Novel Intrinsic Mechanisms of Active Drug Extrusion at the Blood-Brain Barrier: Potential Targets for Enhancing Drug Delivery to the Brain?



Abstract
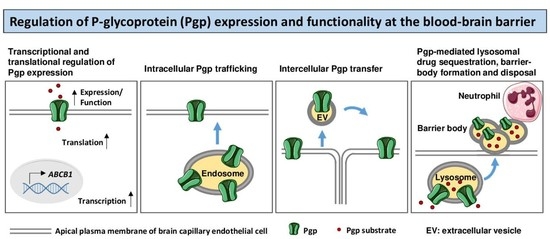
{kind=link}
{kind=link}
{kind=link}
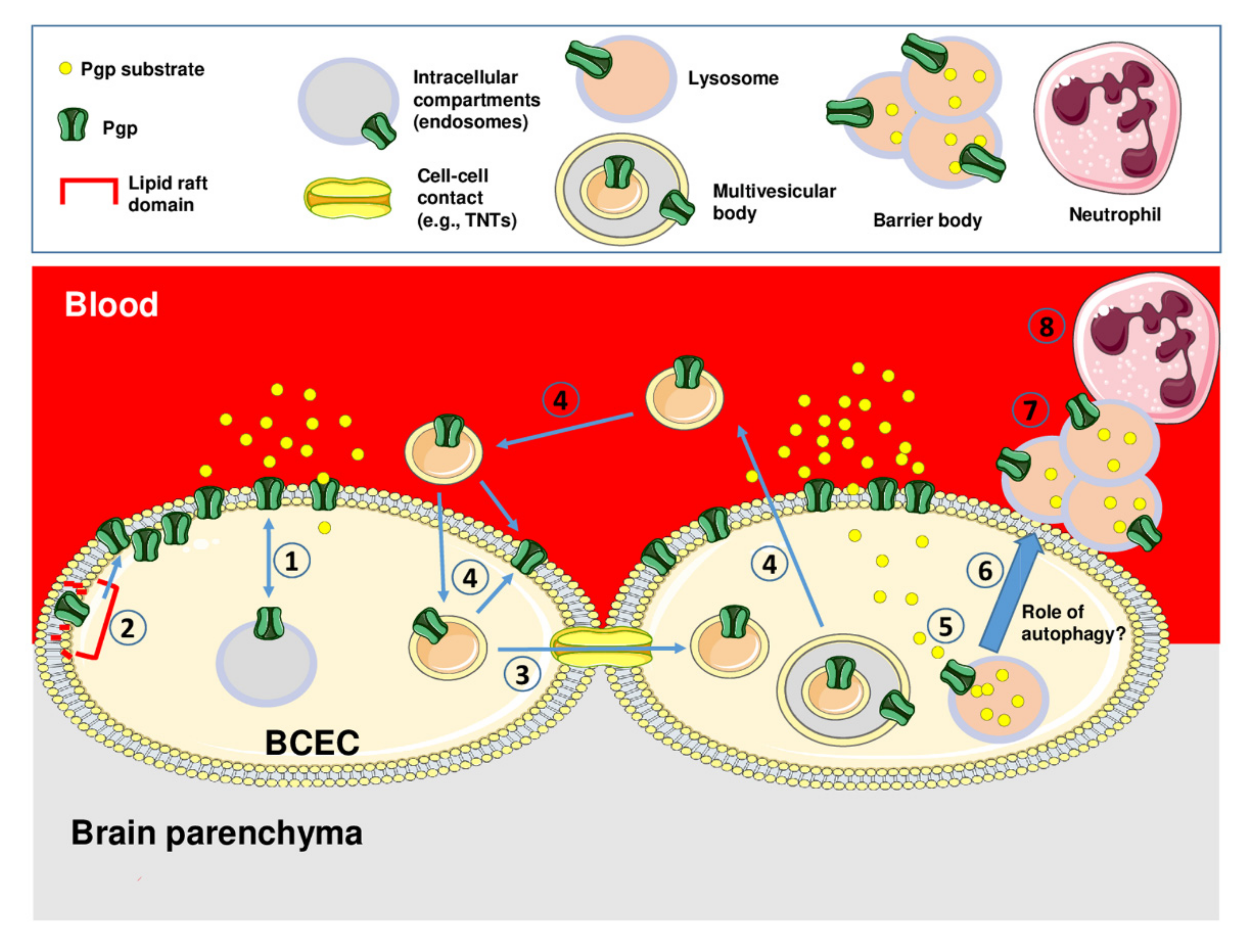{kind=link}
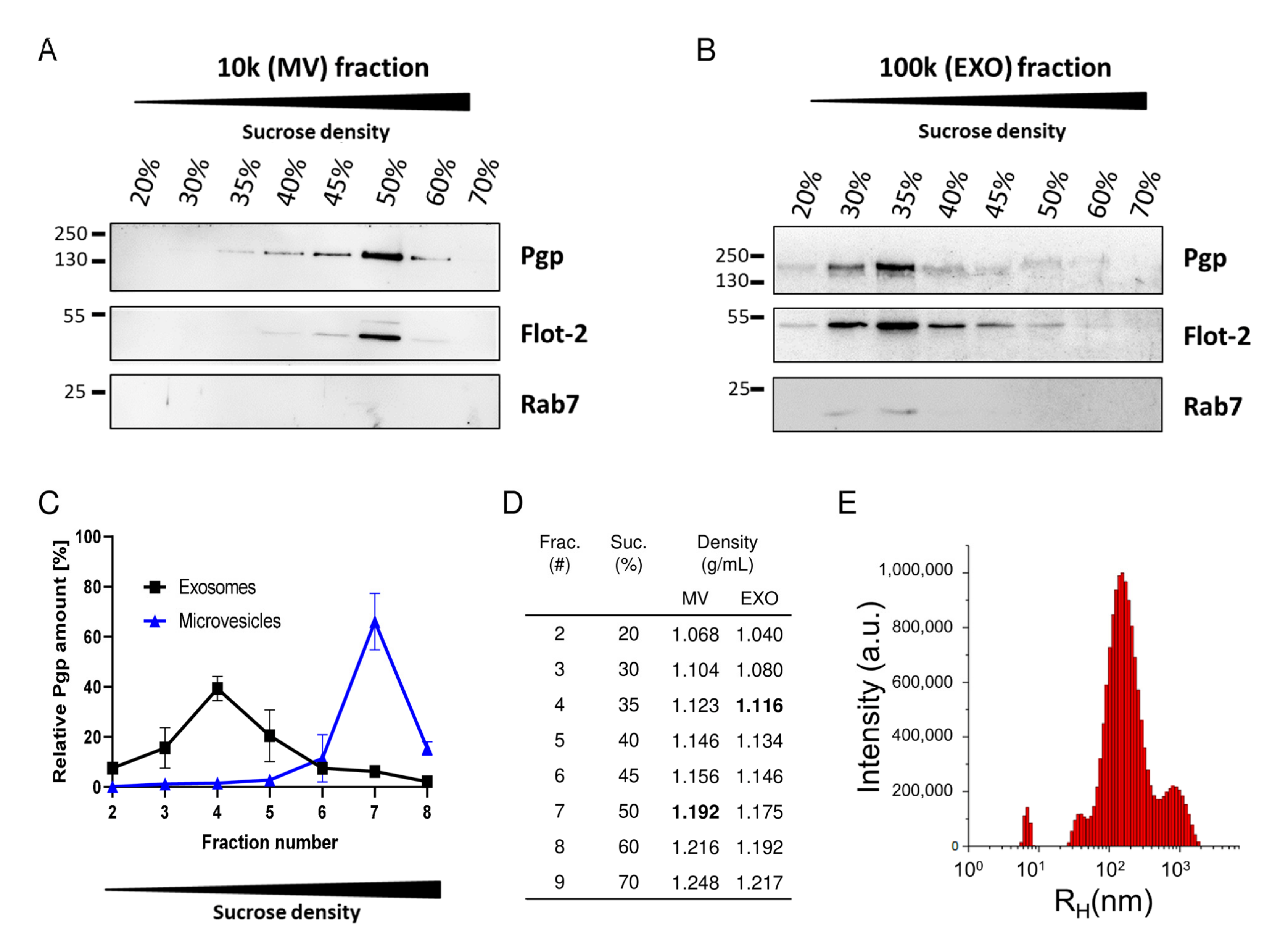{kind=link}
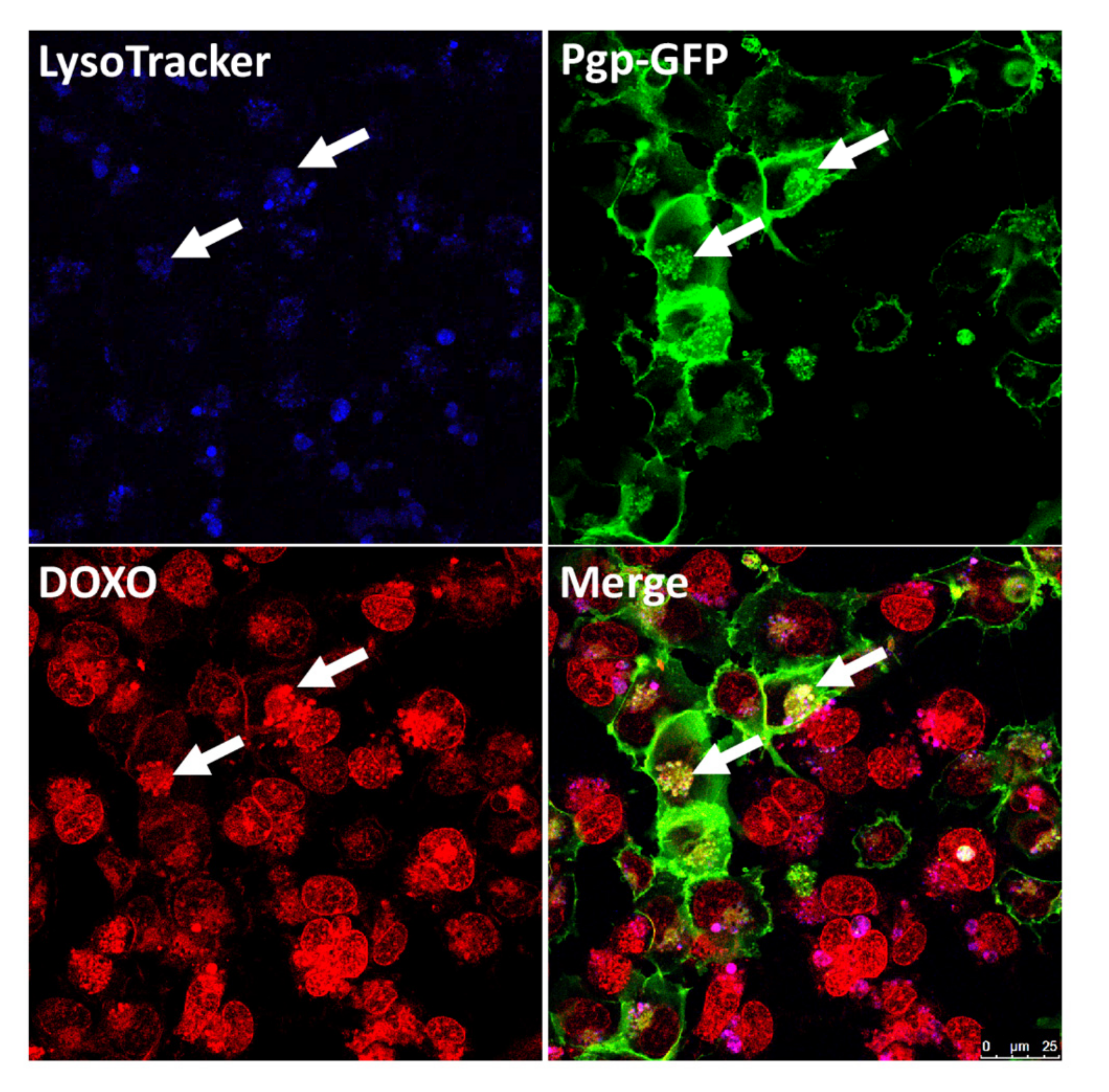{kind=link}
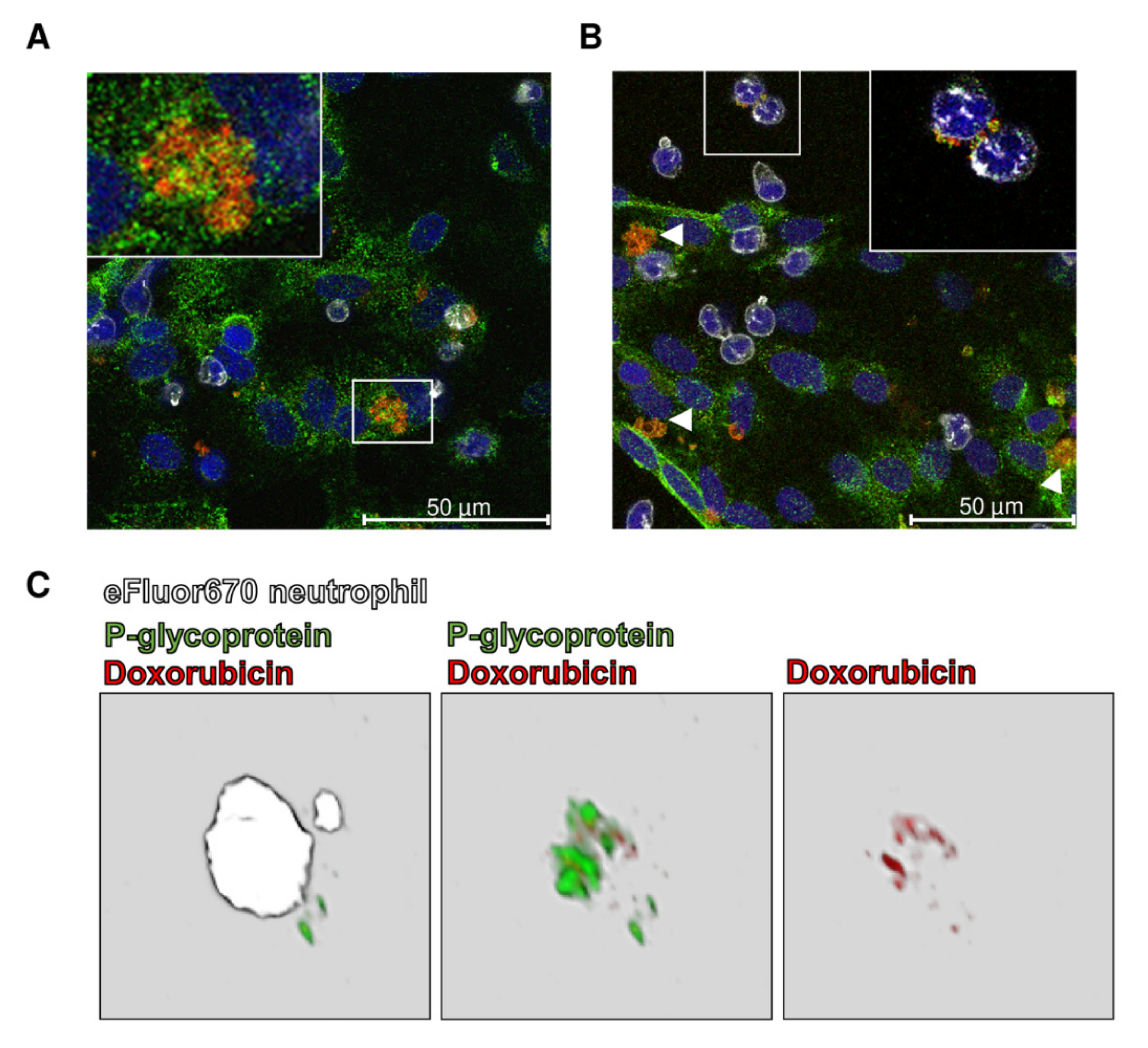{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Regulation of Pgp at the BBB: Conventional Mechanisms
2.1. Transcriptional Activation of Pgp by Ligand-Activated Nuclear Receptors
2.2. Transcriptional Activation of Pgp by Epigenetic Regulation
2.3. Other Mechanisms of Transcriptional Activation of Pgp
2.4. Posttranscriptional Mechanisms in Pgp Adaptation to Xenobiotics
2.5. Posttranslational Mechanisms in Pgp Adaptation to Xenobiotics
2.6. Activation of Membrane-Associated Pgp
2.7. Compensatory Function of BCRP and Other Drug Efflux Transporters at the BBB
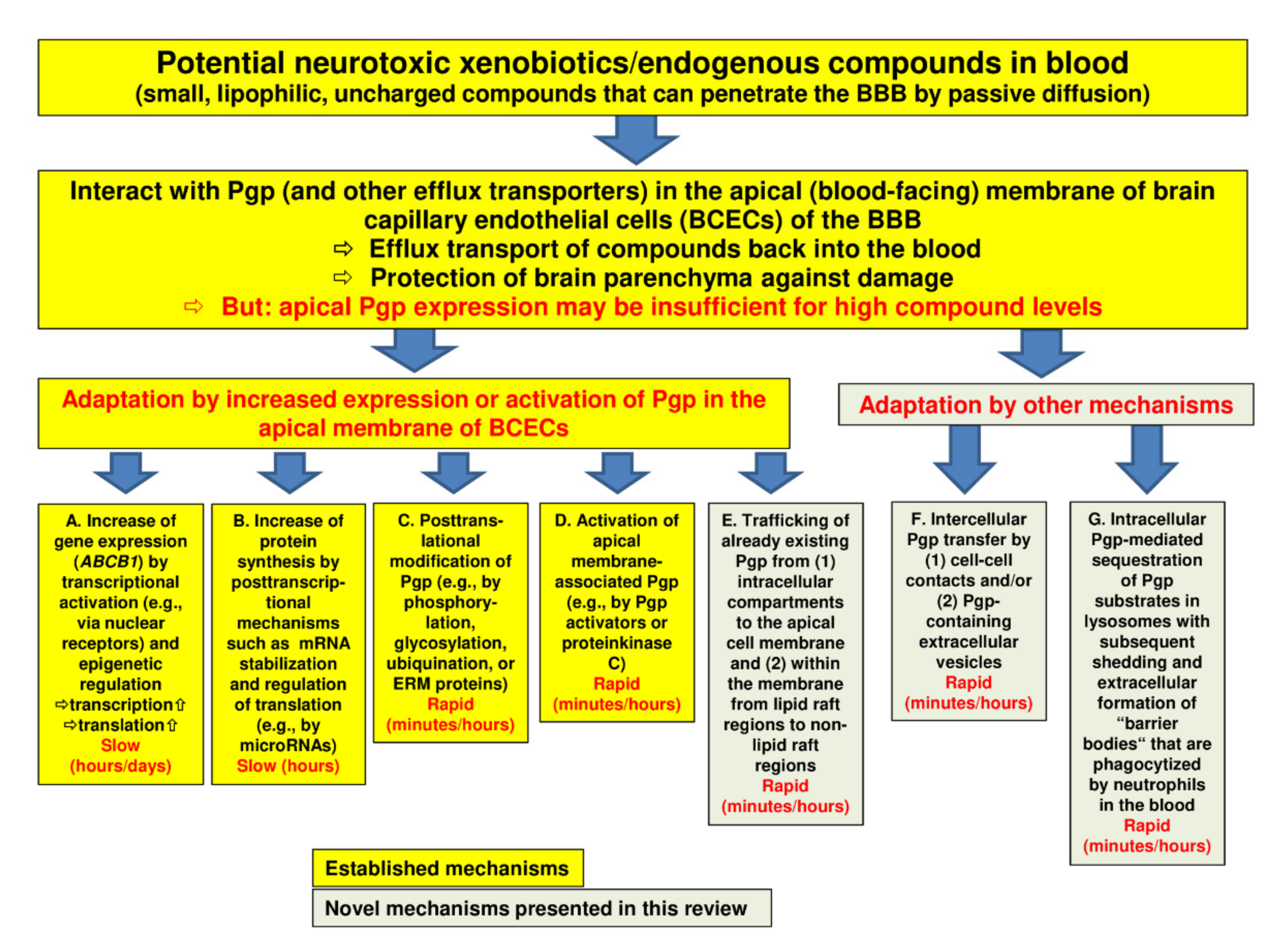
3. Regulation of Pgp at the BBB: Novel Mechanisms
3.1. Intracellular Pgp Trafficking
3.2. Intercellular Pgp Transfer
3.3. Intracellular Sequestration of Pgp Substrates in Lysosomes and Subsequent Disposal
4. Novel Intrinsic Mechanisms of Active Drug Extrusion at the BBB: Potential Targets for Enhancing Drug Delivery to the Brain?
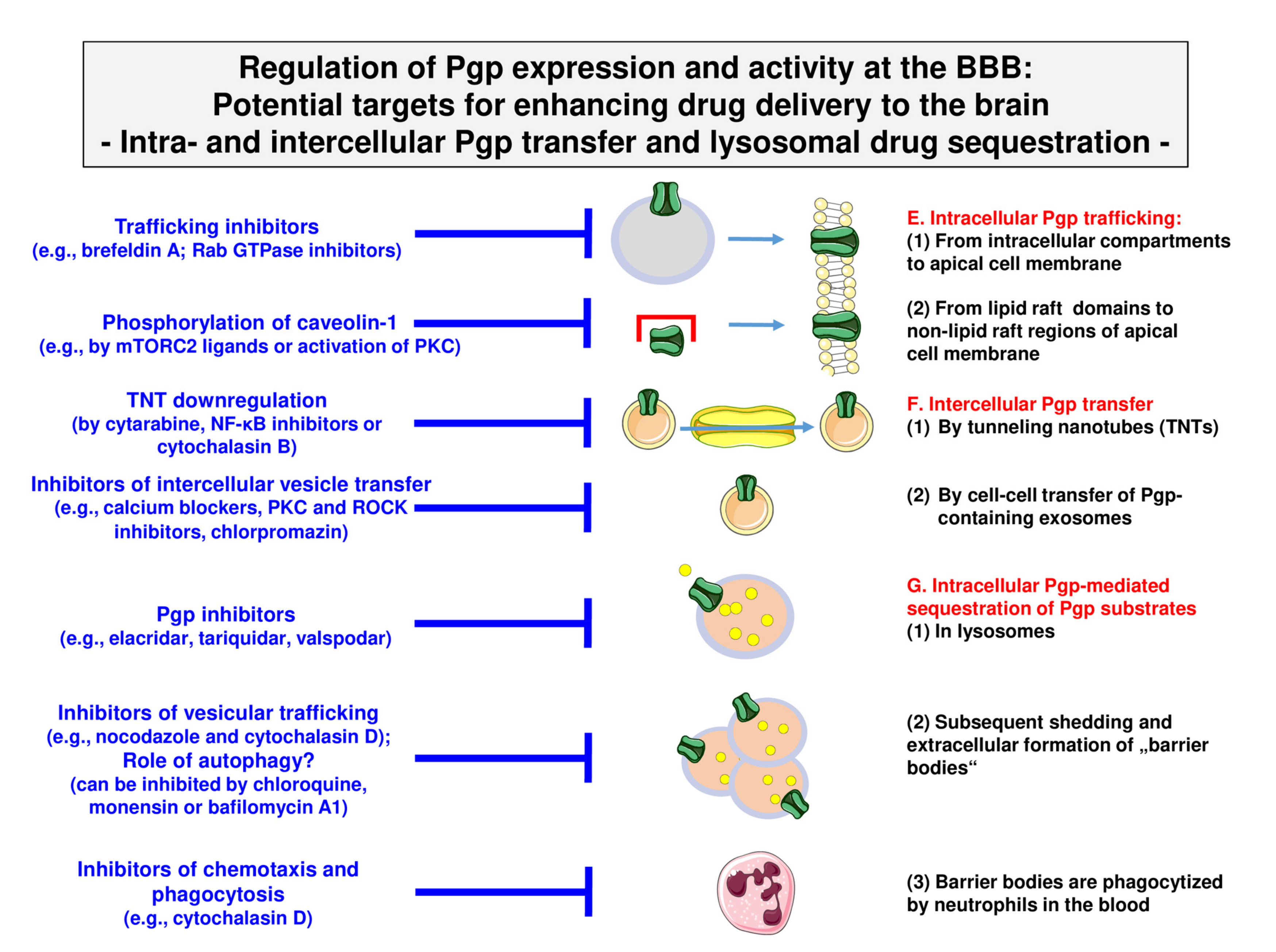
4.1. Targeting Intracellular Pgp Trafficking
4.2. Targeting Intercellular Pgp Transfer
4.3. Targeting Intracellular Sequestration of Pgp Substrates in Lysosomes and Subsequent Disposal
5. Experimental In Vivo Strategies for Translation of In Vitro Findings on Pgp Regulation at the BBB
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Abbott, N.J. Blood-Brain Barrier Structure and Function and the Challenges for CNS Drug Delivery. J. Inherit. Metab. Dis. 2013, 36, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.; Friedman, A. Structural, Molecular and Functional Alterations of the Blood-Brain Barrier during Epileptogenesis and Epilepsy: A Cause, Consequence or Both? Int. J. Mol. Sci. 2020, 21, 591. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and Function of the Blood-Brain Barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Tietz, S.; Engelhardt, B. Brain Barriers: Crosstalk between Complex Tight Junctions and Adherens Junctions. J. Cell Biol. 2015, 209, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.; Potschka, H. Drug Resistance in Brain Diseases and the Role of Drug Efflux Transporters. Nat. Rev. Neurosci. 2005, 6, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the Role of ABC Transporters in Multidrug-Resistant Cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.; Potschka, H.; Sisodiya, S.M.; Vezzani, A. Drug Resistance in Epilepsy: Clinical Impact, Potential Mechanisms, and New Innovative Treatment Options. Pharmacol. Rev. 2020, 72, 606–638. [Google Scholar] [CrossRef]
- Abdullahi, W.; Davis, T.P.; Ronaldson, P.T. Functional Expression of P-Glycoprotein and Organic Anion Transporting Polypeptides at the Blood-Brain Barrier: Understanding Transport Mechanisms for Improved CNS Drug Delivery? AAPS J. 2017, 19, 931–939. [Google Scholar] [CrossRef]
- Hersh, D.S.; Wadajkar, A.S.; Roberts, N.; Perez, J.G.; Connolly, N.P.; Frenkel, V.; Winkles, J.A.; Woodworth, G.F.; Kim, A.J. Evolving Drug Delivery Strategies to Overcome the Blood Brain Barrier. Curr. Pharm. Des. 2016, 22, 1177–1193. [Google Scholar] [CrossRef]
- Waghray, D.; Zhang, Q. Inhibit or Evade Multidrug Resistance P-Glycoprotein in Cancer Treatment. J. Med. Chem. 2018, 61, 5108–5121. [Google Scholar] [CrossRef]
- Schinkel, A.H. P-Glycoprotein, a Gatekeeper in the Blood-Brain Barrier. Adv. Drug Deliv. Rev. 1999, 36, 179–194. [Google Scholar] [CrossRef]
- Miller, D.S.; Bauer, B.; Hartz, A.M.S. Modulation of P-Glycoprotein at the Blood-Brain Barrier: Opportunities to Improve CNS Pharmacotherapy. Pharmacol. Rev. 2008, 60, 196–209. [Google Scholar] [CrossRef] [PubMed]
- Hartz, A.M.; Bauer, B. Regulation of ABC Transporters at the Blood-Brain Barrier: New Targets for CNS Therapy. Mol. Interv. 2010, 10, 293–304. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Miller, D.S. Regulation of P-Glycoprotein and Other ABC Drug Transporters at the Blood-Brain Barrier. Trends Pharmacol. Sci. 2010, 31, 246–254. [Google Scholar] [CrossRef]
- Miller, D.S. Regulation of ABC Transporters Blood-Brain Barrier: The Good, the Bad, and the Ugly. Adv. Cancer Res. 2015, 125, 43–70. [Google Scholar] [PubMed]
- Urquhart, B.L.; Tirona, R.G.; Kim, R.B. Nuclear Receptors and the Regulation of Drug-Metabolizing Enzymes and Drug Transporters: Implications for Interindividual Variability in Response to Drugs. J. Clin. Pharmacol. 2007, 47, 566–578. [Google Scholar] [CrossRef]
- Xu, D.; Huang, S.; Wang, H.; Xie, W. Regulation of Brain Drug Metabolizing Enzymes and Transporters by Nuclear Receptors. Drug Metab. Rev. 2018, 50, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Bauer, B.; Yang, X.; Hartz, A.M.; Olson, E.R.; Zhao, R.; Kalvass, J.C.; Pollack, G.M.; Miller, D.S. In Vivo Activation of Human Pregnane X Receptor Tightens the Blood-Brain Barrier to Methadone Through P-Glycoprotein Up-Regulation. Mol. Pharmacol. 2006, 70, 1212–1219. [Google Scholar] [CrossRef]
- Kim, R.B. Drugs as P-Glycoprotein Substrates, Inhibitors, and Inducers. Drug Metab. Rev. 2002, 34, 47–54. [Google Scholar] [CrossRef]
- Silva, R.; Vilas-Boas, V.; Carmo, H.; Dinis-Oliveira, R.J.; Carvalho, F.; de Lourdes, B.M.; Remiao, F. Modulation of P-Glycoprotein Efflux Pump: Induction and Activation as a Therapeutic Strategy. Pharmacol. Ther. 2015, 149, 1–123. [Google Scholar] [CrossRef]
- Elmeliegy, M.; Vourvahis, M.; Guo, C.; Wang, D.D. Effect of P-Glycoprotein (P-Gp) Inducers on Exposure of P-Gp Substrates: Review of Clinical Drug-Drug Interaction Studies. Clin. Pharmacokinet. 2020, 59, 699–714. [Google Scholar] [CrossRef] [PubMed]
- Ambroziak, K.; Kuteykin-Teplyakov, K.; Luna-Tortós, C.; Al Falah, M.; Fedrowitz, M.; Löscher, W. Exposure to Antiepileptic Drugs Does Not Alter the Functionality of P-Glycoprotein in Brain Capillary Endothelial and Kidney Cell Lines. Eur. J. Pharmacol. 2010, 628, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Alms, D.; Fedrowitz, M.; Römermann, K.; Noack, A.; Löscher, W. Marked Differences in the Effect of Antiepileptic and Cytostatic Drugs on the Functionality of P-Glycoprotein in Human and Rat Brain Capillary Endothelial Cell Lines. Pharm. Res. 2014, 31, 1588–1604. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, K.; Giesy, J.P.; Hu, J. Families of Nuclear Receptors in Vertebrate Models: Characteristic and Comparative Toxicological Perspective. Sci. Rep. 2015, 5, 8554. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.G.; Sikic, B.I. Molecular Pathways: Regulation and Therapeutic Implications of Multidrug Resistance. Clin. Cancer Res. 2012, 18, 1863–1869. [Google Scholar] [CrossRef]
- Eberharter, A.; Becker, P.B. Histone Acetylation: A Switch between Repressive and Permissive Chromatin. Second in Review Series on Chromatin Dynamics. EMBO Rep. 2002, 3, 224–229. [Google Scholar] [CrossRef] [PubMed]
- You, D.; Richardson, J.R.; Aleksunes, L.M. Epigenetic Regulation of Multidrug Resistance Protein 1 and Breast Cancer Resistance Protein Transporters by Histone Deacetylase Inhibition. Drug Metab. Dispos. 2020, 48, 459–480. [Google Scholar] [CrossRef] [PubMed]
- You, D.; Wen, X.; Gorczyca, L.; Morris, A.; Richardson, J.R.; Aleksunes, L.M. Increased MDR1 Transporter Expression in Human Brain Endothelial Cells Through Enhanced Histone Acetylation and Activation of Aryl Hydrocarbon Receptor Signaling. Mol. Neurobiol. 2019, 56, 6986–7002. [Google Scholar] [CrossRef]
- Rothhammer, V.; Quintana, F.J. The Aryl Hydrocarbon Receptor: An Environmental Sensor Integrating Immune Responses in Health and Disease. Nat. Rev. Immunol. 2019, 19, 184–197. [Google Scholar] [CrossRef]
- Wang, X.; Hawkins, B.T.; Miller, D.S. Aryl Hydrocarbon Receptor-Mediated Up-Regulation of ATP-Driven Xenobiotic Efflux Transporters at the Blood-Brain Barrier. FASEB J. 2011, 25, 644–652. [Google Scholar] [CrossRef]
- Phiel, C.J.; Zhang, F.; Huang, E.Y.; Guenther, M.G.; Lazar, M.A.; Klein, P.S. Histone Deacetylase Is a Direct Target of Valproic Acid, a Potent Anticonvulsant, Mood Stabilizer, and Teratogen. J. Biol. Chem. 2001, 276, 36734–36741. [Google Scholar] [CrossRef] [PubMed]
- Noack, A.; Noack, S.; Buettner, M.; Naim, H.Y.; Löscher, W. Intercellular Transfer of P-Glycoprotein in Human Blood-Brain Barrier Endothelial Cells Is Increased by Histone Deacetylase Inhibitors. Sci. Rep. 2016, 6, 29253. [Google Scholar] [CrossRef] [PubMed]
- Hartz, A.M.S.; Rempe, R.G.; Soldner, E.L.B.; Pekcec, A.; Schlichtiger, J.; Kryscio, R.; Bauer, B. Cytosolic Phospholipase A2 Is a Key Regulator of Blood-Brain Barrier Function in Epilepsy. FASEB J. 2019, 33, 14281–14295. [Google Scholar] [CrossRef] [PubMed]
- Potschka, H. Role of CNS Efflux Drug Transporters in Antiepileptic Drug Delivery: Overcoming CNS Efflux Drug Transport. Adv. Drug Deliv. Rev. 2012, 64, 943–952. [Google Scholar] [CrossRef]
- Miller, D.S. ABC Transporter Regulation by Signaling at the Blood-Brain Barrier: Relevance to Pharmacology. Adv. Pharmacol. 2014, 71, 1–24. [Google Scholar]
- Potschka, H. Modulating P-Glycoprotein Regulation: Future Perspectives for Pharmacoresistant Epilepsies? Epilepsia 2010, 51, 1333–1347. [Google Scholar] [CrossRef]
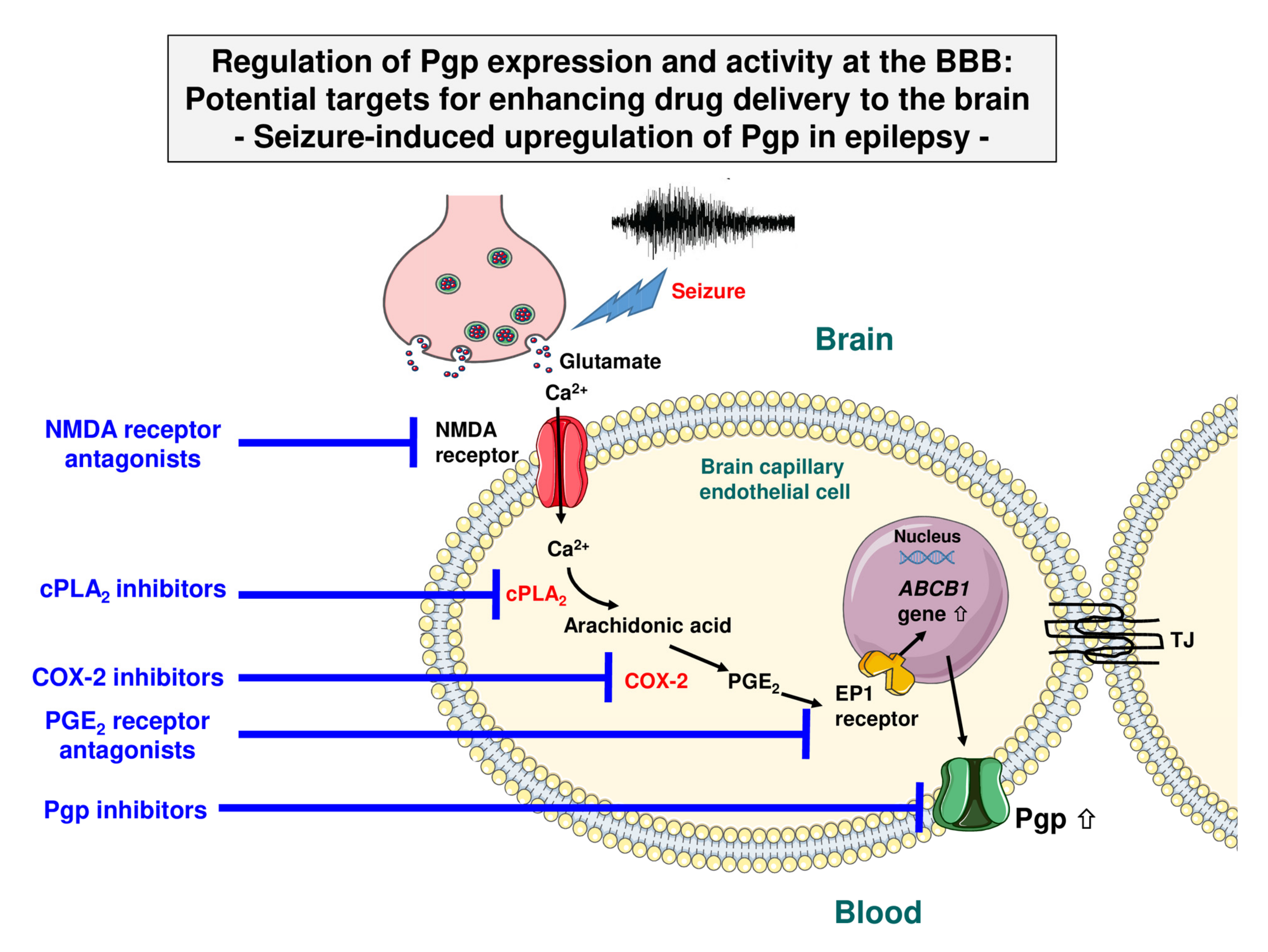
- Bankstahl, J.P.; Hoffmann, K.; Bethmann, K.; Löscher, W. Glutamate Is Critically Involved in Seizure-Induced Overexpression of P-Glycoprotein in the Brain. Neuropharmacology 2008, 54, 1006–1016. [Google Scholar] [CrossRef]
- Bauer, B.; Hartz, A.M.; Pekcec, A.; Toellner, K.; Miller, D.S.; Potschka, H. Seizure-Induced Up-Regulation of P-Glycoprotein at the Blood-Brain Barrier Through Glutamate and Cyclooxygenase-2 Signaling. Mol. Pharmacol. 2008, 73, 1444–1453. [Google Scholar] [CrossRef]
- Zibell, G.; Unkruer, B.; Pekcec, A.; Hartz, A.M.; Bauer, B.; Miller, D.S.; Potschka, H. Prevention of Seizure-Induced Up-Regulation of Endothelial P-Glycoprotein by COX-2 Inhibition. Neuropharmacology 2009, 56, 849–855. [Google Scholar] [CrossRef]
- Schlichtiger, J.; Pekcec, A.; Bartmann, H.; Winter, P.; Fuest, C.; Soerensen, J.; Potschka, H. Celecoxib Treatment Restores Pharmacosensitivity in a Rat Model of Pharmacoresistant Epilepsy. Br. J. Pharmacol. 2010, 160, 1062–1071. [Google Scholar] [CrossRef]
- van Vliet, E.A.; Zibell, G.; Pekcec, A.; Schlichtiger, J.; Edelbroek, P.M.; Holtman, L.; Aronica, E.; Gorter, J.A.; Potschka, H. COX-2 Inhibition Controls P-Glycoprotein Expression and Promotes Brain Delivery of Phenytoin in Chronic Epileptic Rats. Neuropharmacology 2010, 58, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Pekcec, A.; Unkruer, B.; Schlichtiger, J.; Soerensen, J.; Hartz, A.M.; Bauer, B.; van Vliet, E.A.; Gorter, J.A.; Potschka, H. Targeting Prostaglandin E2 EP1 Receptors Prevents Seizure-Associated P-Glycoprotein Up-Regulation. J. Pharmacol. Exp. Ther. 2009, 330, 939–947. [Google Scholar] [CrossRef] [PubMed]
- Brandt, C.; Bethmann, K.; Gastens, A.M.; Löscher, W. The Multidrug Transporter Hypothesis of Drug Resistance in Epilepsy: Proof-of-Principle in a Rat Model of Temporal Lobe Epilepsy. Neurobiol. Dis. 2006, 24, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Avemary, J.; Salvamoser, J.D.; Peraud, A.; Remi, J.; Noachtar, S.; Fricker, G.; Potschka, H. Dynamic Regulation of P-Glycoprotein in Human Brain Capillaries. Mol. Pharm. 2013, 10, 3333–3341. [Google Scholar] [CrossRef]
- Luna-Munguia, H.; Salvamoser, J.D.; Pascher, B.; Pieper, T.; Getzinger, T.; Kudernatsch, M.; Kluger, G.; Potschka, H. Glutamate-Mediated Upregulation of the Multidrug Resistance Protein 2 in Porcine and Human Brain Capillaries. J. Pharmacol. Exp. Ther. 2015, 352, 368–378. [Google Scholar] [CrossRef]
- Salvamoser, J.D.; Avemary, J.; Luna-Munguia, H.; Pascher, B.; Getzinger, T.; Pieper, T.; Kudernatsch, M.; Kluger, G.; Potschka, H. Glutamate-Mediated Down-Regulation of the Multidrug-Resistance Protein BCRP/ABCG2 in Porcine and Human Brain Capillaries. Mol. Pharm. 2015, 12, 2049–2060. [Google Scholar] [CrossRef]
- Hartz, A.M.; Pekcec, A.; Soldner, E.L.; Zhong, Y.; Schlichtiger, J.; Bauer, B. P-Gp Protein Expression and Transport Activity in Rodent Seizure Models and Human Epilepsy. Mol. Pharm. 2017, 14, 999–1011. [Google Scholar] [CrossRef]
- Novel Combination Therapy Treats Neurological Disorders. Available online: https://www.israel21c.org/novel-combination-therapy-treats-neurological-disorders/ (accessed on 3 June 2020).
- Ma, F.; Zhang, X.; Yin, K.J. MicroRNAs in Central Nervous System Diseases: A Prospective Role in Regulating Blood-Brain Barrier Integrity. Exp. Neurol. 2020, 323, 113094. [Google Scholar] [CrossRef]
- Haenisch, S.; Werk, A.N.; Cascorbi, I. MicroRNAs and Their Relevance to ABC Transporters. Br. J. Clin. Pharmacol. 2014, 77, 587–596. [Google Scholar] [CrossRef]
- Xie, Y.; Shao, Y.; Deng, X.; Wang, M.; Chen, Y. MicroRNA-298 Reverses Multidrug Resistance to Antiepileptic Drugs by Suppressing MDR1/P-Gp Expression in Vitro. Front. Neurosci. 2018, 12, 602. [Google Scholar] [CrossRef]
- Ménez, C.; Mselli-Lakhal, L.; Foucaud-Vignault, M.; Balaguer, P.; Alvinerie, M.; Lespine, A. Ivermectin Induces P-Glycoprotein Expression and Function through MRNA Stabilization in Murine Hepatocyte Cell Line. Biochem. Pharmacol. 2012, 83, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Czuba, L.C.; Hillgren, K.M.; Swaan, P.W. Post-Translational Modifications of Transporters. Pharmacol. Ther. 2018, 192, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Yano, K.; Tomono, T.; Ogihara, T. Advances in Studies of P-Glycoprotein and Its Expression Regulators. Biol. Pharm. Bull. 2018, 41, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Kobori, T.; Fujiwara, S.; Miyagi, K.; Harada, S.; Nakamoto, K.; Nakagawa, T.; Takahashi, H.; Narita, M.; Tokuyama, S. Involvement of Moesin in the Development of Morphine Analgesic Tolerance Through P-Glycoprotein at the Blood-Brain Barrier. Drug Metab. Pharmacokinet. 2014, 29, 482–489. [Google Scholar] [CrossRef]
- Hoshi, Y.; Uchida, Y.; Kuroda, T.; Tachikawa, M.; Couraud, P.O.; Suzuki, T.; Terasaki, T. Distinct Roles of Ezrin, Radixin and Moesin in Maintaining the Plasma Membrane Localizations and Functions of Human Blood-Brain Barrier Transporters. J. Cereb. Blood Flow Metab. 2020, 40, 1533–1545. [Google Scholar] [CrossRef]
- Sterz, K.; Möllmann, L.; Jacobs, A.; Baumert, D.; Wiese, M. Activators of P-Glycoprotein: Structure-Activity Relationships and Investigation of Their Mode of Action. ChemMedChem 2009, 4, 1897–1911. [Google Scholar] [CrossRef]
- Martins, E.; Silva, V.; Lemos, A.; Palmeira, A.; Puthongking, P.; Sousa, E.; Rocha-Pereira, C.; Ghanem, C.I.; Carmo, H.; Remiao, F.; et al. Newly Synthesized Oxygenated Xanthones As Potential P-Glycoprotein Activators: In Vitro, Ex Vivo, and In Silico Studies. Molecules 2019, 24, 707. [Google Scholar] [CrossRef]
- Fine, R.L.; Chambers, T.C.; Sachs, C.W. P-Glycoprotein, Multidrug Resistance and Protein Kinase C. Stem Cells 1996, 14, 47–55. [Google Scholar] [CrossRef]
- Ott, M.; Huls, M.; Cornelius, M.G.; Fricker, G. St. John’s Wort Constituents Modulate P-Glycoprotein Transport Activity at the Blood-Brain Barrier. Pharm. Res. 2010, 27, 811–822. [Google Scholar] [CrossRef]
- Uchida, Y.; Ohtsuki, S.; Katsukura, Y.; Ikeda, C.; Suzuki, T.; Kamiie, J.; Terasaki, T. Quantitative Targeted Absolute Proteomics of Human Blood-Brain Barrier Transporters and Receptors. J. Neurochem. 2011, 117, 333–345. [Google Scholar] [CrossRef]
- Agarwal, S.; Hartz, A.M.; Elmquist, W.F.; Bauer, B. Breast Cancer Resistance Protein and P-Glycoprotein in Brain Cancer: Two Gatekeepers Team Up. Curr. Pharm. Des. 2011, 17, 2793–2802. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Römermann, K.; Karch, R.; Wulkersdorfer, B.; Stanek, J.; Philippe, C.; Maier-Salamon, A.; Haslacher, H.; Jungbauer, C.; Wadsak, W.; et al. Pilot PET Study to Assess the Functional Interplay Between ABCB1 and ABCG2 at the Human Blood-Brain Barrier. Clin. Pharmacol. Ther. 2016, 100, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.; Potschka, H. Role of Drug Efflux Transporters in the Brain for Drug Disposition and Treatment of Brain Diseases. Prog. Neurobiol. 2005, 76, 22–76. [Google Scholar] [CrossRef] [PubMed]
- Saidijam, M.; Karimi, D.F.; Sohrabi, S.; Patching, S.G. Efflux Proteins at the Blood-Brain Barrier: Review and Bioinformatics Analysis. Xenobiotica 2018, 48, 506–532. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Arias, I.M. Intracellular Trafficking of P-Glycoprotein. Int. J. Biochem. Cell Biol. 2012, 44, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Kipp, H.; Arias, I.M. Trafficking of Canalicular ABC Transporters in Hepatocytes. Annu. Rev. Physiol. 2002, 64, 595–608. [Google Scholar] [CrossRef]
- Geisslinger, F.; Müller, M.; Vollmar, A.M.; Bartel, K. Targeting Lysosomes in Cancer As Promising Strategy to Overcome Chemoresistance-A Mini Review. Front. Oncol. 2020, 10, 1156. [Google Scholar] [CrossRef] [PubMed]
- Pokharel, D.; Roseblade, A.; Oenarto, V.; Lu, J.F.; Bebawy, M. Proteins Regulating the Intercellular Transfer and Function of P-Glycoprotein in Multidrug-Resistant Cancer. Ecancermedicalscience 2017, 11, 768. [Google Scholar] [CrossRef]
- Jordens, I.; Marsman, M.; Kuijl, C.; Neefjes, J. Rab Proteins, Connecting Transport and Vesicle Fusion. Traffic 2005, 6, 1070–1077. [Google Scholar] [CrossRef]
- Stenmark, H. Rab GTPases as Coordinators of Vesicle Traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef]
- Diekmann, Y.; Seixas, E.; Gouw, M.; Tavares-Cadete, F.; Seabra, M.C.; Pereira-Leal, J.B. Thousands of Rab GTPases for the Cell Biologist. PLoS Comput. Biol. 2011, 7, e1002217. [Google Scholar] [CrossRef] [PubMed]
- Tome, M.E.; Schaefer, C.P.; Jacobs, L.M.; Zhang, Y.; Herndon, J.M.; Matty, F.O.; Davis, T.P. Identification of P-Glycoprotein Co-Fractionating Proteins and Specific Binding Partners in Rat Brain Microvessels. J. Neurochem. 2015, 134, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Ferrándiz-Huertas, C.; Fernández-Carvajal, A.; Ferrer-Montiel, A. Rab4 Interacts With the Human P-Glycoprotein and Modulates Its Surface Expression in Multidrug Resistant K562 Cells. Int. J. Cancer 2011, 128, 192–205. [Google Scholar] [CrossRef] [PubMed]
- Noack, A. University of Veterinary Medicine: Hannover, Germany, Unpublished work. 2015.
- Kipp, H.; Pichetshote, N.; Arias, I.M. Transporters on Demand: Intrahepatic Pools of Canalicular ATP Binding Cassette Transporters in Rat Liver. J. Biol. Chem. 2001, 276, 7218–7224. [Google Scholar] [CrossRef]
- Fu, D.; Bebawy, M.; Kable, E.P.; Roufogalis, B.D. Dynamic and Intracellular Trafficking of P-Glycoprotein-EGFP Fusion Protein: Implications in Multidrug Resistance in Cancer. Int. J. Cancer 2004, 109, 174–181. [Google Scholar] [CrossRef]
- Porcelli, L.; Lemos, C.; Peters, G.J.; Paradiso, A.; Azzariti, A. Intracellular Trafficking of MDR Transporters and Relevance of SNPs. Curr. Top. Med. Chem. 2009, 9, 197–208. [Google Scholar] [CrossRef]
- Davis, T.P.; Sanchez-Covarubias, L.; Tome, M.E. P-Glycoprotein Trafficking As a Therapeutic Target to Optimize CNS Drug Delivery. Adv. Pharmacol. 2014, 71, 25–44. [Google Scholar]
- Hartz, A.M.; Bauer, B.; Fricker, G.; Miller, D.S. Rapid Regulation of P-Glycoprotein at the Blood-Brain Barrier by Endothelin-1. Mol. Pharmacol. 2004, 66, 387–394. [Google Scholar] [CrossRef]
- McCaffrey, G.; Staatz, W.D.; Sanchez-Covarrubias, L.; Finch, J.D.; Demarco, K.; Laracuente, M.L.; Ronaldson, P.T.; Davis, T.P. P-Glycoprotein Trafficking at the Blood-Brain Barrier Altered by Peripheral Inflammatory Hyperalgesia. J. Neurochem. 2012, 122, 962–975. [Google Scholar] [CrossRef]
- Orlowski, S.; Martin, S.; Escargueil, A. P-Glycoprotein and ’Lipid Rafts’: Some Ambiguous Mutual Relationships (Floating on Them, Building Them or Meeting Them by Chance?). Cell. Mol. Life Sci. 2006, 63, 1038–1059. [Google Scholar] [CrossRef]
- Botto, L.; Masserini, M.; Palestini, P. Changes in the Composition of Detergent-Resistant Membrane Domains of Cultured Neurons Following Protein Kinase C Activation. J. Neurosci. Res. 2007, 85, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Reichel, A. Barrier and Transporter Meeting, Bad Herrenalb, Germany. Unpublished work. 2000. [Google Scholar]
- Noack, A.; Noack, S.; Hoffmann, A.; Maalouf, K.; Buettner, M.; Couraud, P.O.; Romero, I.A.; Weksler, B.; Alms, D.; Römermann, K.; et al. Drug-Induced Trafficking of P-Glycoprotein in Human Brain Capillary Endothelial Cells As Demonstrated by Exposure to Mitomycin C. PLoS ONE 2014, 9, e88154. [Google Scholar] [CrossRef] [PubMed]
- Tai, L.M.; Reddy, P.S.; Lopez-Ramirez, M.A.; Davies, H.A.; Male, D.K.; Loughlin, A.J.; Romero, I.A. Polarized P-Glycoprotein Expression by the Immortalised Human Brain Endothelial Cell Line, HCMEC/D3, Restricts Apical-to-Basolateral Permeability to Rhodamine 123. Brain Res. 2009, 1292, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Dauchy, S.; Miller, F.; Couraud, P.O.; Weaver, R.J.; Weksler, B.; Romero, I.A.; Scherrmann, J.M.; De Waziers, I.; Decleves, X. Expression and Transcriptional Regulation of ABC Transporters and Cytochromes P450 in HCMEC/D3 Human Cerebral Microvascular Endothelial Cells. Biochem. Pharmacol. 2009, 77, 897–909. [Google Scholar] [CrossRef] [PubMed]
- Sai, Y.; Nies, A.T.; Arias, I.M. Bile Acid Secretion and Direct Targeting of Mdr1-Green Fluorescent Protein From Golgi to the Canalicular Membrane in Polarized WIF-B Cells. J. Cell Sci. 1999, 112 Pt 24, 4535–4545. [Google Scholar]
- Kipp, H.; Arias, I.M. Intracellular Trafficking and Regulation of Canalicular ATP-Binding Cassette Transporters. Semin. Liver Dis. 2000, 20, 339–351. [Google Scholar] [CrossRef]
- Maitra, R.; Halpin, P.A.; Karlson, K.H.; Page, R.L.; Paik, D.Y.; Leavitt, M.O.; Moyer, B.D.; Stanton, B.A.; Hamilton, J.W. Differential Effects of Mitomycin C and Doxorubicin on P-Glycoprotein Expression. Biochem. J. 2001, 355, 617–624. [Google Scholar] [CrossRef]
- Petriz, J.; Gottesman, M.M.; Aran, J.M. An MDR-EGFP Gene Fusion Allows for Direct Cellular Localization, Function and Stability Assessment of P-Glycoprotein. Curr. Drug Deliv. 2004, 1, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Vogel, M.; Hartmann, T.; Koberle, M.; Treiber, M.; Autenrieth, I.B.; Schumacher, U.K. Rifampicin Induces MDR1 Expression in Candida Albicans. J. Antimicrob. Chemother. 2008, 61, 541–547. [Google Scholar] [CrossRef]
- Zhao, Z.; Liu, W.; Su, Y.; Zhu, J.; Zheng, G.; Luo, Q.; Jin, X. Evaluation of Biodistribution and Safety of Adenovirus Vector Containing MDR1 in Mice. J. Exp. Clin. Cancer Res. 2010, 29, 1–10. [Google Scholar] [CrossRef]
- Ihnat, M.A.; Nervi, A.M.; Anthony, S.P.; Kaltreider, R.C.; Warren, A.J.; Pesce, C.A.; Davis, S.A.; Lariviere, J.P.; Hamilton, J.W. Effects of Mitomycin C and Carboplatin Pretreatment on Multidrug Resistance-Associated P-Glycoprotein Expression and on Subsequent Suppression of Tumor Growth by Doxorubicin and Paclitaxel in Human Metastatic Breast Cancer Xenografted Nude Mice. Oncol. Res. 1999, 11, 303–310. [Google Scholar] [PubMed]
- Huber, O.; Brunner, A.; Maier, P.; Kaufmann, R.; Couraud, P.O.; Cremer, C.; Fricker, G. Localization Microscopy (SPDM) Reveals Clustered Formations of P-Glycoprotein in a Human Blood-Brain Barrier Model. PLoS ONE 2012, 7, e44776. [Google Scholar] [CrossRef] [PubMed]
- Bendayan, R.; Lee, G.; Bendayan, M. Functional Expression and Localization of P-Glycoprotein at the Blood Brain Barrier. Microsc. Res. Tech. 2002, 57, 365–380. [Google Scholar] [CrossRef]
- Szaflarski, W.; Sujka-Kordowska, P.; Januchowski, R.; Wojtowicz, K.; Andrzejewska, M.; Nowicki, M.; Zabel, M. Nuclear Localization of P-Glycoprotein Is Responsible for Protection of the Nucleus from Doxorubicin in the Resistant LoVo Cell Line. Biomed. Pharmacother. 2013, 67, 497–502. [Google Scholar] [CrossRef]
- Tome, M.E.; Herndon, J.M.; Schaefer, C.P.; Jacobs, L.M.; Zhang, Y.; Jarvis, C.K.; Davis, T.P. P-Glycoprotein Traffics From the Nucleus to the Plasma Membrane in Rat Brain Endothelium During Inflammatory Pain. J. Cereb. Blood Flow Metab. 2016, 36, 1913–1928. [Google Scholar] [CrossRef]
- Schaefer, C.P.; Arkwright, N.B.; Jacobs, L.M.; Jarvis, C.K.; Hunn, K.C.; Largent-Milnes, T.M.; Tome, M.E.; Davis, T.P. Chronic Morphine Exposure Potentiates P-Glycoprotein Trafficking From Nuclear Reservoirs in Cortical Rat Brain Microvessels. PLoS ONE 2018, 13, e0192340. [Google Scholar] [CrossRef]
- Ambudkar, S.V.; Sauna, Z.E.; Gottesman, M.M.; Szakacs, G. A Novel Way to Spread Drug Resistance in Tumor Cells: Functional Intercellular Transfer of P-Glycoprotein (ABCB1). Trends Pharmacol. Sci. 2005, 26, 385–387. [Google Scholar] [CrossRef]
- Drab, M.; Stopar, D.; Kralj-Iglic, V.; Iglic, A. Inception Mechanisms of Tunneling Nanotubes. Cells 2019, 8, 626. [Google Scholar] [CrossRef]
- Rustom, A.; Saffrich, R.; Markovic, I.; Walther, P.; Gerdes, H.H. Nanotubular Highways for Intercellular Organelle Transport. Science 2004, 303, 1007–1010. [Google Scholar] [CrossRef]
- Vignais, M.L.; Caicedo, A.; Brondello, J.M.; Jorgensen, C. Cell Connections by Tunneling Nanotubes: Effects of Mitochondrial Trafficking on Target Cell Metabolism, Homeostasis, and Response to Therapy. Stem Cells Int. 2017, 2017, 6917941. [Google Scholar] [CrossRef]
- De Bock, M.; Leybaert, L.; Giaume, C. Connexin Channels at the Glio-Vascular Interface: Gatekeepers of the Brain. Neurochem. Res. 2017, 42, 2519–2536. [Google Scholar] [CrossRef] [PubMed]
- Vader, P.; Breakefield, X.O.; Wood, M.J. Extracellular Vesicles: Emerging Targets for Cancer Therapy. Trends Mol. Med. 2014, 20, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, C.; Cavallini, L.; Spinelli, C.; Yang, J.; Reis-Sobreiro, M.; de Candia, P.; Minciacchi, V.R.; Di Vizio, D. Focus on Extracellular Vesicles: New Frontiers of Cell-to-Cell Communication in Cancer. Int. J. Mol. Sci. 2016, 17, 175. [Google Scholar] [CrossRef] [PubMed]
- Blanc, L.; Vidal, M. New Insights into the Function of Rab GTPases in the Context of Exosomal Secretion. Small GTPases 2018, 9, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Levchenko, A.; Mehta, B.M.; Niu, X.; Kang, G.; Villafania, L.; Way, D.; Polycarpe, D.; Sadelain, M.; Larson, S.M. Intercellular Transfer of P-Glycoprotein Mediates Acquired Multidrug Resistance in Tumor Cells. Proc. Natl. Acad. Sci. USA 2005, 102, 1933–1938. [Google Scholar] [CrossRef]
- Gong, J.; Jaiswal, R.; Mathys, J.M.; Combes, V.; Grau, G.E.; Bebawy, M. Microparticles and Their Emerging Role in Cancer Multidrug Resistance. Cancer Treat. Rev. 2012, 38, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, R.; Luk, F.; Dalla, P.V.; Grau, G.E.; Bebawy, M. Breast Cancer-Derived Microparticles Display Tissue Selectivity in the Transfer of Resistance Proteins to Cells. PLoS ONE 2013, 8, e61515. [Google Scholar] [CrossRef]
- Gong, J.; Jaiswal, R.; Dalla, P.; Luk, F.; Bebawy, M. Microparticles in Cancer: A Review of Recent Developments and the Potential for Clinical Application. Semin. Cell Dev. Biol. 2015, 40, 35–40. [Google Scholar] [CrossRef]
- Taylor, J.; Bebawy, M. Proteins Regulating Microvesicle Biogenesis and Multidrug Resistance in Cancer. Proteomics 2019, 19, e1800165. [Google Scholar] [CrossRef]
- Ni, X.; Li, L.; Pan, G. HDAC Inhibitor-Induced Drug Resistance Involving ATP-Binding Cassette Transporters (Review). Oncol. Lett. 2015, 9, 515–521. [Google Scholar] [CrossRef]
- Löscher, W.; Langer, O. Imaging of P-Glycoprotein Function and Expression to Elucidate Mechanisms of Pharmacoresistance in Epilepsy. Curr. Top. Med. Chem. 2010, 10, 1785–1791. [Google Scholar] [CrossRef] [PubMed]
- Bankstahl, J.P.; Bankstahl, M.; Kuntner, C.; Stanek, J.; Wanek, T.; Meier, M.; Ding, X.Q.; Müller, M.; Langer, O.; Löscher, W. A Novel Positron Emission Tomography Imaging Protocol Identifies Seizure-Induced Regional Overactivity of P-Glycoprotein at the Blood-Brain Barrier. J. Neurosci. 2011, 31, 8803–8811. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, M.; Asselin, M.C.; Liu, J.; Wang, S.; McMahon, A.; Anton-Rodriguez, J.; Walker, M.; Symms, M.; Brown, G.; Hinz, R.; et al. P-Glycoprotein Expression and Function in Patients with Temporal Lobe Epilepsy: A Case-Control Study. Lancet Neurol. 2013, 12, 777–785. [Google Scholar] [CrossRef]
- Angelopoulou, E.; Paudel, Y.N.; Shaikh, M.F.; Piperi, C. Flotillin: A Promising Biomarker for Alzheimer’s Disease. J. Pers. Med. 2020, 10, 20. [Google Scholar] [CrossRef] [PubMed]
- Woodman, P.G.; Futter, C.E. Multivesicular Bodies: Co-Ordinated Progression to Maturity. Curr. Opin. Cell Biol. 2008, 20, 408–414. [Google Scholar] [CrossRef]
- Fu, D. Where Is It and How Does It Get There—Intracellular Localization and Traffic of P-Glycoprotein. Front. Oncol. 2013, 3, 321. [Google Scholar] [CrossRef]
- Katayama, K.; Kapoor, K.; Ohnuma, S.; Patel, A.; Swaim, W.; Ambudkar, I.S.; Ambudkar, S.V. Revealing the Fate of Cell Surface Human P-Glycoprotein (ABCB1): The Lysosomal Degradation Pathway. Biochim. Biophys. Acta 2015, 1853, 2361–2370. [Google Scholar] [CrossRef]
- Preston, J.E.; Joan, A.N.; Begley, D.J. Transcytosis of Macromolecules at the Blood-Brain Barrier. Adv. Pharmacol. 2014, 71, 147–163. [Google Scholar]
- Toth, A.E.; Holst, M.R.; Nielsen, M.S. Vesicular Transport Machinery in Brain Endothelial Cells: What We Know and What We Do Not. Curr. Pharm. Des. 2020, 26, 1405–1416. [Google Scholar] [CrossRef]
- Settembre, C.; Ballabio, A. Lysosomal Adaptation: How the Lysosome Responds to External Cues. Cold Spring Harb. Perspect. Biol. 2014, 6, a016907. [Google Scholar] [CrossRef]
- Larsen, A.K.; Escargueil, A.E.; Skladanowski, A. Resistance Mechanisms Associated With Altered Intracellular Distribution of Anticancer Agents. Pharmacol. Ther. 2000, 85, 217–229. [Google Scholar] [CrossRef]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomes as Mediators of Drug Resistance in Cancer. Drug Resist. Updat. 2016, 24, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Seebacher, N.; Lane, D.J.; Richardson, D.R.; Jansson, P.J. Turning the Gun on Cancer: Utilizing Lysosomal P-Glycoprotein As a New Strategy to Overcome Multi-Drug Resistance. Free Radic. Biol. Med. 2016, 96, 432–445. [Google Scholar] [CrossRef] [PubMed]
- Kubo, Y.; Nakazawa, A.; Akanuma, S.I.; Hosoya, K.I. Blood-to-Retina Transport of Fluorescence-Labeled Verapamil at the Blood-Retinal Barrier. Pharm. Res. 2018, 35, 93. [Google Scholar] [CrossRef]
- Kubo, Y.; Yamada, M.; Konakawa, S.; Akanuma, S.I.; Hosoya, K.I. Uptake Study in Lysosome-Enriched Fraction: Critical Involvement of Lysosomal Trapping in Quinacrine Uptake but Not Fluorescence-Labeled Verapamil Transport at Blood-Retinal Barrier. Pharmaceutics 2020, 12, 747. [Google Scholar] [CrossRef]
- Ferrao, P.; Sincock, P.; Cole, S.; Ashman, L. Intracellular P-Gp Contributes to Functional Drug Efflux and Resistance in Acute Myeloid Leukaemia. Leuk. Res. 2001, 25, 395–405. [Google Scholar] [CrossRef]
- Molinari, A.; Calcabrini, A.; Meschini, S.; Stringaro, A.; Crateri, P.; Toccacieli, L.; Marra, M.; Colone, M.; Cianfriglia, M.; Arancia, G. Subcellular Detection and Localization of the Drug Transporter P-Glycoprotein in Cultured Tumor Cells. Curr. Protein Pept. Sci. 2002, 3, 653–670. [Google Scholar] [CrossRef]
- Rajagopal, A.; Simon, S.M. Subcellular Localization and Activity of Multidrug Resistance Proteins. Mol. Biol. Cell 2003, 14, 3389–3399. [Google Scholar] [CrossRef]
- Yamagishi, T.; Sahni, S.; Sharp, D.M.; Arvind, A.; Jansson, P.J.; Richardson, D.R. P-Glycoprotein Mediates Drug Resistance Via a Novel Mechanism Involving Lysosomal Sequestration. J. Biol. Chem. 2013, 288, 31761–31771. [Google Scholar] [CrossRef]
- Gorden, B.H.; Saha, J.; Khammanivong, A.; Schwartz, G.K.; Dickerson, E.B. Lysosomal Drug Sequestration As a Mechanism of Drug Resistance in Vascular Sarcoma Cells Marked by High CSF-1R Expression. Vasc. Cell 2014, 6, 20. [Google Scholar] [CrossRef]
- Seebacher, N.A.; Lane, D.J.; Jansson, P.J.; Richardson, D.R. Glucose Modulation Induces Lysosome Formation and Increases Lysosomotropic Drug Sequestration Via the P-Glycoprotein Drug Transporter. J. Biol. Chem. 2016, 291, 3796–3820. [Google Scholar] [CrossRef] [PubMed]
- Okura, T.; Hattori, A.; Takano, Y.; Sato, T.; Hammarlund-Udenaes, M.; Terasaki, T.; Deguchi, Y. Involvement of the Pyrilamine Transporter, a Putative Organic Cation Transporter, in Blood-Brain Barrier Transport of Oxycodone. Drug Metab. Dispos. 2008, 36, 2005–2013. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, A.; Okura, T.; Higuchi, K.; Deguchi, Y. Cocktail-Dosing Microdialysis Study to Simultaneously Assess Delivery of Multiple Organic-Cationic Drugs to the Brain. J. Pharm. Sci. 2016, 105, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Kurosawa, T.; Tega, Y.; Higuchi, K.; Yamaguchi, T.; Nakakura, T.; Mochizuki, T.; Kusuhara, H.; Kawabata, K.; Deguchi, Y. Expression and Functional Characterization of Drug Transporters in Brain Microvascular Endothelial Cells Derived From Human Induced Pluripotent Stem Cells. Mol. Pharm. 2018, 15, 5546–5555. [Google Scholar] [CrossRef] [PubMed]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomal Sequestration of Hydrophobic Weak Base Chemotherapeutics Triggers Lysosomal Biogenesis and Lysosome-Dependent Cancer Multidrug Resistance. Oncotarget 2015, 6, 1143–1156. [Google Scholar] [CrossRef]
- Toth, A.E.; Siupka, P.; TJ, P.A.; Venö, S.T.; Thomsen, L.B.; Moos, T.; Lohi, H.T.; Madsen, P.; Lykke-Hartmann, K.; Nielsen, M.S. The Endo-Lysosomal System of Brain Endothelial Cells Is Influenced by Astrocytes In Vitro. Mol. Neurobiol. 2018, 55, 8522–8537. [Google Scholar] [CrossRef]
- Toth, A.E.; Nielsen, S.S.E.; Tomaka, W.; Abbott, N.J.; Nielsen, M.S. The Endo-Lysosomal System of BEnd.3 and HCMEC/D3 Brain Endothelial Cells. Fluids Barriers CNS 2019, 16, 14. [Google Scholar] [CrossRef]
- Villasenor, R.; Ozmen, L.; Messaddeq, N.; Grüninger, F.; Loetscher, H.; Keller, A.; Betsholtz, C.; Freskgård, P.O.; Collin, L. Trafficking of Endogenous Immunoglobulins by Endothelial Cells at the Blood-Brain Barrier. Sci. Rep. 2016, 6, 25658. [Google Scholar] [CrossRef]
- Gali, C.C.; Fanaee-Danesh, E.; Zandl-Lang, M.; Albrecher, N.M.; Tam-Amersdorfer, C.; Stracke, A.; Sachdev, V.; Reichmann, F.; Sun, Y.; Avdili, A.; et al. Amyloid-Beta Impairs Insulin Signaling by Accelerating Autophagy-Lysosomal Degradation of LRP-1 and IR-β in Blood-Brain Barrier Endothelial Cells in Vitro and in 3XTg-AD Mice. Mol. Cell Neurosci. 2019, 99, 103390. [Google Scholar] [CrossRef]
- Noack, A.; Gericke, B.; von Köckritz-Blickwede, M.; Menze, A.; Noack, S.; Gerhauser, I.; Osten, F.; Naim, H.Y.; Löscher, W. A Novel Mechanism of Drug Extrusion by Brain Endothelial Cells Via Lysosomal Drug Trapping and Disposal by Neutrophils. Proc. Natl. Acad. Sci. USA 2018, 115, E9590–E9599. [Google Scholar] [CrossRef]
- Helms, H.C.; Abbott, N.J.; Burek, M.; Cecchelli, R.; Couraud, P.O.; Deli, M.A.; Forster, C.; Galla, H.J.; Romero, I.A.; Shusta, E.V.; et al. In Vitro Models of the Blood-Brain Barrier: An Overview of Commonly Used Brain Endothelial Cell Culture Models and Guidelines for Their Use. J. Cereb. Blood Flow Metab. 2016, 36, 862–890. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Dufour, J.M. Cell Lines: Valuable Tools or Useless Artifacts. Spermatogenesis 2012, 2, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Lebedeva, I.V.; Pande, P.; Patton, W.F. Sensitive and Specific Fluorescent Probes for Functional Analysis of the Three Major Types of Mammalian ABC Transporters. PLoS ONE 2011, 6, e22429. [Google Scholar] [CrossRef] [PubMed]
- Fader, C.M.; Colombo, M.I. Autophagy and Multivesicular Bodies: Two Closely Related Partners. Cell Death. Differ. 2009, 16, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Cocucci, E.; Meldolesi, J. Ectosomes. Curr. Biol. 2011, 21, R940–R941. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Tam, A.; Santi, S.A.; Parissenti, A.M. Role of Autophagy and Lysosomal Drug Sequestration in Acquired Resistance to Doxorubicin in MCF-7 Cells. BMC Cancer 2016, 16, 762. [Google Scholar] [CrossRef]
- Chen, C.; Lu, L.; Yan, S.; Yi, H.; Yao, H.; Wu, D.; He, G.; Tao, X.; Deng, X. Autophagy and Doxorubicin Resistance in Cancer. Anticancer Drugs 2018, 29, 1–9. [Google Scholar] [CrossRef]
- Kannan, P.; Brimacombe, K.R.; Kreisl, W.C.; Liow, J.S.; Zoghbi, S.S.; Telu, S.; Zhang, Y.; Pike, V.W.; Halldin, C.; Gottesman, M.M.; et al. Lysosomal Trapping of a Radiolabeled Substrate of P-Glycoprotein As a Mechanism for Signal Amplification in PET. Proc. Natl. Acad. Sci. USA 2011, 108, 2593–2598. [Google Scholar] [CrossRef]
- Stefan, S.M.; Jansson, P.J.; Kalinowski, D.S.; Anjum, R.; Dharmasivam, M.; Richardson, D.R. The Growing Evidence for Targeting P-Glycoprotein in Lysosomes to Overcome Resistance. Future Med. Chem. 2020, 12, 473–477. [Google Scholar] [CrossRef]
- Saftig, P.; Klumperman, J. Lysosome Biogenesis and Lysosomal Membrane Proteins: Trafficking Meets Function. Nat. Rev. Mol. Cell Biol. 2009, 10, 623–635. [Google Scholar] [CrossRef]
- Chapel, A.; Kieffer-Jaquinod, S.; Sagné, C.; Verdon, Q.; Ivaldi, C.; Mellal, M.; Thirion, J.; Jadot, M.; Bruley, C.; Garin, J.; et al. An Extended Proteome Map of the Lysosomal Membrane Reveals Novel Potential Transporters. Mol. Cell Proteom. 2013, 12, 1572–1588. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.S.; Cannon, R.E. Signaling Pathways That Regulate Basal ABC Transporter Activity at the Blood-Brain Barrier. Curr. Pharm. Des. 2014, 20, 1463–1471. [Google Scholar] [CrossRef] [PubMed]
- Gil-Martins, E.; Barbosa, D.J.; Silva, V.; Remiao, F.; Silva, R. Dysfunction of ABC Transporters at the Blood-Brain Barrier: Role in Neurological Disorders. Pharmacol. Ther. 2020, 213, 107554. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Wang, J.; Wang, X.; Liu, F.; Jiang, B.; Zhang, Y. Targeting Rabs As a Novel Therapeutic Strategy for Cancer Therapy. Drug Discov. Today 2017, 22, 1139–1147. [Google Scholar] [CrossRef]
- Hong, L.; Guo, Y.; BasuRay, S.; Agola, J.O.; Romero, E.; Simpson, D.S.; Schroeder, C.E.; Simons, P.; Waller, A.; Garcia, M.; et al. A Pan-GTPase Inhibitor as a Molecular Probe. PLoS ONE 2015, 10, e0134317. [Google Scholar] [CrossRef]
- Luker, G.D.; Pica, C.M.; Kumar, A.S.; Covey, D.F.; Piwnica-Worms, D. Effects of Cholesterol and Enantiomeric Cholesterol on P-Glycoprotein Localization and Function in Low-Density Membrane Domains. Biochemistry 2000, 39, 7651–7661. [Google Scholar] [CrossRef]
- Rothnie, A.; Theron, D.; Soceneantu, L.; Martin, C.; Traikia, M.; Berridge, G.; Higgins, C.F.; Devaux, P.F.; Callaghan, R. The Importance of Cholesterol in Maintenance of P-Glycoprotein Activity and Its Membrane Perturbing Influence. Eur. Biophys. J. 2001, 30, 430–442. [Google Scholar] [CrossRef]
- Barakat, S.; Demeule, M.; Pilorget, A.; Régina, A.; Gingras, D.; Baggetto, L.G.; Béliveau, R. Modulation of P-Glycoprotein Function by Caveolin-1 Phosphorylation. J. Neurochem. 2007, 101, 1–8. [Google Scholar] [CrossRef]
- Hau, A.M.; Gupta, S.; Leivo, M.Z.; Nakashima, K.; Macias, J.; Zhou, W.; Hodge, A.; Wulfkuhle, J.; Conkright, B.; Bhuvaneshwar, K.; et al. Dynamic Regulation of Caveolin-1 Phosphorylation and Caveolae Formation by Mammalian Target of Rapamycin Complex 2 in Bladder Cancer Cells. Am. J. Pathol. 2019, 189, 1846–1862. [Google Scholar] [CrossRef]
- Oka, N.; Yamamoto, M.; Schwencke, C.; Kawabe, J.; Ebina, T.; Ohno, S.; Couet, J.; Lisanti, M.P.; Ishikawa, Y. Caveolin Interaction With Protein Kinase C. Isoenzyme-Dependent Regulation of Kinase Activity by the Caveolin Scaffolding Domain Peptide. J. Biol. Chem. 1997, 272, 33416–33421. [Google Scholar] [CrossRef]
- Omsland, M.; Bruserud, Ã.; Gjertsen, B.T.; Andresen, V. Tunneling Nanotube (TNT) Formation Is Downregulated by Cytarabine and NF-κB Inhibition in Acute Myeloid Leukemia (AML). Oncotarget 2017, 8, 7946–7963. [Google Scholar] [CrossRef] [PubMed]
- Matejka, N.; Reindl, J. Perspectives of Cellular Communication through Tunneling Nanotubes in Cancer Cells and the Connection to Radiation Effects. Radiat. Oncol. 2019, 14, 218. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, R.; Sedger, L.M. Intercellular Vesicular Transfer by Exosomes, Microparticles and Oncosomes—Implications for Cancer Biology and Treatments. Front. Oncol. 2019, 9, 125. [Google Scholar] [CrossRef] [PubMed]
- Zegers, M.M.; Zaal, K.J.; van IJzendoorn, S.C.; Klappe, K.; Hoekstra, D. Actin Filaments and Microtubules Are Involved in Different Membrane Traffic Pathways That Transport Sphingolipids to the Apical Surface of Polarized HepG2 Cells. Mol. Biol. Cell 1998, 9, 1939–1949. [Google Scholar] [CrossRef]
- Kocaturk, N.M.; Akkoc, Y.; Kig, C.; Bayraktar, O.; Gozuacik, D.; Kutlu, O. Autophagy As a Molecular Target for Cancer Treatment. Eur. J. Pharm. Sci. 2019, 134, 116–137. [Google Scholar] [CrossRef]
- Muller, W.A. Mechanisms of Leukocyte Transendothelial Migration. Annu. Rev. Pathol. 2011, 6, 323–344. [Google Scholar] [CrossRef]
- Schimmel, L.; Heemskerk, N.; van Buul, J.D. Leukocyte Transendothelial Migration: A Local Affair. Small GTPases 2017, 8, 1–15. [Google Scholar] [CrossRef]
- Ploppa, A.; Ayers, D.M.; Johannes, T.; Unertl, K.E.; Durieux, M.E. The Inhibition of Human Neutrophil Phagocytosis and Oxidative Burst by Tricyclic Antidepressants. Anesth. Analg. 2008, 107, 1229–1235. [Google Scholar] [CrossRef]
- Esmann, L.; Idel, C.; Sarkar, A.; Hellberg, L.; Behnen, M.; Möller, S.; van Zandbergen, G.; Klinger, M.; Köhl, J.; Bussmeyer, U.; et al. Phagocytosis of Apoptotic Cells by Neutrophil Granulocytes: Diminished Proinflammatory Neutrophil Functions in the Presence of Apoptotic Cells. J. Immunol. 2010, 184, 391–400. [Google Scholar] [CrossRef]
- Bankstahl, J.P.; Kuntner, C.; Abrahim, A.; Karch, R.; Stanek, J.; Wanek, T.; Wadsak, W.; Kletter, K.; Müller, M.; Löscher, W.; et al. Tariquidar-Induced P-Glycoprotein Inhibition at the Rat Blood-Brain Barrier Studied With (R)-11C-Verapamil and PET. J. Nucl. Med. 2008, 49, 1328–1335. [Google Scholar] [CrossRef][Green Version]
- Dörner, B.; Kuntner, C.; Bankstahl, J.P.; Bankstahl, M.; Stanek, J.; Wanek, T.; Stundner, G.; Mairinger, S.; Löscher, W.; Muller, M.; et al. Synthesis and Small-Animal Positron Emission Tomography Evaluation of [11C]-Elacridar As a Radiotracer to Assess the Distribution of P-Glycoprotein at the Blood-Brain Barrier. J. Med. Chem. 2009, 52, 6073–6082. [Google Scholar] [CrossRef] [PubMed]
- Wagner, C.C.; Bauer, M.; Karch, R.; Feurstein, T.; Kopp, S.; Chiba, P.; Kletter, K.; Löscher, W.; Muller, M.; Zeitlinger, M.; et al. A Pilot Study to Assess the Efficacy of Tariquidar to Inhibit P-Glycoprotein at the Human Blood-Brain Barrier with (R)-11C-Verapamil and PET. J. Nucl. Med. 2009, 50, 1954–1961. [Google Scholar] [CrossRef] [PubMed]
- Bauer, F.; Kuntner, C.; Bankstahl, J.P.; Wanek, T.; Bankstahl, M.; Stanek, J.; Mairinger, S.; Dörner, B.; Löscher, W.; Müller, M.; et al. Synthesis and in Vivo Evaluation of [11C]Tariquidar, a Positron Emission Tomography Radiotracer Based on a Third-Generation P-Glycoprotein Inhibitor. Bioorg. Med. Chem. 2010, 18, 5489–5497. [Google Scholar] [CrossRef] [PubMed]
- Kuntner, C.; Bankstahl, J.P.; Bankstahl, M.; Stanek, J.; Wanek, T.; Stundner, G.; Karch, R.; Brauner, R.; Meier, M.; Ding, X.; et al. Dose-Response Assessment of Tariquidar and Elacridar and Regional Quantification of P-Glycoprotein Inhibition at the Rat Blood-Brain Barrier Using (R)-[(11)C]Verapamil PET. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 942–953. [Google Scholar] [CrossRef]
- Mairinger, S.; Langer, O.; Kuntner, C.; Wanek, T.; Bankstahl, J.P.; Bankstahl, M.; Stanek, J.; Dörner, B.; Bauer, F.; Baumgartner, C.; et al. Synthesis and in Vivo Evaluation of the Putative Breast Cancer Resistance Protein Inhibitor [11C]Methyl 4-((4-(2-(6,7-Dimethoxy-1,2,3,4-Tetrahydroisoquinolin-2-Yl)Ethyl)Phenyl)Amino-Car Bonyl)-2-(Quinoline-2-Carbonylamino)Benzoate. Nucl. Med. Biol. 2010, 37, 637–644. [Google Scholar] [CrossRef][Green Version]
- Bauer, M.; Karch, R.; Neumann, F.; Wagner, C.C.; Kletter, K.; Müller, M.; Löscher, W.; Zeitlinger, M.; Langer, O. Assessment of Regional Differences in Tariquidar-Induced P-Glycoprotein Modulation at the Human Blood-Brain Barrier. J Cereb. Blood Flow Metab. 2010, 30, 510–515. [Google Scholar] [CrossRef]
- Dörner, B.; Kuntner, C.; Bankstahl, J.P.; Wanek, T.; Bankstahl, M.; Stanek, J.; Mullauer, J.; Bauer, F.; Mairinger, S.; Löscher, W.; et al. Radiosynthesis and in Vivo Evaluation of 1-[18F]Fluoroelacridar As a Positron Emission Tomography Tracer for P-Glycoprotein and Breast Cancer Resistance Protein. Bioorg. Med. Chem. 2011, 19, 2190–2198. [Google Scholar] [CrossRef]
- Bauer, M.; Zeitlinger, M.; Karch, R.; Matzneller, P.; Stanek, J.; Jager, W.; Bohmdorfer, M.; Wadsak, W.; Mitterhauser, M.; Bankstahl, J.P.; et al. Pgp-Mediated Interaction Between (R)-[11C]Verapamil and Tariquidar at the Human Blood-Brain Barrier: A Comparison With Rat Data. Clin. Pharmacol. Ther. 2012, 91, 227–233. [Google Scholar] [CrossRef]
- Wanek, T.; Kuntner, C.; Bankstahl, J.P.; Mairinger, S.; Bankstahl, M.; Stanek, J.; Sauberer, M.; Filip, T.; Erker, T.; Müller, M.; et al. A Novel PET Protocol for Visualization of Breast Cancer Resistance Protein Function at the Blood-Brain Barrier. J. Cereb. Blood Flow Metab. 2012, 32, 2002–2011. [Google Scholar] [CrossRef]
- Bankstahl, J.P.; Bankstahl, M.; Römermann, K.; Wanek, T.; Stanek, J.; Windhorst, A.D.; Fedrowitz, M.; Erker, T.; Müller, M.; Löscher, W.; et al. Tariquidar and Elacridar Are Dose-Dependently Transported by P-Glycoprotein and Bcrp at the Blood-Brain Barrier: A Small-Animal Positron Emission Tomography and in Vitro Study. Drug Metab. Dispos. 2013, 41, 754–762. [Google Scholar] [CrossRef]
- Müllauer, J.; Karch, R.; Bankstahl, J.P.; Bankstahl, M.; Stanek, J.; Wanek, T.; Mairinger, S.; Müller, M.; Löscher, W.; Langer, O.; et al. Assessment of Cerebral P-Glycoprotein Expression and Function With PET by Combined [11C]Inhibitor and [11C]Substrate Scans in Rats. Nucl. Med. Biol. 2013, 40, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Römermann, K.; Wanek, T.; Bankstahl, M.; Bankstahl, J.P.; Fedrowitz, M.; Müller, M.; Löscher, W.; Kuntner, C.; Langer, O. (R)-[(11)C]Verapamil Is Selectively Transported by Murine and Human P-Glycoprotein at the Blood-Brain Barrier, and Not by MRP1 and BCRP. Nucl. Med. Biol. 2013, 40, 873–878. [Google Scholar] [CrossRef] [PubMed]
- Wanek, T.; Römermann, K.; Mairinger, S.; Stanek, J.; Sauberer, M.; Filip, T.; Traxl, A.; Kuntner, C.; Pahnke, J.; Bauer, F.; et al. Factors Governing P-Glycoprotein-Mediated Drug-Drug Interactions at the Blood-Brain Barrier Measured With Positron Emission Tomography. Mol. Pharm. 2015, 12, 3214–3225. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.; Potschka, H. Role of Multidrug Transporters in Pharmacoresistance to Antiepileptic Drugs. J. Pharmacol. Exp. Ther. 2002, 301, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Potschka, H.; Fedrowitz, M.; Löscher, W. P-Glycoprotein and Multidrug Resistance-Associated Protein Are Involved in the Regulation of Extracellular Levels of the Major Antiepileptic Drug Carbamazepine in the Brain. Neuroreport 2001, 12, 3557–3560. [Google Scholar] [CrossRef]
- Potschka, H.; Löscher, W. Multidrug Resistance-Associated Protein Is Involved in the Regulation of Extracellular Levels of Phenytoin in the Brain. Neuroreport 2001, 12, 2387–2389. [Google Scholar] [CrossRef]
- Potschka, H.; Fedrowitz, M.; Löscher, W. P-Glycoprotein-Mediated Efflux of Phenobarbital, Lamotrigine, and Felbamate at the Blood-Brain Barrier: Evidence from Microdialysis Experiments in Rats. Neurosci. Lett. 2002, 327, 173–176. [Google Scholar] [CrossRef]
- Potschka, H.; Baltes, S.; Löscher, W. Inhibition of Multidrug Transporters by Verapamil or Probenecid Does Not Alter Blood-Brain Barrier Penetration of Levetiracetam in Rats. Epilepsy Res. 2004, 58, 85–91. [Google Scholar] [CrossRef]
- Baltes, S.; Fedrowitz, M.; Luna-Tortós, C.; Potschka, H.; Löscher, W. Valproic Acid Is Not a Substrate for P-Glycoprotein or Multidrug Resistance Proteins 1 and 2 in a Number of in Vitro and in Vivo Transport Assays. J. Pharmacol. Exp. Ther. 2007, 320, 331–343. [Google Scholar] [CrossRef]
- Sziráki, I.; Erdö, F.; Beéry, E.; Molnár, P.M.; Fazakas, C.; Wilhelm, I.; Makai, I.; Kis, E.; Herédi-Szabó, K.; Abonyi, T.; et al. Quinidine As an ABCB1 Probe for Testing Drug Interactions at the Blood-Brain Barrier: An in Vitro in Vivo Correlation Study. J. Biomol. Screen. 2011, 16, 886–894. [Google Scholar] [CrossRef]
- Sziráki, I.; Erdö, F.; Trampus, P.; Sike, M.; Molnár, P.M.; Rajnai, Z.; Molnár, J.; Wilhelm, I.; Fazakas, C.; Kis, E.; et al. The Use of Microdialysis Techniques in Mice to Study P-Gp Function at the Blood-Brain Barrier. J. Biomol. Screen. 2013, 18, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Burgio, D.E.; Gosland, M.P.; McNamara, P.J. Effects of P-Glycoprotein Modulators on Etoposide Elimination and Central Nervous System Distribution. J. Pharmacol. Exp. Ther. 1998, 287, 911–917. [Google Scholar] [PubMed]
- Römermann, K.; Fedrowitz, M.; Hampel, P.; Kaczmarek, E.; Töllner, K.; Erker, T.; Sweet, D.H.; Löscher, W. Multiple Blood-Brain Barrier Transport Mechanisms Limit Bumetanide Accumulation, and Therapeutic Potential, in the Mammalian Brain. Neuropharmacology 2017, 117, 182–194. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.; Baena, V.; Ge, S.; Jiang, X.; Jellison, E.R.; Kiprono, T.; Agalliu, D.; Pachter, J.S. Appearance of Claudin-5(+) Leukocytes in the Central Nervous System During Neuroinflammation: A Novel Role for Endothelial-Derived Extracellular Vesicles. J. Neuroinflammation 2016, 13, 292. [Google Scholar] [CrossRef] [PubMed]
- Callaghan, R.; Luk, F.; Bebawy, M. Inhibition of the Multidrug Resistance P-Glycoprotein: Time for a Change of Strategy? Drug Metab. Dispos. 2014, 42, 623–631. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Löscher, W.; Gericke, B. Novel Intrinsic Mechanisms of Active Drug Extrusion at the Blood-Brain Barrier: Potential Targets for Enhancing Drug Delivery to the Brain? Pharmaceutics 2020, 12, 966. https://doi.org/10.3390/pharmaceutics12100966
Löscher W, Gericke B. Novel Intrinsic Mechanisms of Active Drug Extrusion at the Blood-Brain Barrier: Potential Targets for Enhancing Drug Delivery to the Brain? Pharmaceutics. 2020; 12(10):966. https://doi.org/10.3390/pharmaceutics12100966
Chicago/Turabian StyleLöscher, Wolfgang, and Birthe Gericke. 2020. "Novel Intrinsic Mechanisms of Active Drug Extrusion at the Blood-Brain Barrier: Potential Targets for Enhancing Drug Delivery to the Brain?" Pharmaceutics 12, no. 10: 966. https://doi.org/10.3390/pharmaceutics12100966
APA StyleLöscher, W., & Gericke, B. (2020). Novel Intrinsic Mechanisms of Active Drug Extrusion at the Blood-Brain Barrier: Potential Targets for Enhancing Drug Delivery to the Brain? Pharmaceutics, 12(10), 966. https://doi.org/10.3390/pharmaceutics12100966