Silica-Polymer Composites as the Novel Antibiotic Delivery Systems for Bone Tissue Infection

 , , and
, , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Mesoporous Silica Materials (MCM-41)
2.3. Ciprofloxacin Adsorption
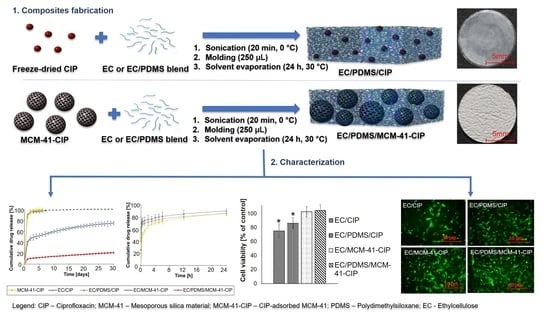
2.4. Composites Fabrication
2.5. Composites Physicochemical Characterization
2.6. Ciprofloxacin In Vitro Release
2.7. Composites Biological Evaluation
2.7.1. Composites Sterilization
2.7.2. Antimicrobial Activity
2.7.3. Cytotoxicity Assay
3. Results and Discussion
3.1. The Synthesis of MCM-41 and CIP Adsorption onto Its Surface
3.2. Fabrication of Composites
3.3. Composites Physicochemical Properties
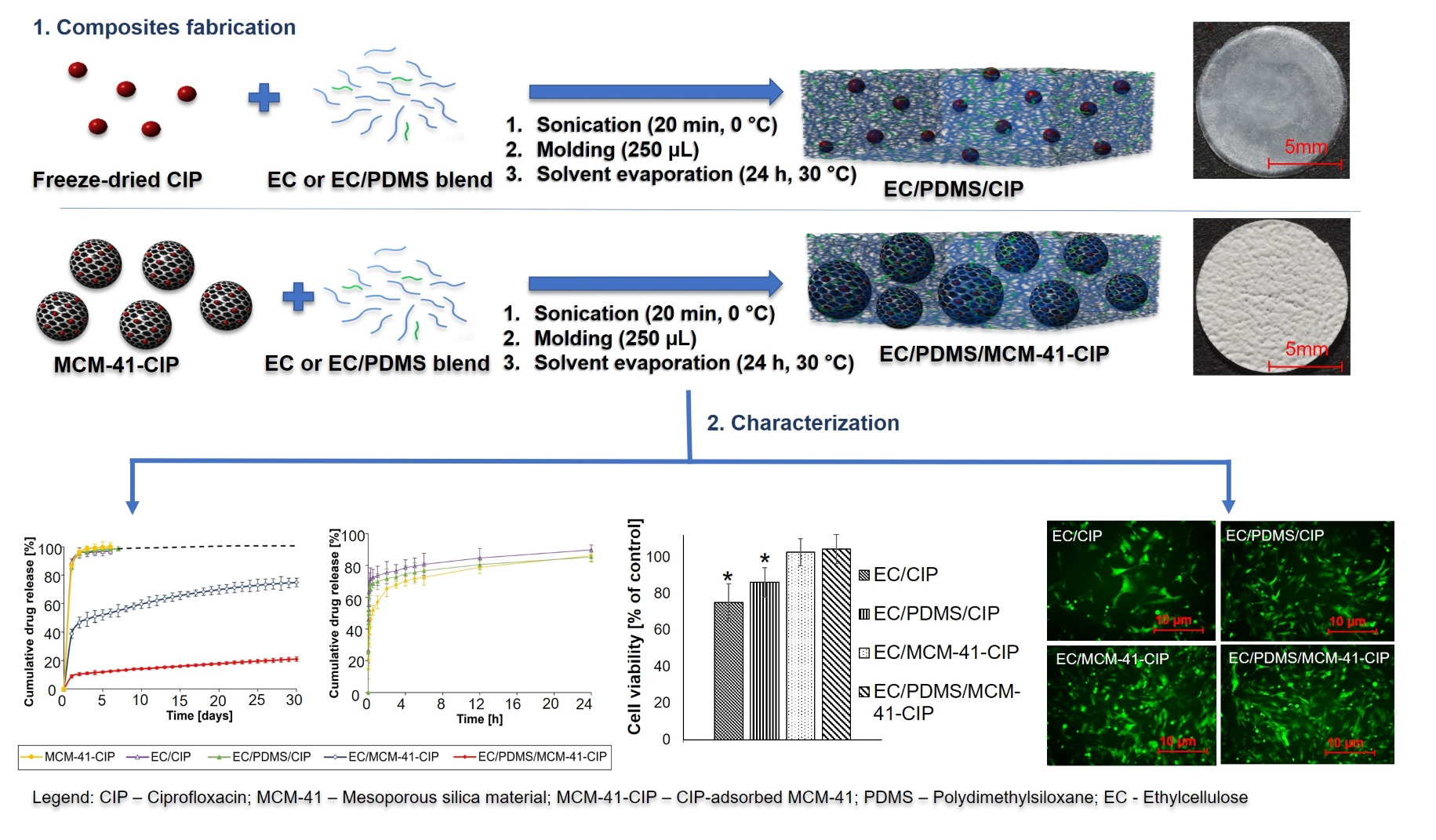
3.3.1. Molecular Structure
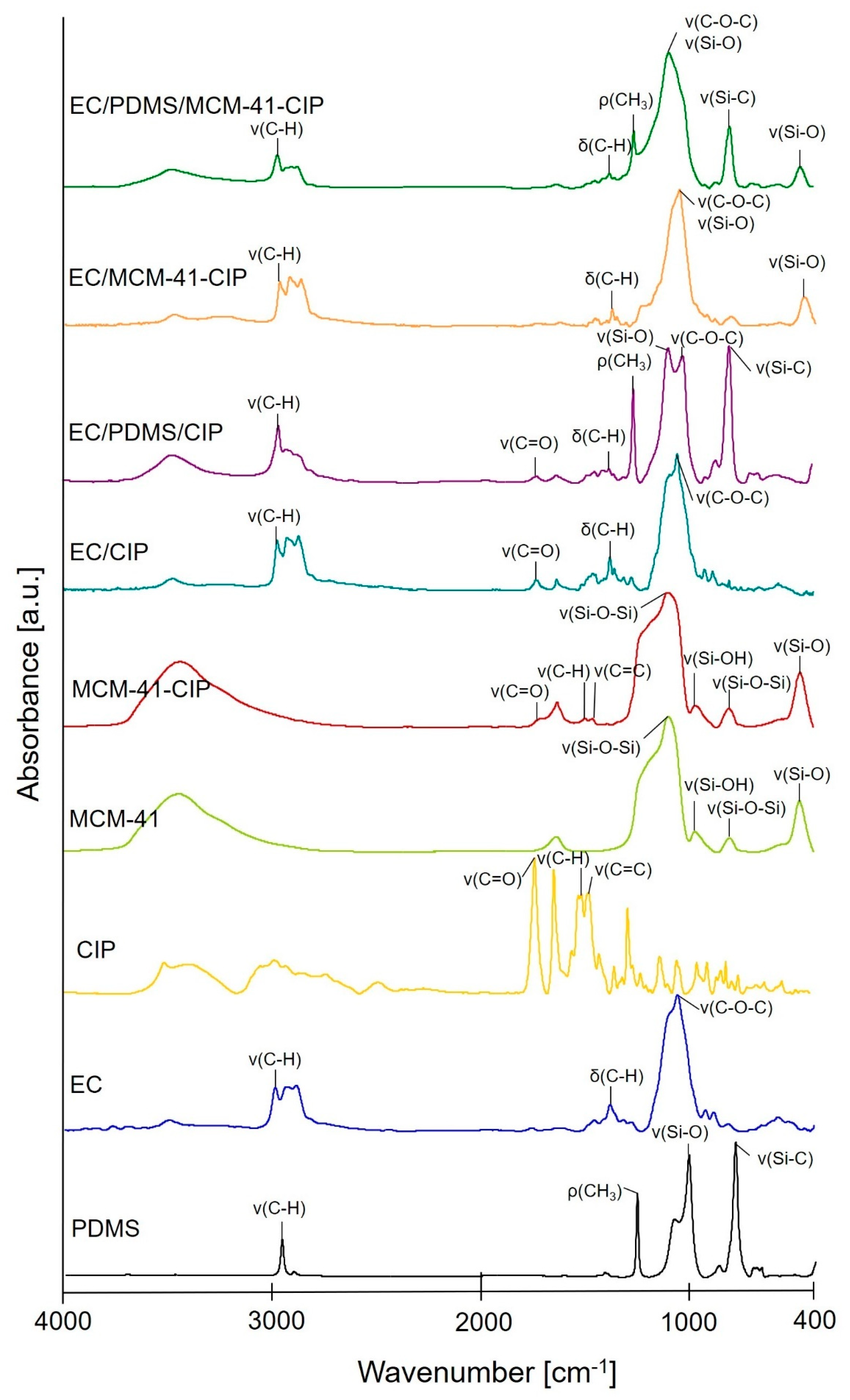
3.3.2. Morphological and Structural Analysis
3.3.3. Texture Analysis
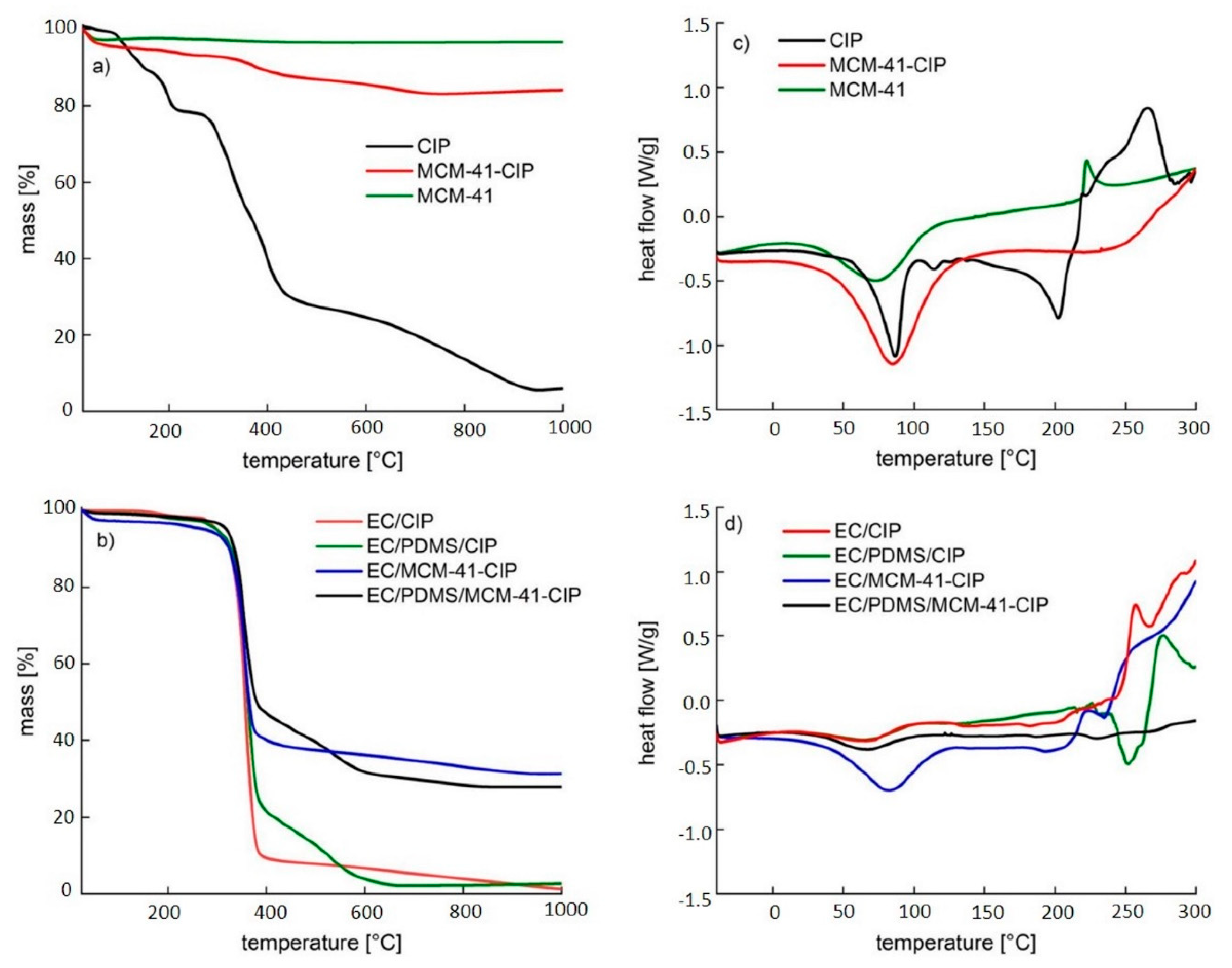
3.3.4. Thermal Analysis
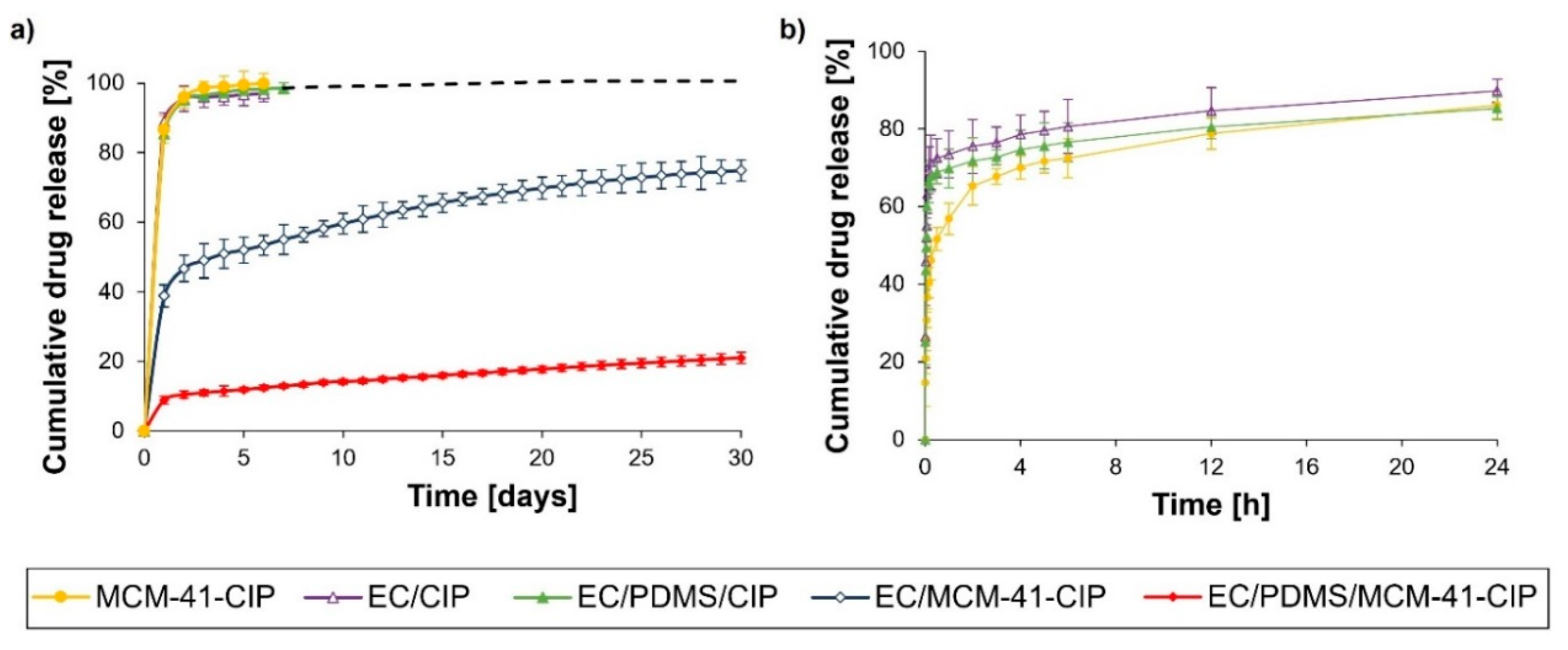
3.3.5. In Vitro Ciprofloxacin Release
3.4. Composites Biological Evaluation
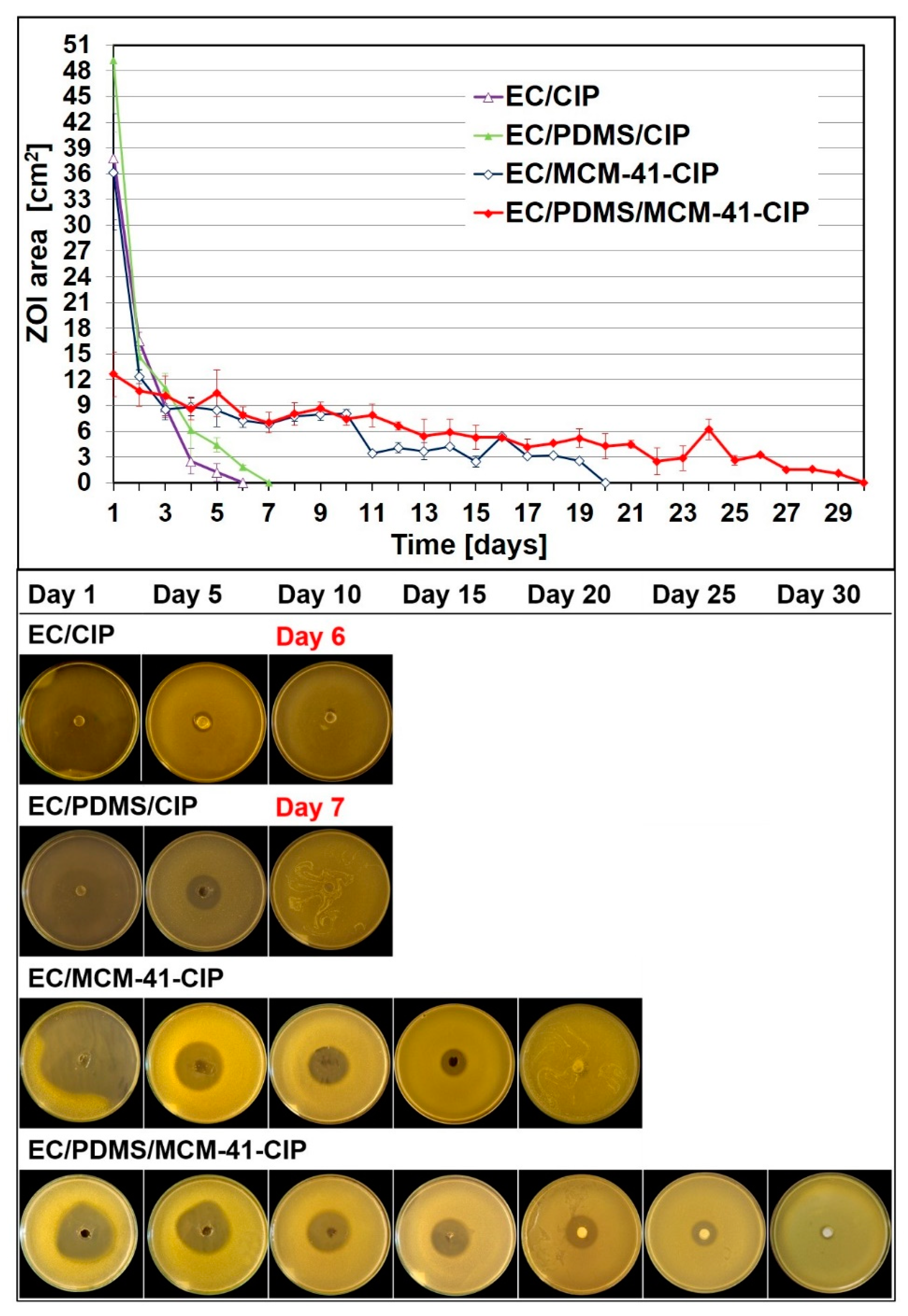
3.4.1. Bacterial Growth Inhibition Assay
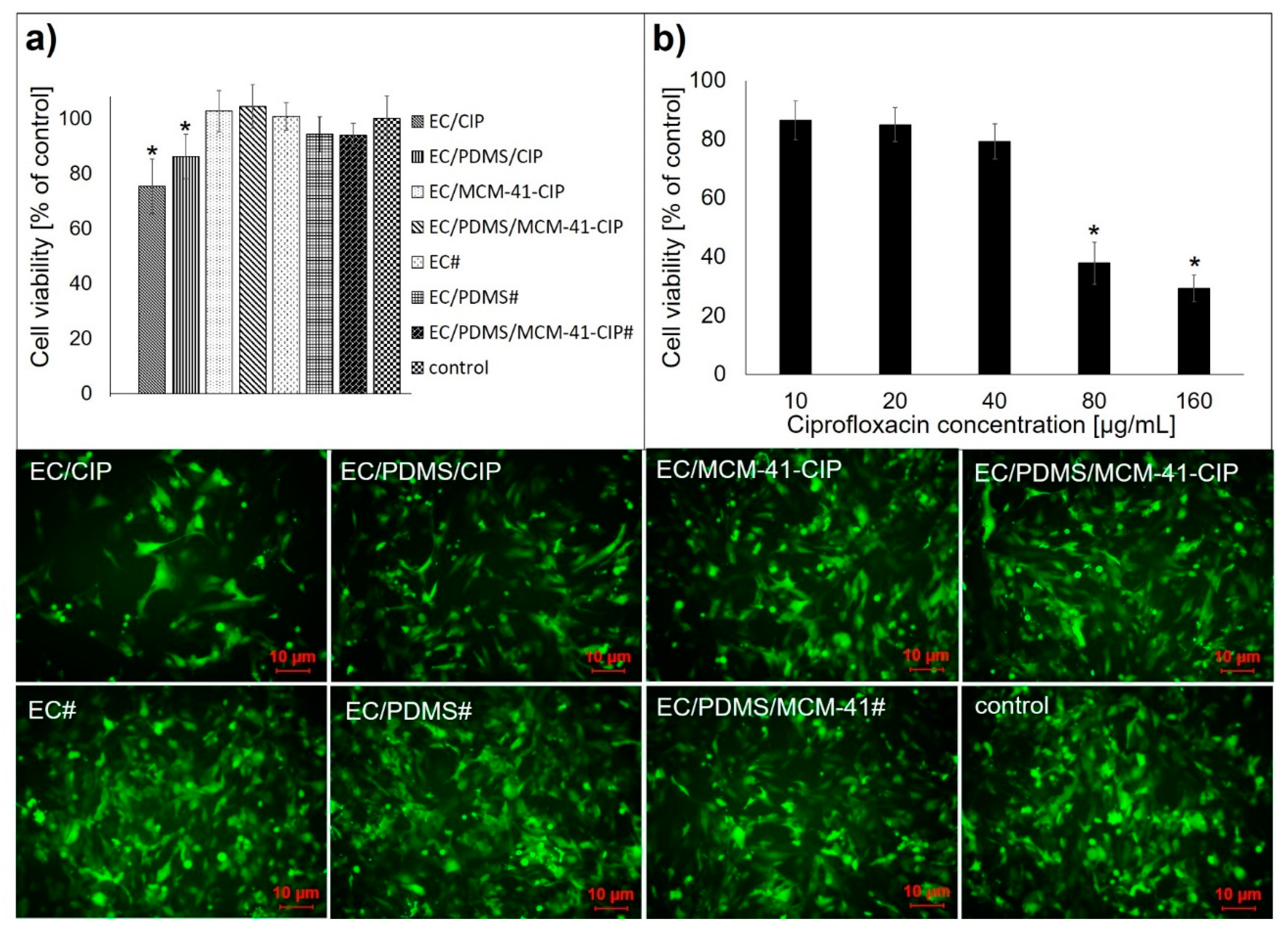
3.4.2. Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shah, M.Q.; Zardad, M.S.; Khan, A.; Ahmed, S.; Awan, A.S.; Mohammad, T. Surgical site infection in orthopaedic implants and its common bacteria with their sensitivities to antibiotics, in open reduction internal fixation. J. Ayub Med. Coll. Abbottabad 2017, 29, 50–53. [Google Scholar]
- Tsourvakas, S. Local antibiotic therapy in the treatment of bone and soft tissue infections. In Selected Topics in Plastic Reconstructive Surgery; Danilla, S., Ed.; IntechOpen: London, UK, 2012; pp. 17–46. [Google Scholar]
- Zalavras, C.G.; Patzakis, M.J.; Holtom, P. Local antibiotic therapy in the treatment of open fractures and osteomyelitis. Clin. Orthop. Relat. Res. 2004, 427, 86–93. [Google Scholar] [CrossRef]
- Soundrapandian, C.; Basu, D.; Sa, B.; Datta, S. Local drug delivery system for the treatment of osteomyelitis: In vitro evaluation. Drug Dev. Ind. Pharm. 2011, 37, 538–546. [Google Scholar] [CrossRef]
- Thabit, A.K.; Fatani, D.F.; Bamakhrama, M.S.; Barnawi, O.A.; Basudan, L.O.; Alhejaili, S.F. Antibiotic penetration into bone and joints: An updated review. Int. J. Infect. Dis. 2019, 81, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Mantripragada, V.P.; Jayasuriya, A.C. Effect of dual delivery of antibiotics (vancomycin and cefazolin) and BMP-7 from chitosan microparticles on Staphylococcus epidermidis and pre-osteoblasts in vitro. Mater. Sci. Eng. C 2016, 67, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Duewelhenke, N.; Krut, O.; Eysel, P. Influence on mitochondria and cytotoxicity of different antibiotics administered in high concentrations on primary human osteoblasts and cell lines. Antimicrob. Agents Chemother. 2007, 51, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Rathbone, C.R.; Cross, J.D.; Brown, K.V.; Murray, C.K.; Wenke, J.C. Effect of various concentrations of antibiotics on osteogenic cell viability and activity. J. Orthop. Res. 2011, 29, 1070–1074. [Google Scholar] [CrossRef]
- Martínez-Carmona, M.; Gunko, Y.K.; Vallet-Regí, M. Mesoporous silica materials as drug delivery: “The nightmare” of bacterial infection. Pharmaceutics 2018, 10, 279. [Google Scholar] [CrossRef]
- Beck, G.R.; Ha, S.W.; Camalier, C.E.; Yamaguchi, M.; Li, Y.; Lee, J.K.; Weitzmann, M.N. Bioactive silica-based nanoparticles stimulate bone-forming osteoblasts, suppress bone-resorbing osteoclasts, and enhance bone mineral density in vivo. Nanomed. Nanotechnol. Biol. Med. 2012, 8, 793–803. [Google Scholar] [CrossRef]
- Nairi, V.; Medda, L.; Monduzzi, M.; Salis, A. Adsorption and release of ampicillin antibiotic from ordered mesoporous silica. J. Colloid Interface Sci. 2017, 497, 217–225. [Google Scholar] [CrossRef]
- Shi, X.; Wang, Y.; Ren, L.; Zhao, N.; Gong, Y.; Wang, D.A. Novel mesoporous silica-based antibiotic releasing scaffold for bone repair. Acta Biomater. 2009, 5, 1697–1707. [Google Scholar] [CrossRef]
- Narayan, R.; Nayak, U.Y.; Raichur, A.M.; Garg, S. Mesoporous silica nanoparticles: A comprehensive review on synthesis and recent advances. Pharmaceutics 2018, 10, 118. [Google Scholar] [CrossRef]
- Izquierdo-Barba, I.; Sousa, E.; Doadrio, J.C.; Doadrio, A.L.; Pariente, J.P.; Martínez, A.; Babonneau, F.; Vallet-Regí, M. Influence of mesoporous structure type on the controlled delivery of drugs: Release of ibuprofen from MCM-48, SBA-15 and functionalized SBA-15. J. Sol-Gel Sci. Technol. 2009, 50, 421–429. [Google Scholar] [CrossRef]
- Natarajan, S.K.; Selvaraj, S. Mesoporous silica nanoparticles: Importance of surface modifications and its role in drug delivery. RSC Adv. 2014, 4, 14328–14334. [Google Scholar] [CrossRef]
- Gounani, Z.; Asadollahi, M.A.; Pedersen, J.N.; Lyngsø, J.; Skov Pedersen, J.; Arpanaei, A.; Meyer, R.L. Mesoporous silica nanoparticles carrying multiple antibiotics provide enhanced synergistic effect and improved biocompatibility. Colloids Surf. B Biointerfaces 2019, 175, 498–508. [Google Scholar] [CrossRef]
- Szewczyk, A.; Prokopowicz, M.; Sawicki, W.; Majda, D.; Walker, G. Aminopropyl-functionalized mesoporous silica SBA-15 as drug carrier for cefazolin: Adsorption profiles, release studies, and mineralization potential. Microporous Mesoporous Mater. 2019, 274, 113–126. [Google Scholar] [CrossRef]
- Varache, M.; Bezverkhyy, I.; Weber, G.; Saviot, L.; Chassagnon, R.; Baras, F.; Bouyer, F. Loading of cisplatin into mesoporous silica nanoparticles: Effect of surface functionalization. Langmuir 2019, 35, 8984–8995. [Google Scholar] [CrossRef]
- Yang, H.; Cheng, B.; Li, Z.; Su, K.; Guo, Q.; Han, P. Organically modified MCM-type material preparation and its usage in controlled ibuprofen delivery. Adv. Mater. Res. 2010, 342, 607–613. [Google Scholar] [CrossRef]
- Doadrio, J.C.; Sousa, E.M.B.; Izquierdo-Barba, I.; Doadrio, A.L.; Perez-Pariente, J.; Vallet-Regí, M. Functionalization of mesoporous materials with long alkyl chains as a strategy for controlling drug delivery pattern. J. Mater. Chem. 2006, 16, 462–466. [Google Scholar] [CrossRef]
- Martín, A.; Morales, V.; Ortiz-Bustos, J.; Pérez-Garnes, M.; Bautista, L.F.; García-Muñoz, R.A.; Sanz, R. Modelling the adsorption and controlled release of drugs from the pure and amino surface-functionalized mesoporous silica hosts. Microporous Mesoporous Mater. 2018, 262, 23–34. [Google Scholar] [CrossRef]
- Maria, G.; Stoica, A.I.; Luta, I.; Stirbet, D.; Radu, G.L. Cephalosporin release from functionalized MCM-41 supports interpreted by various models. Microporous Mesoporous Mater. 2012, 162, 80–90. [Google Scholar] [CrossRef]
- Zhou, X.; Weng, W.; Chen, B.; Feng, W.; Wang, W.; Nie, W.; Chen, L.; Mo, X.; Su, J.; He, C. Mesoporous silica nanoparticles/gelatin porous composite scaffolds with localized and sustained release of vancomycin for treatment of infected bone defects. J. Mater. Chem. B 2018, 6, 740–752. [Google Scholar] [CrossRef]
- Chen, X.; Xu, C.; He, H. Electrospinning of silica nanoparticles-entrapped nanofibers for sustained gentamicin release. Biochem. Biophys. Res. Commun. 2019, 516, 1085–1089. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.M.; Shi, M. PLGA/mesoporous silica hybrid structure for controlled drug release. J. Control. Release 2004, 98, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Zanjanizadeh Ezazi, N.; Shahbazi, M.A.; Shatalin, Y.V.; Nadal, E.; Mäkilä, E.; Salonen, J.; Kemell, M.; Correia, A.; Hirvonen, J.; Santos, H.A. Conductive vancomycin-loaded mesoporous silica polypyrrole-based scaffolds for bone regeneration. Int. J. Pharm. 2018, 536, 241–250. [Google Scholar] [CrossRef]
- Lee, D.W.; Yoo, B.R. Advanced silica/polymer composites: Materials and applications. J. Ind. Eng. Chem. 2016, 38, 1–12. [Google Scholar] [CrossRef]
- Masters, E.A.; Trombetta, R.P.; de Mesy Bentley, K.L.; Boyce, B.F.; Gill, A.L.; Gill, S.R.; Nishitani, K.; Ishikawa, M.; Morita, Y.; Ito, H.; et al. Evolving concepts in bone infection: Redefining “biofilm”, “acute vs. chronic osteomyelitis”, “the immune proteome” and “local antibiotic therapy”. Bone Res. 2019, 7, 20. [Google Scholar]
- Nandi, S.K.; Mukherjee, P.; Roy, S.; Kundu, B.; De, D.K.; Basu, D. Local antibiotic delivery systems for the treatment of osteomyelitis—A review. Mater. Sci. Eng. C 2009, 29, 2478–2485. [Google Scholar] [CrossRef]
- Nayak, A.K.; Bhattacharyya, A.; Sen, K.K. In Vivo ciprofloxacin release from hydroxyapatite-based bone implants in rabbit tibia: A preliminary study. ISRN Orthop. 2011, 2011, 420549. [Google Scholar] [CrossRef]
- Dajcs, J.J.; Thibodeaux, B.A.; Marquart, M.E.; Girgis, D.O.; Traidej, M.; O’Callaghan, R.J. Effectiveness of ciprofloxacin, levofloxacin, or moxifloxacin for treatment of experimental Staphylococcus aureus keratitis. Antimicrob. Agents Chemother. 2004, 48, 1948–1952. [Google Scholar] [CrossRef]
- Tran, T.T.D.; Tran, P.A. Controlled release film forming systems in drug delivery: The potential for efficient drug delivery. Pharmaceutics 2019, 11, 290. [Google Scholar] [CrossRef] [PubMed]
- Adeleke, O.A. Premium ethylcellulose polymer based architectures at work in drug delivery. Int. J. Pharm. X 2019, 1, 100023. [Google Scholar] [CrossRef] [PubMed]
- Regdon, G.; Hegyesi, D.; Pintye-Hódi, K. Thermal study of ethyl cellulose coating films used for modified release (MR) dosage forms. J. Therm. Anal. Calorim. 2012, 108, 347–352. [Google Scholar] [CrossRef]
- Kim, S.H.; Moon, J.H.; Kim, J.H.; Jeong, S.M.; Lee, S.H. Flexible, stretchable and implantable PDMS encapsulated cable for implantable medical device. Biomed. Eng. Lett. 2011, 1, 199–203. [Google Scholar] [CrossRef]
- Raczkowska, J.; Prauzner-Bechcicki, S.; Lukes, J.; Sepitka, J.; Bernasik, A.; Awsiuk, K.; Paluszkiewicz, C.; Pabijan, J.; Lekka, M.; Budkowski, A. Physico-chemical properties of PDMS surfaces suitable as substrates for cell cultures. Appl. Surf. Sci. 2016, 389, 247–254. [Google Scholar] [CrossRef]
- Prokopowicz, M.; Szewczyk, A.; Łunio, R.; Sawicki, W. Monolithic polydimethylsiloxane-modified silica composites prepared by a low-temperature sol–gel micromolding technique for controlled drug release. React. Funct. Polym. 2017, 114, 136–145. [Google Scholar] [CrossRef]
- Prokopowicz, M. Correlation between physicochemical properties of doxorubicin-loaded silica/polydimethylsiloxane xerogel and in vitro release of drug. Acta Biomater. 2009, 5, 193–207. [Google Scholar] [CrossRef]
- Nahrup, J.S.; Gao, Z.M.; Mark, J.E.; Sakr, A. Poly(dimethylsiloxane) coatings for controlled drug release—Polymer modifications. Int. J. Pharm. 2004, 270, 199–208. [Google Scholar] [CrossRef]
- Skwira, A.; Szewczyk, A.; Prokopowicz, M. The effect of polydimethylsiloxane-ethylcellulose coating blends on the surface characterization and drug release of ciprofloxacin-loaded mesoporous silica. Polymers 2019, 11, 1450. [Google Scholar] [CrossRef]
- Mesallati, H.; Umerska, A.; Paluch, K.J.; Tajber, L. Amorphous polymeric drug salts as ionic solid dispersion forms of ciprofloxacin. Mol. Pharm. 2017, 14, 2209–2223. [Google Scholar] [CrossRef]
- Council of Europe. Methods of preparation of sterile products. In European Pharmacopoeia, 9th ed.; Council of Europe: Strasbourg, France, 2017; pp. 4333–4336. [Google Scholar]
- White, J.F.; Verma, S.K. Isolation of endophytic microbes. In Seed Endophytes; Springer: Cham, Switzerland, 2019; pp. 57–63. [Google Scholar]
- Council of Europe. Biological tests. In European Pharmacopoeia, 9th ed.; Council of Europe: Strasbourg, France, 2017; pp. 185–188. [Google Scholar]
- Howlin, R.P.; Brayford, M.J.; Webb, J.S.; Cooper, J.J.; Aiken, S.S.; Stoodley, P. Antibiotic-loaded synthetic calcium sulfate beads for prevention of bacterial colonization and biofilm formation in periprosthetic infections. Antimicrob. Agents Chemother. 2015, 59, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, I.; Hilpert, K.; Hancock, R.E.W. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008, 3, 163–175. [Google Scholar] [CrossRef] [PubMed]
- International Organization for Standardization. Biological Evaluation of Medical Devices—Part 5: Tests for In Vitro Cytotoxicity; ISO 10993-5:2009; ISO: Geneva, Switzerland, 2012. [Google Scholar]
- Qi, R.; Shen, M.; Cao, X.; Guo, R.; Tian, X.; Yu, J.; Shi, X. Exploring the dark side of MTT viability assay of cells cultured onto electrospun PLGA-based composite nanofibrous scaffolding materials. Analyst 2011, 136, 2897–2903. [Google Scholar] [CrossRef] [PubMed]
- Miller, F.; Hinze, U.; Chichkov, B.; Leibold, W.; Lenarz, T.; Paasche, G. Validation of eGFP fluorescence intensity for testing in vitro cytotoxicity according to ISO 10993-5. J. Biomed. Mater. Res. B Appl. Biomater. 2017, 105, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Kecht, J.; Bein, T. Oxidative removal of template molecules and organic functionalities in mesoporous silica nanoparticles by H2O2 treatment. Microporous Mesoporous Mater. 2008, 116, 123–130. [Google Scholar] [CrossRef]
- Roik, N.V.; Belyakova, L.A.; Dziazko, M.O. Adsorption of antitumor antibiotic doxorubicin on MCM-41-type silica surface. Adsorpt. Sci. Technol. 2017, 35, 1–16. [Google Scholar] [CrossRef]
- Moodley, T.; Singh, M. Polymeric mesoporous silica nanoparticles for enhanced delivery of 5-Fluorouracil in vitro. Pharmaceutics 2019, 11, 288. [Google Scholar] [CrossRef]
- Hung, C.C.; Huang, W.C.; Lin, Y.W.; Yu, T.W.; Chen, H.H.; Lin, S.C.; Chiang, W.H.; Chiu, H.C. Active tumor permeation and uptake of surface charge-switchable theranostic nanoparticles for imaging-guided photothermal/chemo combinatorial therapy. Theranostics 2016, 6, 302–317. [Google Scholar] [CrossRef]
- Li, X.; Han, C.; Zhu, W.; Ma, W.; Luo, Y.; Zhou, Y.; Yu, J.; Wei, K. Cr(VI) removal from aqueous by adsorption on amine-functionalized mesoporous silica prepared from silica fume. J. Chem. 2014, 2014, 765856. [Google Scholar] [CrossRef]
- Durgapal, S.; Mukhopadhyay, S.; Goswami, L. Preparation, characterization and evaluation of floating microparticles of ciprofloxacin. Int. J. Appl. Pharm. 2017, 9, 1–8. [Google Scholar] [CrossRef]
- Trivedi, M.K.; Branton, A.; Trivedi, D.; Nayak, G.; Mishra, R.K.; Jana, S. Characterization of physicochemical and thermal properties of biofield treated ethyl cellulose and methyl cellulose. Int. J. Biomed. Mater. Res. 2015, 3, 83–91. [Google Scholar]
- Hanoosh, W.S.; Abdelrazaq, E.M. Polydimethyl siloxane toughened epoxy resins: Tensile strength and dynamic mechanical analysis. Malays. Polym. J. 2009, 4, 52–61. [Google Scholar]
- Siddiqui, S.; Siddiqui, Z.N. Strontium doped MCM-41: A highly efficient, recyclable and heterogeneous catalyst for the synthesis of phenoxy pyrazolyl pyrazolines. Catal. Lett. 2018, 148, 3628–3645. [Google Scholar] [CrossRef]
- Turel, I.; Bukovec, P. Comparison of the thermal stability of ciprofloxacin and its compounds. Thermochim. Acta 1996, 287, 311–318. [Google Scholar] [CrossRef]
- Galarneau, A.; Nader, M.; Guenneau, F.; Di Renzo, F.; Gedeon, A. Understanding the stability in water of mesoporous SBA-15 and M CM-41. J. Phys. Chem. C 2007, 111, 8268–8277. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of Composite | 1 EC Content [µL] | PDMS Content [µL] | 2 MCM-41-CIP Content [mg] | 2 CIP Content [mg] |
|---|---|---|---|---|
| EC/CIP | 250 | - | - | 0.79 ± 0.05 |
| EC/PDMS/CIP | 245 | 5 | - | |
| EC/MCM-41-CIP | 250 | - | 6.0 ± 0.02 | |
| EC/PDMS/MCM-41-CIP | 245 | 5 |
| Name of Composite | Rupture Force [N] | Elasticity [mm] | Firmness [N mm−1] | Weight [mg] | Thickness [μm] |
|---|---|---|---|---|---|
| EC/CIP | 0.48 ± 0.16 | 0.58 ± 0.12 | 0.68 ± 0.22 | 10.84 ± 0.16 | 42 ± 2 |
| EC/PDMS/CIP | 0.66 ± 0.06 | 0.80 ± 0.33 | 0.97 ± 0.41 | 14.81 ± 0.18 | 44 ± 1 |
| EC/MCM-41-CIP | 1.25 ± 0.34 | 0.50 ± 0.21 | 2.5 ± 0.76 | 16.12 ± 0.22 | 52 ± 3 |
| EC/PDMS/MCM-41-CIP | 1.97 ± 0.21 | 0.62 ± 0.14 | 3.22 ± 0.81 | 20.58 ± 0.32 | 53 ± 2 |
| EC | 0.54 ± 0.28 | 0.80 ± 0.18 | 0.67 ± 0.18 | 10.02 ± 0.11 | 38 ± 2 |
| EC/PDMS | 0.71 ± 0.14 | 1.03 ± 0.22 | 0.72 ± 0.01 | 14.64 ± 0.28 | 40 ± 4 |
| Name of Composite | Linear Regression Equation | R2 | k0 (%/day) | 1 Estimated Dose of Drug Released (µg/day) |
|---|---|---|---|---|
| EC/CIP | y = 0.35x + 94.9 | 0.919 | 0.35 | 2.8 |
| EC/PDMS/CIP | y = 0.64x + 94.4 | 0.932 | 0.64 | 5.1 |
| EC/MCM-41-CIP | y = 0.99x + 48.6 | 0.954 | 0.99 | 7.8 |
| EC/PDMS/MCM-41-CIP | y = 0.73x + 10.2 | 0.995 | 0.73 | 5.8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skwira, A.; Szewczyk, A.; Konopacka, A.; Górska, M.; Majda, D.; Sądej, R.; Prokopowicz, M. Silica-Polymer Composites as the Novel Antibiotic Delivery Systems for Bone Tissue Infection. Pharmaceutics 2020, 12, 28. https://doi.org/10.3390/pharmaceutics12010028
Skwira A, Szewczyk A, Konopacka A, Górska M, Majda D, Sądej R, Prokopowicz M. Silica-Polymer Composites as the Novel Antibiotic Delivery Systems for Bone Tissue Infection. Pharmaceutics. 2020; 12(1):28. https://doi.org/10.3390/pharmaceutics12010028
Chicago/Turabian StyleSkwira, Adrianna, Adrian Szewczyk, Agnieszka Konopacka, Monika Górska, Dorota Majda, Rafał Sądej, and Magdalena Prokopowicz. 2020. "Silica-Polymer Composites as the Novel Antibiotic Delivery Systems for Bone Tissue Infection" Pharmaceutics 12, no. 1: 28. https://doi.org/10.3390/pharmaceutics12010028
APA StyleSkwira, A., Szewczyk, A., Konopacka, A., Górska, M., Majda, D., Sądej, R., & Prokopowicz, M. (2020). Silica-Polymer Composites as the Novel Antibiotic Delivery Systems for Bone Tissue Infection. Pharmaceutics, 12(1), 28. https://doi.org/10.3390/pharmaceutics12010028