Therapeutic Interventions into Innate Immune Diseases by Means of Aptamers




Abstract
1. Introduction
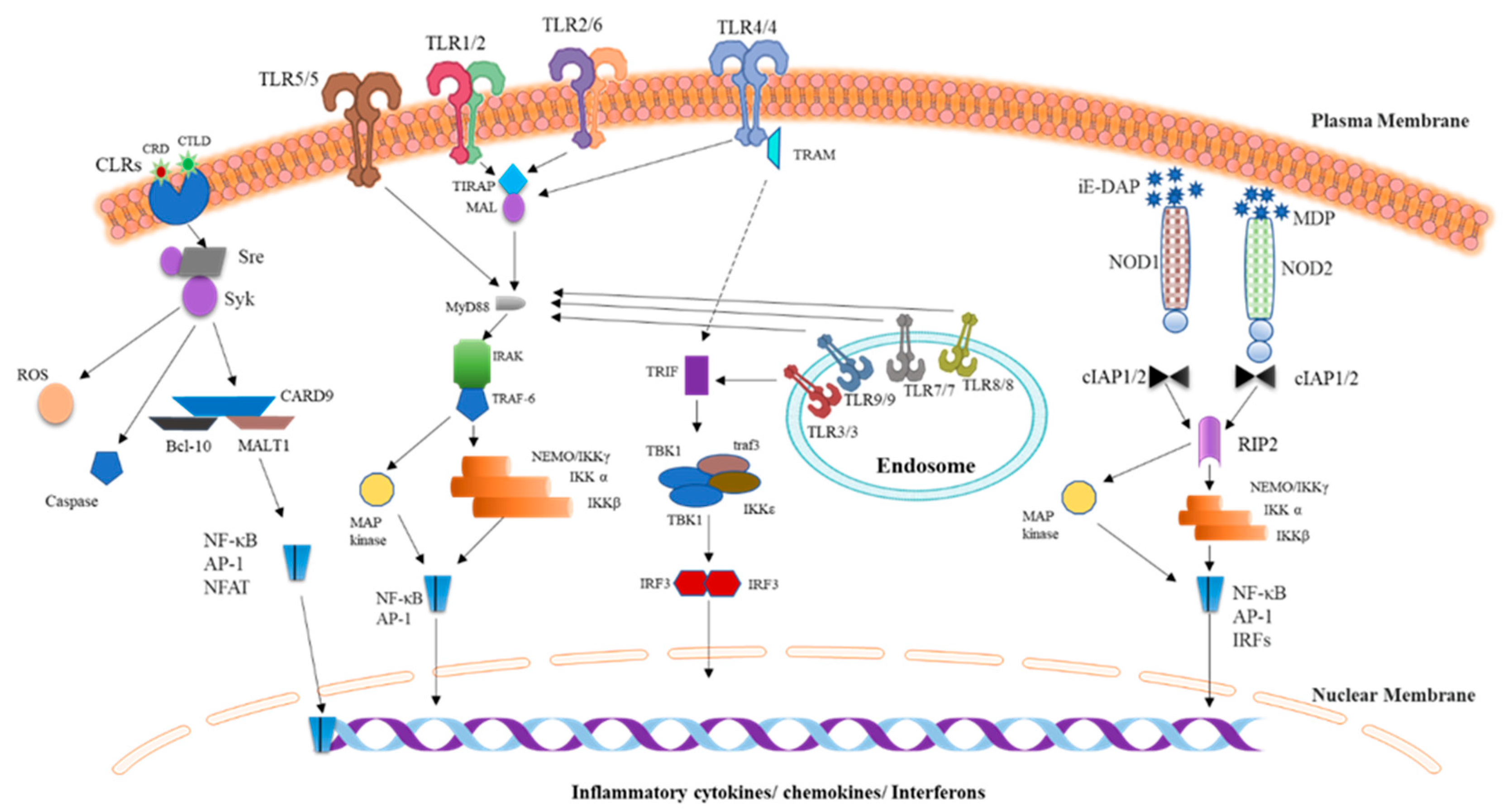
2. Representative Components of Innate Immunity
2.1. TLRs
2.2. Costimulatory Molecules/Receptors
2.3. Complement System
2.3.1. Classical Pathway
2.3.2. Lectin Pathway
2.3.3. Alternative Pathway
2.4. Interleukins
2.4.1. IL-8 Signaling Pathway
2.4.2. IL-10 Signaling Pathway
2.4.3. IL-6 Signaling Pathway
3. Aptamers
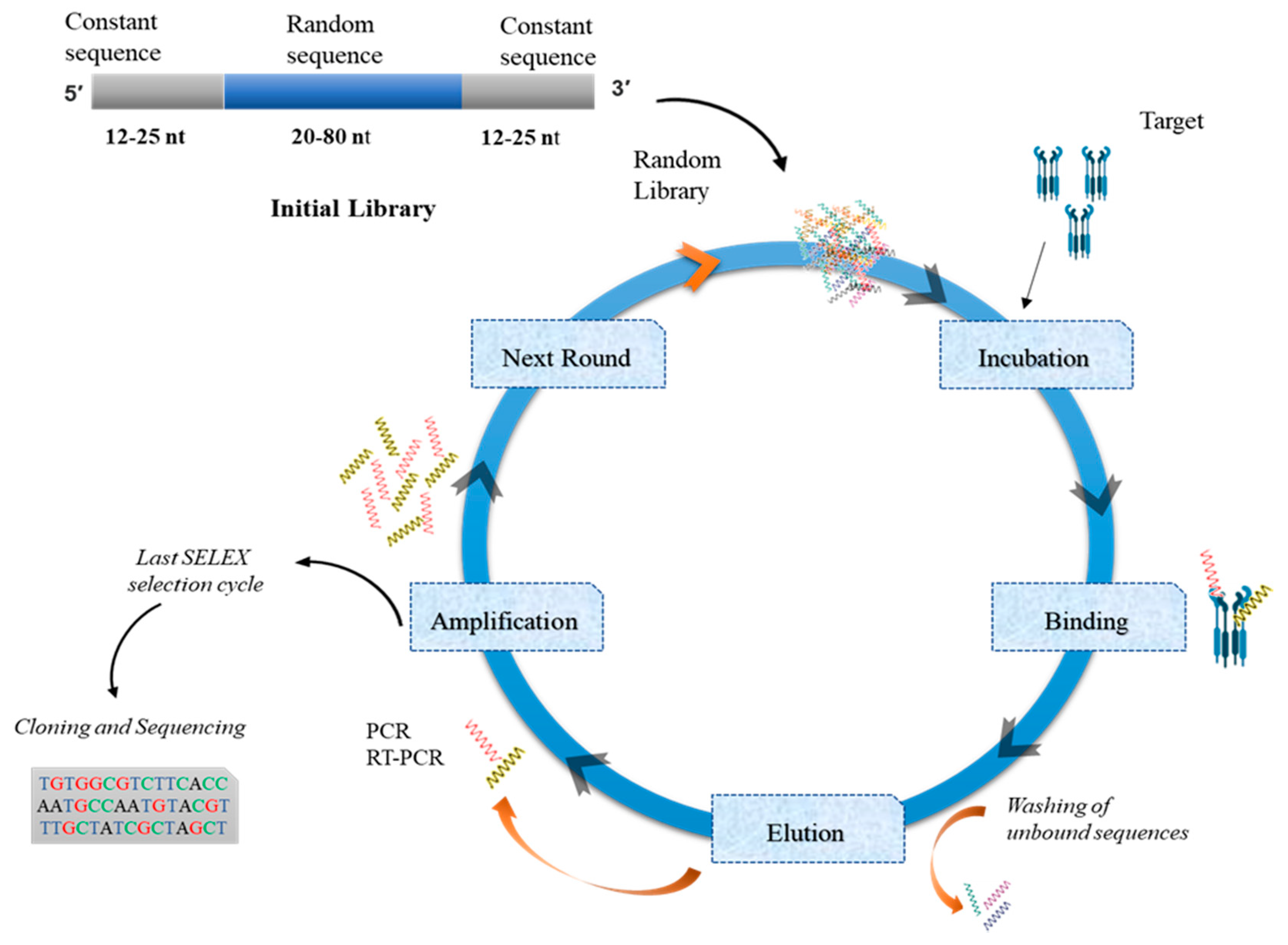
4. Aptamer Production
4.1. SELEX
4.1.1. Randomization
4.1.2. Random-Region Length
4.1.3. Aptamer Library Chemistry
4.1.4. Techniques to Monitor SELEX Rounds
4.1.5. Limitations of SELEX
5. Metabolism and Pharmacokinetics of Therapeutic Aptamers
5.1. Nuclease-Driven Degradation
5.2. Modified Nucleotide Bases
5.2.1. In-SELEX Technique
5.2.2. Post-SELEX Techniques
5.3. Renal Filtration
5.4. Toxicity
6. Aptamers as Therapeutic Agents
6.1. Aptamers Related to Cell Surface Proteins
6.2. Costimulatory Receptors
6.3. Costimulatory Molecules
6.4. Aptamers for the Complement System
6.5. Aptamers for Cytokines
7. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AP-1 | Activated Protein 1 |
| BCR | B-Cell Receptor |
| BMP | Bone Morphogenetic Protein |
| C5 | Compliment component 5 |
| CD | Cluster of Differentiation |
| CLR | C-type Lectin Receptor |
| CTLA-4 | Cytotoxic T Lymphocyte Associated Antigen 4 |
| DAMP | Damage-associated Molecular Pattern |
| C5 | Compliment component 5 |
| CD | Cluster of Differentiation |
| CLR | C-type Lectin Receptor |
| CTLA-4 | Cytotoxic T Lymphocyte Associated Antigen 4 |
| DAMP | Damage-associated Molecular Pattern |
| DNA | Deoxy Ribonucleic Acid |
| ELONA | Enzyme Linked Oligonucleotide Assay |
| EPO | Erythropoietin |
| FACS | Fluorescent Activated Cell Sorting |
| GM-CSF | Granulocyte Macrophage Colony Stimulating Factor |
| HPLC | High Performance Liquid Chromatography |
| IFN-γ | Interferon γ |
| Ig | Immunoglobulin |
| IKK | Inhibitor of κB kinase |
| IL | Interleukin |
| IRAK | Interleukin Receptor Associated Kinase |
| IRF | Interferon Response Factor |
| IκB | Inhibitor of κB |
| KD | Dissociation Constant |
| LNA | Locked Nucleic Acid |
| LRR | Leucine-Rich Repeat |
| MAC | Membrane Attack Complex |
| MAL | MyD88 Adaptor Like |
| MAP | Mitogen-Activated Protein |
| MAPK | Mitogen-Activated Protein Kinase |
| MASP | MBL-Associated Serine Protease |
| MBL | Mannose-Binding Lectin |
| MHC | Major Histocompatibility Complex |
| MyD88 | Myeloid Differentiation primary response 88 |
| NEMO | NF-κB Essential Modulator |
| NF-κB | Nuclear Factor-κB |
| NLR: | NOD-Like Receptor |
| NMR | Nuclear Magnetic Resonance |
| NOD | Nucleotide Oligomerization Domain |
| PAMP | Pathogen Associated Molecular Pattern |
| PCR | Polymerase Chain Reaction |
| PDL1 | Programmed Death-Ligand 1 |
| PEG | Polyethylene Glycol |
| PRR | Pattern Recognition Receptor |
| qPCR | Quantitative PCR |
| RIP | Receptor Interacting Protein |
| RNA | Ribonucleic Acid |
| RT | Reverse Transcription |
| SELEX | Systematic Evolution of Ligands by Exponential Enrichment |
| siRNA | Small Interfering RNA |
| STAT3: | Signal Transducer and Activator of Transcription 3 |
| TAB | TAK1-Binding Protein |
| TAK1 | Transforming Growth Factor β–Activated Kinase 1 |
| TBK1 | TANK-Binding Kinase 1 |
| TIR | Toll/Interleukin-1 Receptor |
| TIRAP | TIR domain-containing Adaptor Protein |
| TLR | Toll-Like Receptor |
| TNFR | Tumor Necrosis Factor Receptor |
| TNFα | Tumor Necrosis Factor α |
| TRAF | Tumor Necrosis Factor Receptor Associated Factor |
| TRAM | TRIF-Related Adaptor Molecule |
| TRIF | TIR-domain-containing Adapter-inducing Interferon-β |
| C1q | Complement Component 1 |
| CXCL12 | C-X-C Motif Chemokine Ligand 12 |
| IL-8 | Interleukin-8 |
| vWF | von Willebrand Factor |
References
- Immunology: Mucosal and body surface defences. Choice Rev. Online 2012, 49, 49. [CrossRef]
- Litman, G.W.; Cannon, J.P.; Dishaw, L.J. Reconstructing immune phylogeny: New perspectives. Nat. Rev. Immunol. 2005, 5, 866–879. [Google Scholar] [CrossRef] [PubMed]
- Firestein, G.S.; Budd, R.C.; Gabriel, S.; McInnes, I.B.; O’Dell, J.R. Kelley and Firestein’s Textbook of Rheumatology, 10th ed.; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Castiglioni, A.; Canti, V.; Rovere-Querini, P.; Manfredi, A.A. High-mobility group box 1 (HMGB1) as a master regulator of innate immunity. Cell Tissue Res. 2010, 343, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, H.; Kono, H.; Patel, Z.; Rock, K.L. Evaluation of the Contribution of Multiple DAMPs and DAMP Receptors in Cell Death-Induced Sterile Inflammatory Responses. PLoS ONE 2014, 9, e104741. [Google Scholar] [CrossRef] [PubMed]
- Janeway, C.A.; Medzhitov, R. Innateimmunerecognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Blasius, A.L.; Beutler, B.A. Intracellular Toll-like Receptors. Immunity 2010, 32, 305–315. [Google Scholar] [CrossRef]
- Medzhitov, R. Recognition of microorganisms and activation of the immune response. Nat. Cell Biol. 2007, 449, 819–826. [Google Scholar] [CrossRef]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen recognition in the innate immune response. Biochem. J. 2009, 420, 1–16. [Google Scholar] [CrossRef]
- Koenderman, L.; Buurman, W.A.; Daha, M.R. The innate immune response. Immunol. Lett. 2014, 162, 95–102. [Google Scholar] [CrossRef]
- Tosi, M.F. Innate immune responses to infection. J. Allergy Clin. Immunol. 2005, 116, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B. ScienceDirect—Molecular Immunology: Innate immunity: An overview. Mol. Immunol. 2004, 40, 845–859. [Google Scholar] [CrossRef] [PubMed]
- Lemaitre, B.; Nicolas, E.; Michaut, L.; Reichhart, J.-M.; Hoffmann, J.A. The Dorsoventral Regulatory Gene Cassette spätzle/Toll/cactus Controls the Potent Antifungal Response in Drosophila Adults. Cell 1996, 86, 973–983. [Google Scholar] [CrossRef]
- Kaisho, T.; Akira, S. Toll-like receptors and their signaling mechanism in innate immunity. Acta Odontol. Scand. 2001, 59, 124–130. [Google Scholar] [CrossRef]
- Li, K.; Qu, S.; Chen, X.; Wu, Q.; Shi, M. Promising Targets for Cancer Immunotherapy: TLRs, RLRs, and STING-Mediated Innate Immune Pathways. Int. J. Mol. Sci. 2017, 18, 404. [Google Scholar] [CrossRef]
- Brodsky, I.E.; Medzhitov, R. Targeting of immune signalling networks by bacterial pathogens. Nat. Cell Biol. 2009, 11, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef]
- Kagan, J.C.; Su, T.; Horng, T.; Chow, A.; Akira, S.; Medzhitov, R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-β. Nat. Immunol. 2008, 9, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Tabeta, K.; Hoebe, K.; Janssen, E.M.; Du, X.; Georgel, P.; Crozat, K.; Mudd, S.; Mann, N.; Sovath, S.; Goode, J.; et al. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat. Immunol. 2006, 7, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-M.; Brinkmann, M.M.; Paquet, M.-E.; Ploegh, H.L. UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nat. Cell Biol. 2008, 452, 234–238. [Google Scholar] [CrossRef]
- Brinkmann, M.M.; Spooner, E.; Hoebe, K.; Beutler, B.; Ploegh, H.L.; Kim, Y.-M. The interaction between the ER membrane protein UNC93B and TLR3, 7, and 9 is crucial for TLR signaling. J. Cell Biol. 2007, 177, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, A.H. Mechanisms of costimulation. Immunol. Rev. 2009, 229, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Elgueta, R.; Benson, M.J.; De Vries, V.C.; Wasiuk, A.; Guo, Y.; Noelle, R.J. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol. Rev. 2009, 229, 152–172. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; Ledbetter, J.A.; Linsley, P.S.; Thompson, C.B. Role of the CD28 receptor in T-cell activation. Immunol. Today 1990, 11, 211–216. [Google Scholar] [CrossRef]
- Brunet, J.-F.; Denizot, F.; Luciani, M.-F.; Roux-Dosseto, M.; Suzan, M.; Mattei, M.-G.; Golstein, P. A new member of the immunoglobulin superfamily—CTLA-4. Nat. Cell Biol. 1987, 328, 267–270. [Google Scholar] [CrossRef]
- Croft, M.; So, T.; Duan, W.; Soroosh, P. The significance of OX40 and OX40L to T-cell biology and immune disease. Immunol. Rev. 2009, 229, 173–191. [Google Scholar] [CrossRef]
- Epstein, F.H.; Schifferli, J.A.; Ng, Y.C.; Peters, D.K. The Role of Complement and Its Receptor in the Elimination of Immune Complexes. New Engl. J. Med. 1986, 315, 488–495. [Google Scholar] [CrossRef]
- Walport, M.J. Complement. New Engl. J. Med. 2001, 344, 1140–1144. [Google Scholar] [CrossRef]
- Arumugam, T.V.; Magnus, T.; Woodruff, T.M.; Proctor, L.M.; Shiels, I.A.; Taylor, S.M. Complement mediators in ischemia–reperfusion injury. Clin. Chim. Acta 2006, 374, 33–45. [Google Scholar] [CrossRef]
- Barrington, R.; Zhang, M.; Fischer, M.; Carroll, M.C. The role of complement in inflammation and adaptive immunity. Immunol. Rev. 2001, 180, 5–15. [Google Scholar] [CrossRef]
- Gasque, P. Complement: A unique innate immune sensor for danger signals. Mol. Immunol. 2004, 41, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Ehrnthaller, C.; Ignatius, A.; Gebhard, F.; Huber-Lang, M. New insights of an old defense system: Structure, function, and clinical relevance of the complement system. Mol. Med. 2011, 17, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Garred, P.; Honoré, C.; Ma, Y.J.; Munthe-Fog, L.; Hummelshøj, T. MBL2, FCN1, FCN2 and FCN3—The genes behind the initiation of the lectin pathway of complement. Mol. Immunol. 2009, 46, 2737–2744. [Google Scholar] [CrossRef]
- Noris, M.; Remuzzi, G. Overview of Complement Activation and Regulation. Semin. Nephrol. 2013, 33, 479–492. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.O.; Skoog, F.; Okumura, F.S.; Von Saltza, M.H.; Strong, F.M. Structure and Synthesis of Kinetin1. J. Am. Chem. Soc. 1955, 77, 2662–2663. [Google Scholar] [CrossRef]
- Fields, J.K.; Günther, S.; Sundberg, E.J. Structural Basis of IL-1 Family Cytokine Signaling. Front. Immunol. 2019, 10, 1412. [Google Scholar] [CrossRef]
- Tsutsui, H.; Cai, X.; Hayashi, S. Interleukin-1 Family Cytokines in Liver Diseases. Mediat. Inflamm. 2015, 2015, 1–19. [Google Scholar] [CrossRef]
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2017, 281, 8–27. [Google Scholar] [CrossRef]
- Mantovani, A.; Dinarello, C.A.; Molgora, M.; Garlanda, C. Interleukin-1 and Related Cytokines in the Regulation of Inflammation and Immunity. Immunity 2019, 50, 778–795. [Google Scholar] [CrossRef]
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The Interleukin-1 Family: Back to the Future. Immunity 2013, 39, 1003–1018. [Google Scholar] [CrossRef]
- Schröder, J.M.; Sticherling, H.H.; Henneicke, W.C.; Preissner, E. Christophers, IL-1 alpha or tumor necrosis factor-alpha stimulate release of three NAP-1/IL-8-related neutrophil chemotactic proteins in human dermal fibroblasts. J. Immunol. 1991, 74, 60–67. [Google Scholar]
- Gimbrone, M.; Obin, M.; Brock, A.; Luis, E.; Hass, P.; Hebert, C.; Yip, Y.; Leung, D.; Lowe, D.; Kohr, W.; et al. Endothelial interleukin-8: A novel inhibitor of leukocyte-endothelial interactions. Science 1989, 246, 1601–1603. [Google Scholar] [CrossRef]
- Bazzoni, F.; Cassatella, M.A.; Rossi, F.; Ceska, M.; Dewald, B.; Baggiolini, M. Phagocytosing neutrophils produce and release high amounts of the neutrophil-activating peptide 1/interleukin 8. J. Exp. Med. 1991, 173, 771–774. [Google Scholar] [CrossRef]
- Gregory, H.; Young, J.; Schröder, J.-M.; Mrowietz, U.; Christophers, E. Structure determination of a human lymphocyte derived neutrophil activating peptide (LYNAP). Biochem. Biophys. Res. Commun. 1988, 151, 883–890. [Google Scholar] [CrossRef]
- Koch, A.E.; Polverini, P.J.; Kunkel, S.L.; Harlow, L.A.; DiPietro, L.A.; Elner, V.M.; Elner, S.G.; Strieter, R.M. Interleukin-8 as a macrophage-derived mediator of angiogenesis. Science 1992, 258, 1798–1801. [Google Scholar] [CrossRef]
- Kobilka, B.K. G protein coupled receptor structure and activation. Biochim. Biophys. Acta (BBA) Biomembr. 2007, 1768, 794–807. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, S.K.; Murphy, P.M. The CXC Chemokines Growth-regulated Oncogene (GRO) α, GROβ, GROγ, Neutrophil-activating Peptide-2, and Epithelial Cell-derived Neutrophil-activating Peptide-78 Are Potent Agonists for the Type B, but Not the Type A, Human Interleukin-8 Receptor. J. Boil. Chem. 1996, 271, 20545–20550. [Google Scholar] [CrossRef]
- Park, S.H.; Casagrande, F.; Cho, L.; Albrecht, L.; Opella, S.J. Interactions of Interleukin-8 with the Human Chemokine Receptor CXCR1 in Phospholipid Bilayers by NMR Spectroscopy. J. Mol. Biol. 2011, 414, 194–203. [Google Scholar] [CrossRef][Green Version]
- Stillie, R.; Farooq, S.M.; Gordon, J.R.; Stadnyk, A.W. The functional significance behind expressing two IL-8 receptor types on PMN. J. Leukoc. Biol. 2009, 86, 529–543. [Google Scholar] [CrossRef] [PubMed]
- Cohenhillel, E.; Yron, I.; Meshel, T.; Soria, G.; Attal, H.; Benbaruch, A. CXCL8-induced FAK phosphorylation via CXCR1 and CXCR2: Cytoskeleton- and integrin-related mechanisms converge with FAK regulatory pathways in a receptor-specific manner. Cytokine 2006, 33, 1–16. [Google Scholar] [CrossRef]
- Donnelly, R.P.; Sheikh, F.; Kotenko, S.V.; Dickensheets, H. The expanded family of class II cytokines that share the IL-10 receptor-2 (IL-10R2) chain. J. Leukoc. Biol. 2004, 76, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J. The JAK-STAT Signaling Pathway: Input and Output Integration. J. Immunol. 2007, 178, 2623–2629. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Murray, P.J. Cytokine Signaling Modules in Inflammatory Responses. Immunity 2008, 28, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Denys, A.; Udalova, I.A.; Smith, C.; Williams, L.M.; Ciesielski, C.J.; Campbell, J.; Andrews, C.; Kwiatkowski, D.; Foxwell, B.M.J. Evidence for a Dual Mechanism for IL-10 Suppression of TNF-α Production That Does Not Involve Inhibition of p38 Mitogen-Activated Protein Kinase or NF-κB in Primary Human Macrophages. J. Immunol. 2002, 168, 4837–4845. [Google Scholar] [CrossRef] [PubMed]
- Williams, L.M.; Ricchetti, G.; Sarma, U.; Smallie, T.; Foxwell, B.M.J. Interleukin-10 suppression of myeloid cell activation - a continuing puzzle. Immunology 2004, 113, 281–292. [Google Scholar] [CrossRef]
- Mihara, M.; Hashizume, M.; Yoshida, H.; Suzuki, M.; Shiina, M. IL-6/IL-6 receptor system and its role in physiological and pathological conditions. Clin. Sci. 2011, 122, 143–159. [Google Scholar] [CrossRef]
- Kishimoto, T. IL-6: From its discovery to clinical applications. Int. Immunol. 2010, 22, 347–352. [Google Scholar] [CrossRef]
- Fischer, P.; Hilfiker-Kleiner, D. Survival pathways in hypertrophy and heart failure: The gp130-STAT axis. Basic Res. Cardiol. 2007, 102, 393–411. [Google Scholar] [CrossRef]
- Tau, G.; Rothman, P. Biologic functions of the IFN-gamma receptors. Allergy 1999, 54, 1233–1251. [Google Scholar] [CrossRef]
- Parameswaran, N.; Patial, S. Tumor Necrosis Factor-α Signaling in Macrophages. Crit. Rev. Eukaryot. Gene Expr. 2010, 20, 87–103. [Google Scholar] [CrossRef]
- Bouchard, P.; Hutabarat, R.; Thompson, K. Discovery and Development of Therapeutic Aptamers. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 237–257. [Google Scholar] [CrossRef]
- Darfeuille, F.; Reigadas, S.; Hansen, J.B.; Orum, H.; Di Primo, C.; Toulmé, J.-J. Aptamers Targeted to an RNA Hairpin Show Improved Specificity Compared to that of Complementary Oligonucleotides. Biochemistry 2006, 45, 12076–12082. [Google Scholar] [CrossRef] [PubMed]
- Kalra, P.; Dhiman, A.; Cho, W.C.; Bruno, J.G.; Sharma, T.K. Simple Methods and Rational Design for Enhancing Aptamer Sensitivity and Specificity. Front. Mol. Biosci. 2018, 5. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Rossi, J. Erratum: Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 440. [Google Scholar] [CrossRef] [PubMed]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Mayer, G. The Chemical Biology of Aptamers. Angew. Chem. Int. Ed. 2009, 48, 2672–2689. [Google Scholar] [CrossRef]
- Gelinas, A.D.; Davies, D.R.; Janjic, N. Embracing proteins: Structural themes in aptamer–protein complexes. Curr. Opin. Struct. Biol. 2016, 36, 122–132. [Google Scholar] [CrossRef]
- Jenison, R.D.; Gill, S.C.; Pardi, A.; Polisky, B. High-resolution molecular discrimination by RNA. Science 1994, 263, 1425–1429. [Google Scholar] [CrossRef]
- Chen, L.; Rashid, F.; Shah, A.; Awan, H.M.; Wu, M.; Liu, A.; Wang, J.; Zhu, T.; Luo, Z.; Shan, G. The isolation of an RNA aptamer targeting to p53 protein with single amino acid mutation. Proc. Natl. Acad. Sci. USA 2015, 112, 10002–10007. [Google Scholar] [CrossRef]
- Bunka, D.H.J.; Stockley, P.G. Aptamers come of age—At last. Nat. Rev. Genet. 2006, 4, 588–596. [Google Scholar] [CrossRef]
- Hidding, J. A therapeutic battle: Antibodies vs. Aptamers. Nanosci. Master Progr. 2017, 16, 181–202. [Google Scholar]
- Cataldo, R.; Ciriaco, F.; Alfinito, E. A validation strategy for in silico generated aptamers. Comput. Biol. Chem. 2018, 77, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, T.C.; Davies, U.R.; Hisaminato, A.; Resnicow, D.I.; Gupta, S.; Waugh, S.M.; Nagabukuro, A.; Wadatsu, T.; Hishigaki, H.; Gawande, B.; et al. Non-helical DNA Triplex Forms a Unique Aptamer Scaffold for High Affinity Recognition of Nerve Growth Factor. Structure 2015, 23, 1293–1304. [Google Scholar] [CrossRef] [PubMed]
- Rohloff, J.C.; Gelinas, A.D.; Jarvis, T.C.; Ochsner, U.A.; Schneider, D.J.; Gold, L.; Janjic, N. Nucleic Acid Ligands With Protein-like Side Chains: Modified Aptamers and Their Use as Diagnostic and Therapeutic Agents. Mol. Ther. Nucleic Acids 2014, 3, e201. [Google Scholar] [CrossRef] [PubMed]
- Hermanna, T.; Westhof, E. Non-Watson-Crick base pairs in RNA-protein recognition. Chem. Biol. 1999, 6, R335–R343. [Google Scholar] [CrossRef]
- Nithin, C.; Mukherjee, S.; Bahadur, R.P. A non-redundant protein-RNA docking benchmark version 2.0. Proteins Struct. Funct. Bioinform. 2016, 85, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.E.; Medley, C.D.; Tang, Z.; Shangguan, D.; Lofton, C.; Tan, W. Aptamer-Conjugated Nanoparticles for the Collection and Detection of Multiple Cancer Cells. Anal. Chem. 2007, 79, 3075–3082. [Google Scholar] [CrossRef]
- Prodeus, A.; Cydzik, M.; Abdul-Wahid, A.; Huang, E.; Khatri, I.; Gorczynski, R.; Gariépy, J. Agonistic CD200R1 DNA Aptamers Are Potent Immunosuppressants That Prolong Allogeneic Skin Graft Survival. Mol. Ther. Nucleic Acids 2014, 3, e190. [Google Scholar] [CrossRef]
- Wiegand, T.W.; Williams, P.B.; Dreskin, S.C.; Jouvin, M.H.; Kinet, J.P.; Tasset, D. High-affinity oligonucleotide ligands to human IgE inhibit binding to Fc epsilon receptor I. J. Immunol. 1996, 157, 221–230. [Google Scholar]
- Lakhin, A.V.; Tarantul, V.Z.; Gening, L.V. Aptamers: Problems, Solutions and Prospects. Acta Naturae 2013, 5, 34–43. [Google Scholar] [CrossRef]
- Zhu, Q.; Liu, G.; Kai, M. DNA Aptamers in the Diagnosis and Treatment of Human Diseases. Molecules 2015, 20, 20979–20997. [Google Scholar] [CrossRef] [PubMed]
- Bock, L.C.; Griffin, L.C.; Latham, J.A.; Vermaas, E.H.; Toole, J.J. Selection of single-stranded DNA molecules that bind and inhibit human thrombin. Nat. Cell Biol. 1992, 355, 564–566. [Google Scholar] [CrossRef] [PubMed]
- Soutschek, J.; Akinc, A.; Bramlage, B.; Charisse, K.; Constien, R.; Donoghue, M.; Elbashir, S.; Gelck, A.; Hadwiger, P.; Harborth, J.; et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature 2004, 432, 173–178. [Google Scholar] [CrossRef]
- Robertson, D.L.; Joyce, G.F. Selection in vitro of an RNA enzyme that specifically cleaves single-stranded DNA. Nat. Cell Biol. 1990, 344, 467–468. [Google Scholar] [CrossRef]
- Gold, L. Oligonucleotides as Research, Diagnostic, and Therapeutic Agents. J. Biol. Chem. 1995, 270, 13581–13584. [Google Scholar] [CrossRef]
- Kuwahara, M.; Sugimoto, N. Molecular Evolution of Functional Nucleic Acids with Chemical Modifications. Molecules 2010, 15, 5423–5444. [Google Scholar] [CrossRef]
- Sawai, H.; Ozaki, A.N.; Satoh, F.; Ohbayashi, T.; Masud, M.M.; Ozaki, H. Expansion of structural and functional diversities of DNA using new 5-substituted deoxyuridine derivatives by PCR with superthermophilic KOD Dash DNA polymerase. Chem. Commun. 2001, 1, 2604–2605. [Google Scholar] [CrossRef]
- Kuwahara, M.; Nagashima, J.-I.; Hasegawa, M.; Tamura, T.; Kitagata, R.; Hanawa, K.; Hososhima, S.-I.; Kasamatsu, T.; Ozaki, H.; Sawai, H. Systematic characterization of 2′-deoxynucleoside- 5′-triphosphate analogs as substrates for DNA polymerases by polymerase chain reaction and kinetic studies on enzymatic production of modified DNA. Nucleic Acids Res. 2006, 34, 5383–5394. [Google Scholar] [CrossRef] [PubMed]
- Srivatsan, S.G.; Tor, Y. Enzymatic incorporation of emissive pyrimidine ribonucleotides. Chem. Asian J. 2009, 4, 419–427. [Google Scholar] [CrossRef]
- Liu, E.; Lam, C.H.; Perrin, D.M. Synthesis and Enzymatic Incorporation of Modified Deoxyuridine Triphosphates. Molecules 2015, 20, 13591–13602. [Google Scholar] [CrossRef]
- Hall, B.; Micheletti, J.M.; Satya, P.; Ogle, K.; Pollard, J.; Ellington, A.D. Design, Synthesis, and Amplification of DNA Pools for In Vitro Selection. Curr. Protoc. Mol. Biol. 2009, 88, 24.2.1–24.2.27. [Google Scholar] [CrossRef] [PubMed]
- Kulbachinskiy, A. Methods for selection of aptamers to protein targets. Biochemistry (Moscow) 2007, 72, 1505–1518. [Google Scholar] [CrossRef] [PubMed]
- Padilla, R.; Sousa, R. A Y639F/H784A T7 RNA polymerase double mutant displays superior properties for synthesizing RNAs with non-canonical NTPs. Nucleic Acids Res. 2002, 30, e138. [Google Scholar] [CrossRef] [PubMed]
- Förster, C.; Zydek, M.; Rothkegel, M.; Wu, Z.; Gallin, C.; Geßner, R.; Lisdat, F.; Fürste, J.P. Properties of an ‘LNA’-modified ricin RNA aptamer. Biochem. Biophys. Res. Commun. 2012, 419, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Ozer, A.; Pagano, J.M.; Lis, J.T. New Technologies Provide Quantum Changes in the Scale, Speed, and Success of SELEX Methods and Aptamer Characterization. Mol. Ther. Nucleic Acids 2014, 3, e183. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Yin, W.; Alshamaileh, H.; Zhang, Y.; Tran, P.H.; Nguyen, T.N.-G.; Li, Y.; Chen, K.; Sun, M.; Hou, Y.; et al. A Detailed Protein-SELEX Protocol Allowing Visual Assessments of Individual Steps for a High Success Rate. Hum. Gene Ther. Methods 2019, 30, 1–16. [Google Scholar] [CrossRef]
- Levay, A.; Brenneman, R.; Hoinka, J.; Sant, D.; Cardone, M.; Trinchieri, G.; Przytycka, T.M.; Berezhnoy, A. Identifying high-affinity aptamer ligands with defined cross-reactivity using high-throughput guided systematic evolution of ligands by exponential enrichment. Nucleic Acids Res. 2015, 43, e82. [Google Scholar] [CrossRef]
- Thiel, W.H.; Bair, T.; Thiel, K.W.; Dassie, J.P.; Rockey, W.M.; Howell, C.A.; Liu, X.Y.; Dupuy, A.J.; Huang, L.; Owczarzy, R.; et al. Nucleotide Bias Observed with a Short SELEX RNA Aptamer Library. Nucleic Acid Ther. 2011, 21, 253–263. [Google Scholar] [CrossRef]
- Thiel, W.H.; Bair, T.; Peek, A.S.; Liu, X.; Dassie, J.; Stockdale, K.R.; Behlke, M.A.; Miller, F.J.; Giangrande, P.H. Rapid Identification of Cell-Specific, Internalizing RNA Aptamers with Bioinformatics Analyses of a Cell-Based Aptamer Selection. PLoS ONE 2012, 7, e43836. [Google Scholar] [CrossRef]
- Spiga, F.M.; Maietta, P.; Guiducci, C. More DNA–Aptamers for Small Drugs: A Capture–SELEX Coupled with Surface Plasmon Resonance and High-Throughput Sequencing. ACS Comb. Sci. 2015, 17, 326–333. [Google Scholar] [CrossRef]
- Jia, W.; Li, H.; Wilkop, T.; Liu, X.; Yu, X.; Cheng, Q.; Xu, D.; Chen, H.-Y. Silver decahedral nanoparticles empowered SPR imaging-SELEX for high throughput screening of aptamers with real-time assessment. Biosens. Bioelectron. 2018, 109, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Fang, X. Aptamers Selected by Cell-SELEX for Theranostics; Springer Science and Business Media LLC: Berlin, Germany, 2015. [Google Scholar]
- Kunii, T.; Ogura, S.-I.; Mie, M.; Kobatake, E. Selection of DNA aptamers recognizing small cell lung cancer using living cell-SELEX. Analyst 2011, 136, 1310–1312. [Google Scholar] [CrossRef]
- Nabavinia, M.S.; Charbgoo, F.; Alibolandi, M.; Mosaffa, F.; Gholoobi, A.; Ramezani, M.; Abnous, K. Comparison of Flow Cytometry and ELASA for Screening of Proper Candidate Aptamer in Cell-SELEX Pool. Appl. Biochem. Biotechnol. 2017, 184, 444–452. [Google Scholar] [CrossRef]
- Dastjerdi, K.; Tabar, G.H.; Dehghani, H.; Haghparast, A. Generation of an enriched pool of DNA aptamers for an HER2-overexpressing cell line selected by Cell SELEX. Biotechnol. Appl. Biochem. 2011, 58, 226–230. [Google Scholar] [CrossRef]
- Avci-Adali, M.; Wilhelm, N.; Perle, N.; Stoll, H.; Schlensak, C.; Wendel, H.-P. Absolute Quantification of Cell-Bound DNA Aptamers During SELEX. Nucleic Acid Ther. 2013, 23, 125–130. [Google Scholar] [CrossRef]
- Mayer, G.; Ahmed, M.-S.L.; Dolf, A.; Endl, E.; Knolle, P.A.; Famulok, M. Fluorescence-activated cell sorting for aptamer SELEX with cell mixtures. Nat. Protoc. 2010, 5, 1993–2004. [Google Scholar] [CrossRef] [PubMed]
- Stoltenburg, R.; Nikolaus, N.; Strehlitz, B. Capture-SELEX: Selection of DNA Aptamers for Aminoglycoside Antibiotics. J. Anal. Methods Chem. 2012, 2012, 1–14. [Google Scholar] [CrossRef]
- Vanbrabant, J.; Leirs, K.; Vanschoenbeek, K.; Vanbrabant, J.; Michiels, L. reMelting curve analysis as a tool for enrichment monitoring in the SELEX process. Analyst 2014, 139, 589–595. [Google Scholar] [CrossRef]
- Charlton, J.; Smith, D. Estimation of SELEX pool size by measurement of DNA renaturation rates. RNA 1999, 5, 1326–1332. [Google Scholar] [CrossRef][Green Version]
- Schütze, T.; Arndt, P.F.; Menger, M.; Wochner, A.; Vingron, M.; Erdmann, V.A.; Lehrach, H.; Kaps, C.; Glökler, J. A calibrated diversity assay for nucleic acid libraries using DiStRO—A Diversity Standard of Random Oligonucleotides. Nucleic Acids Res. 2009, 38, e23. [Google Scholar] [CrossRef]
- Luo, Z.; He, L.; Wang, J.; Fang, X.; Zhang, L. Developing a combined strategy for monitoring the progress of aptamer selection. Analyst 2017, 142, 3136–3139. [Google Scholar] [CrossRef]
- Müller, J.; El-Maarri, O.; Oldenburg, J.; Pötzsch, B.; Mayer, G. Monitoring the progression of the in vitro selection of nucleic acid aptamers by denaturing high-performance liquid chromatography. Anal. Bioanal. Chem. 2007, 390, 1033–1037. [Google Scholar] [CrossRef][Green Version]
- Schütze, T.; Wilhelm, B.; Greiner, N.; Braun, H.; Peter, F.; Mörl, M.; Erdmann, V.A.; Lehrach, H.; Konthur, Z.; Menger, M.; et al. Probing the SELEX Process with Next-Generation Sequencing. PLoS ONE 2011, 6, e29604. [Google Scholar] [CrossRef] [PubMed]
- Amano, R.; Aoki, K.; Miyakawa, S.; Nakamura, Y.; Kozu, T.; Kawai, G.; Sakamoto, T. NMR monitoring of the SELEX process to confirm enrichment of structured RNA. Sci. Rep. 2017, 7, 283. [Google Scholar] [CrossRef]
- Mencin, N.; Šmuc, T.; Vranicar, M.; Mavri, J.; Hren, M.; Galeša, K.; Krkoc, P.; Ulrich, H.; Šolar, B. Optimization of SELEX: Comparison of different methods for monitoring the progress of in vitro selection of aptamers. J. Pharm. Biomed. Anal. 2014, 91, 151–159. [Google Scholar] [CrossRef]
- Gu, G.; Wang, T.; Yang, Y.; Xu, X.; Wang, J. An Improved SELEX-Seq Strategy for Characterizing DNA-Binding Specificity of Transcription Factor: NF-κB as an Example. PLoS ONE 2013, 8, e76109. [Google Scholar] [CrossRef]
- Encyclopedia of Analytical Chemistry. Available online: https://onlinelibrary.wiley.com/page/book/10.1002/9780470027318/homepage/editor_highlights.htm (accessed on 1 August 2020).
- Lou, X.; Qian, J.; Xiao, Y.; Viel, L.; Gerdon, A.E.; Lagally, E.T.; Atzberger, P.J.; Tarasow, T.M.; Heeger, A.J.; Soh, H.T. Micromagnetic selection of aptamers in microfluidic channels. Proc. Natl. Acad. Sci. USA 2009, 106, 2989–2994. [Google Scholar] [CrossRef]
- Elshafey, R.; Siaj, M.; Zourob, M. In Vitro Selection, Characterization, and Biosensing Application of High-Affinity Cylindrospermopsin-Targeting Aptamers. Anal. Chem. 2014, 86, 9196–9203. [Google Scholar] [CrossRef]
- Kim, J.W.; Kim, E.Y.; Kim, S.Y.; Byun, S.K.; Lee, D.; Oh, K.-J.; Kim, W.K.; Han, B.S.; Chi, S.-W.; Lee, S.C.; et al. Identification of DNA aptamers toward epithelial cell adhesion molecule via cell-SELEX. Mol. Cells 2014, 37, 742–746. [Google Scholar] [CrossRef]
- Lin, H.-I.; Wu, C.-C.; Yang, C.-H.; Chang, K.-W.; Lee, G.-B.; Shiesh, S.-C. Selection of aptamers specific for glycated hemoglobin and total hemoglobin using on-chip SELEX. Lab Chip 2015, 15, 486–494. [Google Scholar] [CrossRef]
- Gening, L.V.; Klincheva, S.A.; Reshetnjak, A.; Grollman, A.P.; Miller, H. RNA aptamers selected against DNA polymerase inhibit the polymerase activities of DNA polymerases and. Nucleic Acids Res. 2006, 34, 2579–2586. [Google Scholar] [CrossRef]
- Yu, R.; Geary, R.; Levin, A. Basic Principles of the Pharmacokinetics of Antisense Oligonucleotide Drugs. In Antisense Drug Technology; Informa UK Limited: London, UK, 2007; pp. 183–215. [Google Scholar]
- Wu, H.; MacLeod, A.; Lima, W.F.; Crooke, S.T. Identification and Partial Purification of Human Double Strand RNase Activity. J. Boil. Chem. 1998, 273, 2532–2542. [Google Scholar] [CrossRef]
- Jäger, S.; Rasched, G.; Kornreich-Leshem, H.; Engeser, M.; Thum, A.O.; Famulok, M. A Versatile Toolbox for Variable DNA Functionalization at High Density. J. Am. Chem. Soc. 2005, 127, 15071–15082. [Google Scholar] [CrossRef]
- Hirao, I.; Kimoto, M.; Mitsui, T.; Fujiwara, T.; Kawai, R.; Sato, A.; Harada, Y.; Yokoyama, S. An unnatural hydrophobic base pair system: Site-specific incorporation of nucleotide analogs into DNA and RNA. Nat. Methods 2006, 3, 729–735. [Google Scholar] [CrossRef]
- Burmeister, P.E.; Lewis, S.D.; Silva, R.F.; Preiss, J.R.; Horwitz, L.R.; Pendergrast, P.S.; McCauley, T.G.; Kurz, J.C.; Epstein, D.M.; Wilson, C.; et al. Direct In Vitro Selection of a 2′-O-Methyl Aptamer to VEGF. Chem. Biol. 2005, 12, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Qiu, Q.; Gill, S.C.; Jayasena, S.D. Modified RNA sequence pools forin vitroselection. Nucleic Acids Res. 1994, 22, 5229–5234. [Google Scholar] [CrossRef]
- Kuwahara, M.; Obika, S. In vitro selection of BNA (LNA) aptamers. Artif. DNA PNA XNA 2013, 4, 39–48. [Google Scholar] [CrossRef]
- Veedu, R.N.; Wengel, J. Locked nucleic acid nucleoside triphosphates and polymerases: On the way towards evolution of LNA aptamers. Mol. BioSyst. 2009, 5, 787–792. [Google Scholar] [CrossRef]
- Ruckman, J.; Green, L.S.; Beeson, J.; Waugh, S.; Gillette, W.L.; Henninger, D.D.; Claesson-Welsh, L.; Janjic, N. 2′-Fluoropyrimidine RNA-based Aptamers to the 165-Amino Acid Form of Vascular Endothelial Growth Factor (VEGF165). J. Boil. Chem. 1998, 273, 20556–20567. [Google Scholar] [CrossRef]
- Lee, Y.; Urban, J.H.; Xu, L.; Sullenger, B.A.; Lee, J. 2′Fluoro Modification Differentially Modulates the Ability of RNAs to Activate Pattern Recognition Receptors. Nucleic Acid Ther. 2016, 26, 173–182. [Google Scholar] [CrossRef]
- Eaton, B.E.; Gold, L.; Hicke, B.J.; Janjié, N.; Jucker, F.M.; Sebesta, D.P.; Tarasow, T.M.; Willis, M.C.; Zichi, D.A. Post-SELEX combinatorial optimization of aptamers. Bioorganic Med. Chem. 1997, 5, 1087–1096. [Google Scholar] [CrossRef]
- Aaldering, L.J.; Tayeb, H.; Krishnan, S.; Fletcher, S.; Wilton, S.D.; Veedu, R.N. Smart functional nucleic acid chimeras: Enabling tissue specific RNA targeting therapy. RNA Biol. 2015, 12, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Maasch, C.; Buchner, K.; Eulberg, D.; Vonhoff, S.; Klussmann, S. Physicochemical Stability of NOX-E36, a 40mer L-RNA (Spiegelmer) for Therapeutic Applications. Nucleic Acids Symp. Ser. 2008, 52, 61–62. [Google Scholar] [CrossRef] [PubMed]
- Guo, P. The emerging field of RNA nanotechnology. Nat. Nanotechnol. 2010, 5, 833–842. [Google Scholar] [CrossRef]
- Zhou, J.; Soontornworajit, B.; Martin, J.; Sullenger, B.A.; Gilboa, E.; Wang, Y. A Hybrid DNA Aptamer-Dendrimer Nanomaterial for Targeted Cell Labeling. Macromol. Biosci. 2009, 9, 831–835. [Google Scholar] [CrossRef]
- Rusconi, C.P.; Roberts, J.D.; Pitoc, G.A.; Nimjee, S.M.; White, R.R.; Quick, G.; Scardino, E.; Fay, W.P.; Sullenger, B.A. Antidote-mediated control of an anticoagulant aptamer in vivo. Nat. Biotechnol. 2004, 22, 1423–1428. [Google Scholar] [CrossRef]
- Lee, C.H.; Lee, S.-H.; Kim, J.H.; Noh, Y.-H.; Noh, G.-J.; Lee, S.-W. Pharmacokinetics of a Cholesterol-conjugated Aptamer Against the Hepatitis C Virus (HCV) NS5B Protein. Mol. Ther. Nucleic Acids 2015, 4, e254. [Google Scholar] [CrossRef]
- Tucker, C.E.; Chen, L.-S.; Judkins, M.B.; Farmer, J.A.; Gill, S.C.; Drolet, D.W. Detection and plasma pharmacokinetics of an anti-vascular endothelial growth factor oligonucleotide-aptamer (NX1838) in rhesus monkeys. J. Chromatogr. B Biomed. Sci. Appl. 1999, 732, 203–212. [Google Scholar] [CrossRef]
- Hayashi, Y. Rapid Spine Delivery and Redistribution of AMPA Receptors After Synaptic NMDA Receptor Activation. Science 1999, 284, 1811–1816. [Google Scholar] [CrossRef]
- Vorobyeva, M.; Vorobjev, P.; Venyaminova, A. Multivalent Aptamers: Versatile Tools for Diagnostic and Therapeutic Applications. Molecules 2016, 21, 1613. [Google Scholar] [CrossRef]
- Borbas, K.E.; Ferreira, C.S.M.; Perkins, A.; Bruce, J.I.; Missailidis, S. Design and Synthesis of Mono- and Multimeric Targeted Radiopharmaceuticals Based on Novel Cyclen Ligands Coupled to Anti-MUC1 Aptamers for the Diagnostic Imaging and Targeted Radiotherapy of Cancer. Bioconjugate Chem. 2007, 18, 1205–1212. [Google Scholar] [CrossRef]
- Choi, D.Y.; Ortube, M.C.; Mccannel, C.A.; Sarraf, D.; Hubschman, J.-P.; Mccannel, T.A.; Gorin, M.B. Sustained Elevated Intraocular Pressures After Intravitreal Injection Of Bevacizumab, Ranibizumab, and Pegaptanib. Retina 2011, 31, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Steffensmeier, A.C.; Azar, A.E.; Fuller, J.J.; Muller, B.A.; Russell, S.R. Vitreous Injections of Pegaptanib Sodium Triggering Allergic Reactions. Am. J. Ophthalmol. 2007, 143, 512–513. [Google Scholar] [CrossRef]
- Falavarjani, K.G.; Nguyen, Q.D. Adverse events and complications associated with intravitreal injection of anti-VEGF agents: A review of literature. Eye 2013, 27, 787–794. [Google Scholar] [CrossRef]
- Farman, C.A.; Kornbrust, D.J. Oligodeoxynucleotide Studies in Primates: Antisense and Immune Stimulatory Indications. Toxicol. Pathol. 2003, 31, 119–122. [Google Scholar] [CrossRef]
- Swayze, E.; Siwkowski, A.M.; Wancewicz, E.V.; Migawa, M.T.; Wyrzykiewicz, T.K.; Hung, G.; Monia, B.P.; Bennett, A.C.F.; Bennett, C.F. Antisense oligonucleotides containing locked nucleic acid improve potency but cause significant hepatotoxicity in animals. Nucleic Acids Res. 2006, 35, 687–700. [Google Scholar] [CrossRef] [PubMed]
- Lincoff, A.M.; Mehran, R.; Povsic, T.J.; Zelenkofske, S.L.; Huang, Z.; Armstrong, P.W.; Steg, P.G.; Bode, C.; Cohen, M.G.; Buller, C.; et al. Effect of the REG1 anticoagulation system versus bivalirudin on outcomes after percutaneous coronary intervention (REGULATE-PCI): A randomised clinical trial. Lancet 2016, 387, 349–356. [Google Scholar] [CrossRef]
- Bruno, J.G. Potential Inherent Stimulation of the Innate Immune System by Nucleic Acid Aptamers and Possible Corrective Approaches. Pharmaceuticals 2018, 11, 62. [Google Scholar] [CrossRef]
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nat. Cell Biol. 1990, 346, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Dua, P.; Kim, S.; Lee, N.-K. Patents on SELEX and therapeutic aptamers. Recent Patents DNA Gene Seq. 2008, 2, 172–186. [Google Scholar] [CrossRef]
- Gupta, S.; Hirota, M.; Waugh, S.M.; Murakami, I.; Suzuki, T.; Muraguchi, M.; Shibamori, M.; Ishikawa, Y.; Jarvis, T.C.; Carter, J.D.; et al. Chemically Modified DNA Aptamers Bind Interleukin-6 with High Affinity and Inhibit Signaling by Blocking Its Interaction with Interleukin-6 Receptor. J. Biol. Chem. 2014, 289, 8706–8719. [Google Scholar] [CrossRef]
- Liu, Y.; Kwa, T.; Revzin, A. Simultaneous detection of cell-secreted TNF-α and IFN-γ using micropatterned aptamer-modified electrodes. Biomaterials 2012, 33, 7347–7355. [Google Scholar] [CrossRef] [PubMed]
- Gay, N.J.; Gangloff, M. Structure and Function of Toll Receptors and Their Ligands. Ann. Rev. Biochem. 2007, 76, 141–165. [Google Scholar] [CrossRef]
- Trivedi, S.; Greidinger, E.L. Endosomal Toll-like receptors in autoimmunity: Mechanisms for clinical diversity. Therapy 2009, 6, 433–442. [Google Scholar] [CrossRef]
- Frazão, J.B.; Errante, P.R.; Condino-Neto, A. Toll-Like Receptors’ Pathway Disturbances are Associated with Increased Susceptibility to Infections in Humans. Arch. Immunol. Ther. Exp. 2013, 61, 427–443. [Google Scholar] [CrossRef]
- Seth, R.B.; Sun, L.; Chen, Z.J. Antiviral innate immunity pathways. Cell Res. 2006, 16, 141–147. [Google Scholar] [CrossRef]
- Chang, Y.-C.; Kao, W.-C.; Wang, W.-Y.; Wang, W.-Y.; Yang, R.-B.; Peck, K. Identification and characterization of oligonucleotides that inhibit Toll-like receptor 2-associated immune responses. FASEB J. 2009, 23, 3078–3088. [Google Scholar] [CrossRef]
- Avci-Adali, M.; Steinle, H.; Michel, T.; Schlensak, C.; Wendel, H.-P. Potential Capacity of Aptamers to Trigger Immune Activation in Human Blood. PLoS ONE 2013, 8, e68810. [Google Scholar] [CrossRef]
- Wu, C.N.; Sabet, M.; Hayashi, T.; Tawatao, R.; Fierer, J.; Carson, D.A.; Guiney, D.G.; Corr, M. In vivo efficacy of a phosphodiester TLR-9 aptamer and its beneficial effect in a pulmonary anthrax infection model. Cell. Immunol. 2008, 251, 78–85. [Google Scholar] [CrossRef]
- Andréola, M.-L.; Calmels, C.; Michel, J.; Toulmé, J.-J.; Litvak, S. Towards the selection of phosphorothioate aptamers. JBIC J. Biol. Inorg. Chem. 2000, 267, 5032–5040. [Google Scholar] [CrossRef]
- Fukuda, K.; Tsujita, T.; Matsumoto, M.; Seya, T.; Sakiyama, H.; Nishikawa, F.; Hasegawa, T. Analysis of the interaction between human TLR3 ectodomain and nucleic acids. Nucleic Acids Symp. Ser. 2006, 50, 249–250. [Google Scholar] [CrossRef][Green Version]
- Pratico, E.D.; Sullenger, B.A.; Nair, S.K. Identification and Characterization of an Agonistic Aptamer Against the T Cell Costimulatory Receptor, OX40. Nucleic Acid Ther. 2013, 23, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Dollins, C.M.; Nair, S.; Boczkowski, D.; Lee, J.; Layzer, J.M.; Gilboa, E.; Sullenger, B.A. Assembling OX40 Aptamers on a Molecular Scaffold to Create a Receptor-Activating Aptamer. Chem. Biol. 2008, 15, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Mallikaratchy, P.R.; Ruggiero, A.; Gardner, J.R.; Kuryavyi, V.; Maguire, W.F.; Heaney, M.L.; McDevitt, M.R.; Patel, D.J.; Scheinberg, D. A multivalent DNA aptamer specific for the B-cell receptor on human lymphoma and leukemia. Nucleic Acids Res. 2010, 39, 2458–2469. [Google Scholar] [CrossRef] [PubMed]
- Ford, M.L.; Adams, A.B.; Pearson, T.C. Targeting co-stimulatory pathways: Transplantation and autoimmunity. Nat. Rev. Nephrol. 2013, 10, 14–24. [Google Scholar] [CrossRef]
- Yeung, M.Y.; Najafian, N.; Sayegh, M.H. Targeting CD28 to prevent transplant rejection. Expert Opin. Ther. Targets 2013, 18, 225–242. [Google Scholar] [CrossRef]
- Leung, J.; Suh, W.-K. The CD28-B7 Family in Anti-Tumor Immunity: Emerging Concepts in Cancer Immunotherapy. Immune Netw. 2014, 14, 265–276. [Google Scholar] [CrossRef]
- Herrmann, A.; Priceman, S.J.; Kujawski, M.; Xin, H.; Cherryholmes, G.A.; Zhang, W.; Zhang, C.; Lahtz, C.; Kowolik, C.; Forman, S.J.; et al. CTLA4 aptamer delivers STAT3 siRNA to tumor-associated and malignant T cells. J. Clin. Investig. 2014, 124, 2977–2987. [Google Scholar] [CrossRef]
- Pastor, F.; Soldevilla, M.M.; Villanueva, H.; Kolonias, D.; Inoges, S.; De Cerio, A.L.; Kandzia, R.; Klimyuk, V.; Gleba, Y.; Gilboa, E.; et al. CD28 Aptamers as Powerful Immune Response Modulators. Mol. Ther. Nucleic Acids 2013, 2, e98. [Google Scholar] [CrossRef]
- Sarma, J.V.; Ward, P.A. The complement system. Cell Tissue Res. 2010, 343, 227–235. [Google Scholar] [CrossRef]
- Mooijaart, S.P.; Koeijvoets, K.M.; Sijbrands, E.J.; Daha, M.R.; Westendorp, R.G. Complement Factor H polymorphism Y402H associates with inflammation, visual acuity, and cardiovascular mortality in the elderly population at large. Exp. Gerontol. 2007, 42, 1116–1122. [Google Scholar] [CrossRef]
- Hoehlig, K.; Maasch, C.; Shushakova, N.; Buchner, K.; Huber-Lang, M.; Purschke, W.G.; Vater, A.; Klussmann, S. A Novel C5a-neutralizing Mirror-image (l-)Aptamer Prevents Organ Failure and Improves Survival in Experimental Sepsis. Mol. Ther. 2013, 21, 2236–2246. [Google Scholar] [CrossRef]
- Stecker, J.R.; Savage, A.A.; Bruno, J.G.; García, D.M.; Koke, J.R. Dynamics and Visualization of MCF7 Adenocarcinoma Cell Death by Aptamer-C1q-Mediated Membrane Attack. Nucleic Acid Ther. 2012, 22, 275–282. [Google Scholar] [CrossRef]
- Dinarello, C.A. Proinflammatory Cytokines. Chest 2000, 118, 503–508. [Google Scholar] [CrossRef]
- Martin, P.; Leibovich, S. Inflammatory cells during wound repair: The good, the bad and the ugly. Trends Cell Boil. 2005, 15, 599–607. [Google Scholar] [CrossRef]
- Karin, N.; Wildbaum, G. The Role of Chemokines in Shaping the Balance Between CD4+ T Cell Subsets and Its Therapeutic Implications in Autoimmune and Cancer Diseases. Front. Immunol. 2015, 6, 609. [Google Scholar] [CrossRef]
- Belladonna, M.L.; Grohmann, U. Bioengineering heterodimeric cytokines: Turning promiscuous proteins into therapeutic agents. Biotechnol. Genet. Eng. Rev. 2013, 29, 149–174. [Google Scholar] [CrossRef]
- Francis, G.E.; Fisher, D.; Delgado, C.; Malik, F.; Gardiner, A.; Neale, D. PEGylation of cytokines and other therapeutic proteins and peptides: The importance of biological optimisation of coupling techniques. Int. J. Hematol. 1998, 68, 1–18. [Google Scholar] [CrossRef]
- Brocker, C.; Thompson, D.; Matsumoto, A.; Nebert, D.W.; Vasiliou, V. Evolutionary divergence and functions of the human interleukin (IL) gene family. Hum. Genom. 2010, 5, 30–55. [Google Scholar] [CrossRef]
- Sung, H.J.; Choi, S.; Lee, J.W.; Ok, C.Y.; Bae, Y.-S.; Kim, Y.-H.; Lee, W.; Heo, K.; Kim, I.-H. Inhibition of human neutrophil activity by an RNA aptamer bound to interleukin-8. Biomaterials 2014, 35, 578–589. [Google Scholar] [CrossRef]
- Berezhnoy, A.; Stewart, C.A.; Ii, J.O.M.; Thiel, W.H.; Giangrande, P.; Trinchieri, G.; Gilboa, E. Isolation and Optimization of Murine IL-10 Receptor Blocking Oligonucleotide Aptamers Using High-throughput Sequencing. Mol. Ther. 2012, 20, 1242–1250. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.; Han, Y.; Hughart, N.; McCarra, J.; Alpini, G.; Meng, F. Interleukin-6 and its receptor, key players in hepatobiliary inflammation and cancer. Transl. Gastrointest. Cancer 2012, 1, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Febbraio, M.A.; Rose-John, S.; Pedersen, B.K. Is Interleukin-6 Receptor Blockade the Holy Grail for Inflammatory Diseases? Clin. Pharmacol. Ther. 2010, 87, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Hirota, M.; Murakami, I.; Ishikawa, Y.; Suzuki, T.; Sumida, S.-I.; Ibaragi, S.; Kasai, H.; Horai, N.; Drolet, D.W.; Gupta, S.; et al. Chemically Modified Interleukin-6 Aptamer Inhibits Development of Collagen-Induced Arthritis in Cynomolgus Monkeys. Nucleic Acid Ther. 2016, 26, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Hu, Y.; Duan, J.; Ma, J.; Xu, D.; Yang, X.-D. Selection of a Novel DNA Aptamer for Assay of Intracellular Interferon-Gamma. PLoS ONE 2014, 9, e98214. [Google Scholar] [CrossRef] [PubMed]
- Orava, E.W.; Jarvik, N.; Shek, Y.L.; Sidhu, S.S.; Gariépy, J. A Short DNA Aptamer That Recognizes TNFα and Blocks Its Activityin Vitro. ACS Chem. Biol. 2012, 8, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.N.; Lee, J.; Raz, E.; Corr, M.; Carson, D.A. Necessity of Oligonucleotide Aggregation for Toll-like Receptor 9 Activation. J. Boil. Chem. 2004, 279, 33071–33078. [Google Scholar] [CrossRef]
- Prodeus, A.; Abdul-Wahid, A.; Fischer, N.W.; Huang, E.H.-B.; Cydzik, M.; Gariépy, J. Targeting the PD-1/PD-L1 Immune Evasion Axis With DNA Aptamers as a Novel Therapeutic Strategy for the Treatment of Disseminated Cancers. Mol. Ther. Nucleic Acids 2015, 4, e237. [Google Scholar] [CrossRef]
- Faryammanesh, R.; Lange, T.; Magbanua, E.; Haas, S.; Meyer, C.; Wicklein, D.; Schumacher, U.; Hahn, U. SDA, a DNA Aptamer Inhibiting E- and P-Selectin Mediated Adhesion of Cancer and Leukemia Cells, the First and Pivotal Step in Transendothelial Migration during Metastasis Formation. PLoS ONE 2014, 9, e93173. [Google Scholar] [CrossRef]
- Mittelberger, F.; Meyer, C.; Waetzig, G.H.; Zacharias, M.; Valentini, E.; Svergun, D.I.; Berg, K.; Lorenzen, I.; Grötzinger, J.; Rose-John, S.; et al. RAID3—An interleukin-6 receptor-binding aptamer with post-selective modification-resistant affinity. RNA Biol. 2015, 12, 1043–1053. [Google Scholar] [CrossRef]
- Meyer, C.; Berg, K.; Eydeler-Haeder, K.; Lorenzen, I.; Grötzinger, J.; Rose-John, S.; Hahn, U. Stabilized Interleukin-6 receptor binding RNA aptamers. RNA Biol. 2013, 11, 57–65. [Google Scholar] [CrossRef]
- Meyer, C.; Eydeler, K.; Magbanua, E.; Živković, T.; Piganeau, N.; Lorenzen, I.; Grötzinger, J.; Mayer, G.; Rose-John, S.; Hahn, U. Interleukin-6 receptor specific RNA aptamers for cargo delivery into target cells. RNA Biol. 2012, 9, 67–80. [Google Scholar] [CrossRef]
- Biesecker, G.; Dihel, L.; Enney, K.; Bendele, R.A. Derivation of RNA aptamer inhibitors of human complement C5. Immunopharmacology 1999, 42, 219–230. [Google Scholar] [CrossRef]
- Bates, P.J.; Laber, D.A.; Miller, D.M.; Thomas, S.D.; Trent, J.O. Discovery and development of the G-rich oligonucleotide AS1411 as a novel treatment for cancer. Exp. Mol. Pathol. 2009, 86, 151–164. [Google Scholar] [CrossRef]
- Vater, A.; Sahlmann, J.; Kröger, N.; Zöllner, S.; Lioznov, M.; Maasch, C.; Buchner, K.; Vossmeyer, D.; Schwoebel, F.; Purschke, W.G.; et al. Hematopoietic Stem and Progenitor Cell Mobilization in Mice and Humans by a First-in-Class Mirror-Image Oligonucleotide Inhibitor of CXCL12. Clin. Pharmacol. Ther. 2013, 94, 150–157. [Google Scholar] [CrossRef]
- Schwoebel, F.; Van Eijk, L.T.; Zboralski, D.; Sell, S.; Buchner, K.; Maasch, C.; Purschke, W.G.; Humphrey, M.; Zöllner, S.; Eulberg, D.; et al. The effects of the anti-hepcidin Spiegelmer NOX-H94 on inflammation-induced anemia in cynomolgus monkeys. Blood 2013, 121, 2311–2315. [Google Scholar] [CrossRef]
- Diener, J.L.; Lagasse, H.A.D.; Duerschmied, D.; Merhi, Y.; Tanguay, J.-F.F.; Hutabarat, R.; Gilbert, J.; Wagner, D.D.; Schaub, R.G. Inhibition of von Willebrand factor-mediated platelet activation and thrombosis by the anti-von Willebrand factor A1-domain aptamer ARC1779. J. Thromb. Haemost. 2009, 7, 1155–1162. [Google Scholar] [CrossRef]
- Knöbl, P.; Jilma, B.; Gilbert, J.C.; Hutabarat, R.M.; Wagner, P.G.; Jilma-Stohlawetz, P. Anti-von Willebrand factor aptamer ARC1779 for refractory thrombotic thrombocytopenic purpura. Transfusion 2009, 49, 2181–2185. [Google Scholar] [CrossRef]
- Troisi, R.; Napolitano, V.; Spiridonova, V.; Krauss, I.R.; Sica, F. Several structural motifs cooperate in determining the highly effective anti-thrombin activity of NU172 aptamer. Nucleic Acids Res. 2018, 46, 12177–12185. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Name of Aptamer | Target | Nature of Aptamer | Clinical Phase | Chemical Modification | References |
|---|---|---|---|---|---|
| PO R10-60 | TLR9 | RNA | phosphodiester | [163,191] | |
| MP7 | (PD1) | DNA | Preclinical | unmodified | [192] |
| SDA | E-selectin and P-selectin | DNA | Preclinical | unmodified | [193] |
| ESTA-1 | E-selectin | DNA | Preclinical | only one thio-modified nucleotide | [193] |
| 9C7 | OX40 | RNA | Preclinical | Modified initial 2-fluoro (2-F) | [166,167] |
| MUC1-5TR-1 | C1q | DNA | Preclinical | 5- Alexa Fluor 546 (AF546) or 5′-biotin modifications | [177] |
| 8A-35 | IL-8 | RNA | Preclinical | 2′-fluoro-pyrimidine modified | [184] |
| AIR-3 | Interleukin 6 receptor | RNA | Preclinical | Shortening of FAIR-6 or 2′-F-Py modified AIR-3 | [194,195,196] |
| SL1026 | Interleukin 6 | RNA | Preclinical | a hexylamine modification | [155] |
| ARC1950 | Human complement C5 | RNA | Phase 2 | 2’-O-methyl purine and 2’-fluoropyrimidine substitutions | [197] |
| AS1411 | Nucleolin | DNA | Phase 2 | unmodified (phosphodiester) DNA backbone | [198] |
| NOX A12 | CXCL12 | RNA | Phase 2 | L-configuration containing branched 40 kDa PEG | [199] |
| NOX-H94 | Hepcidin | RNA | Phase 2 | 5’ end with 40 kDa PEG tail | [200] |
| ARC1779 | A1 domain of vWF | DNA/RNA | Phase 2 | phosphorothioate | [201,202] |
| NU172 | Thrombin | DNA | Phase 2 | unmodified 26-nucleotide DNA aptamer | [203] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yasmeen, F.; Seo, H.; Javaid, N.; Kim, M.S.; Choi, S. Therapeutic Interventions into Innate Immune Diseases by Means of Aptamers. Pharmaceutics 2020, 12, 955. https://doi.org/10.3390/pharmaceutics12100955
Yasmeen F, Seo H, Javaid N, Kim MS, Choi S. Therapeutic Interventions into Innate Immune Diseases by Means of Aptamers. Pharmaceutics. 2020; 12(10):955. https://doi.org/10.3390/pharmaceutics12100955
Chicago/Turabian StyleYasmeen, Farzana, Hana Seo, Nasir Javaid, Moon Suk Kim, and Sangdun Choi. 2020. "Therapeutic Interventions into Innate Immune Diseases by Means of Aptamers" Pharmaceutics 12, no. 10: 955. https://doi.org/10.3390/pharmaceutics12100955
APA StyleYasmeen, F., Seo, H., Javaid, N., Kim, M. S., & Choi, S. (2020). Therapeutic Interventions into Innate Immune Diseases by Means of Aptamers. Pharmaceutics, 12(10), 955. https://doi.org/10.3390/pharmaceutics12100955