] [CrossRef]
- Gao, S.; Wang, S.; Ma, J.; Wu, Y.; Fu, X.; Marella, R.K.; Liu, K.; Fang, Y. Salt Tunable Rheology of Thixotropic Supramolecular Organogels and Their Applications for Crystallization of Organic Semiconductors. Langmuir 2016, 32, 12805–12813. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Briseno, A.L.; Mannsfeld, S.C.B.; You, W.; Locklin, J.; Lee, H.W.; Xia, Y.; Bao, Z. Selective Crystallization of Organic Semiconductors on Patterned Templates of Carbon Nanotubes. Adv. Funct. Mater. 2007, 17, 2891–2896. [Google Scholar] [CrossRef]
- Wang, L.; Liu, X.-Y. Kinetic Analysis of Protein Crystal Nucleation in Gel Matrix. Biophys. J. 2008, 95, 5931–5940. [Google Scholar] [CrossRef]
- Tanabe, K.; Hirose, M.; Murai, R.; Sugiyama, S.; Shimizu, N.; Maruyama, M.; Takahashi, Y.; Adachi, H.; Takano, K.; Murakami, S.; et al. Promotion of Crystal Nucleation of Protein by Semi-Solid Agarose Gel. Appl. Phys. Express 2009, 2, 125501. [Google Scholar] [CrossRef]
- Chayen, N.E.; Saridakis, E.; Sear, R.P. Experiment and theory for heterogeneous nucleation of protein crystals in a porous medium. Proc. Natl. Acad. Sci. USA 2006, 103, 597–601. [Google Scholar] [CrossRef]
- Stack, A.G.; Fernandez-Martinez, A.; Allard, L.F.; Bañuelos, J.L.; Rother, G.; Anovitz, L.M.; Cole, D.R.; Waychunas, G.A. Pore-Size-Dependent Calcium Carbonate Precipitation Controlled by Surface Chemistry. Environ. Sci. Technol. 2014, 48, 6177–6183. [Google Scholar] [CrossRef]
- Lai, T.-T.C.; Cornevin, J.; Ferguson, S.; Li, N.; Trout, B.L.; Myerson, A.S. Control of Polymorphism in Continuous Crystallization via Mixed Suspension Mixed Product Removal Systems Cascade Design. Cryst. Growth Des. 2015, 15, 3374–3382. [Google Scholar] [CrossRef]
- Nicoud, L.; Licordari, F.; Myerson, A.S. Polymorph Control in MSMPR Crystallizers. A Case Study with Paracetamol. Org. Process Res. Dev. 2019, 23, 794–806. [Google Scholar] [CrossRef]
- Agnew, L.R.; McGlone, T.; Wheatcroft, H.P.; Robertson, A.; Parsons, A.R.; Wilson, C.C. Continuous Crystallization of Paracetamol (Acetaminophen) Form II: Selective Access to a Metastable Solid Form. Cryst. Growth Des. 2017, 17, 2418–2427. [Google Scholar] [CrossRef]
- Powell, K.A.; Bartolini, G.; Wittering, K.E.; Saleemi, A.N.; Wilson, C.C.; Rielly, C.D.; Nagy, Z.K. Toward Continuous Crystallization of Urea-Barbituric Acid: A Polymorphic Co-Crystal System. Cryst. Growth Des. 2015, 15, 4821–4836. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banerjee, M.; Brettmann, B. Combining Surface Templating and Confinement for Controlling Pharmaceutical Crystallization. Pharmaceutics 2020, 12, 995. https://doi.org/10.3390/pharmaceutics12100995
Banerjee M, Brettmann B. Combining Surface Templating and Confinement for Controlling Pharmaceutical Crystallization. Pharmaceutics. 2020; 12(10):995. https://doi.org/10.3390/pharmaceutics12100995
Chicago/Turabian StyleBanerjee, Manali, and Blair Brettmann. 2020. "Combining Surface Templating and Confinement for Controlling Pharmaceutical Crystallization" Pharmaceutics 12, no. 10: 995. https://doi.org/10.3390/pharmaceutics12100995
APA StyleBanerjee, M., & Brettmann, B. (2020). Combining Surface Templating and Confinement for Controlling Pharmaceutical Crystallization. Pharmaceutics, 12(10), 995. https://doi.org/10.3390/pharmaceutics12100995