Design and Evaluation of a Poly(Lactide-co-Glycolide)-Based In Situ Film-Forming System for Topical Delivery of Trolamine Salicylate


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Development of Film-Forming Polymeric Solutions
2.2.1. Screening of Components
2.2.2. Optimization of Formulations
2.2.3. Effect of Plasticizer PEG 400, Types of PLGA, and Various Concentration of PLGA for Topical Delivery of TS
2.3. Evaluation of the Formulations
2.3.1. Crystallization Study and Solvent Evaporation Study
2.3.2. In Vitro TS Release Study
2.3.3. In Vitro Permeation Study
Skin Preparation
Evaluation of Skin Integrity
2.4. Quantitative Analysis
2.5. Statistical Analyses
3. Results
3.1. In Situ Film-Forming Behavior
3.2. Crystallization Study and Solvent Evaporation Study
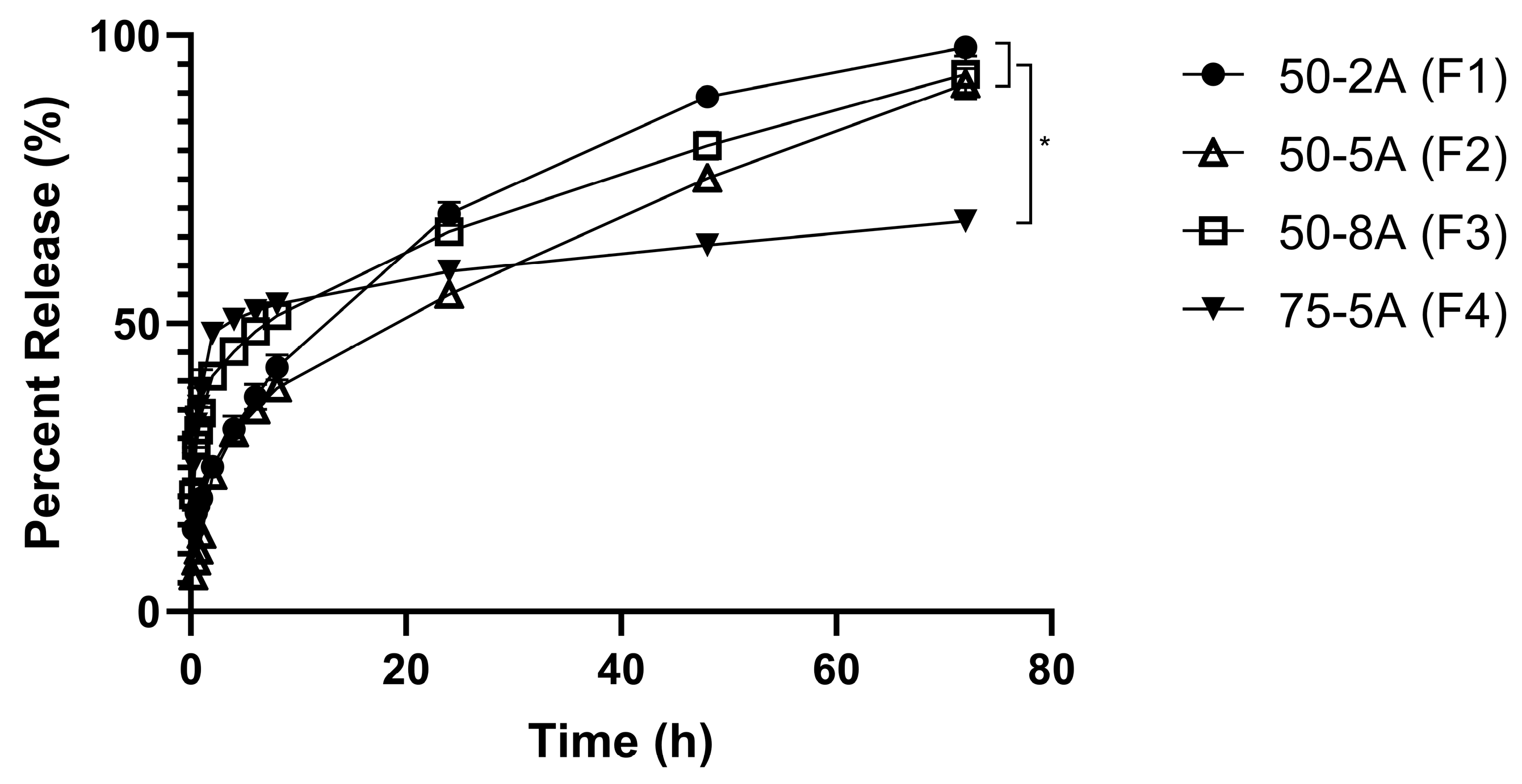
3.3. In Vitro Release Study for Different Types of PLGA
3.4. Effects of Each Ingredient and Permeation Study
3.4.1. Effects of Plasticizer
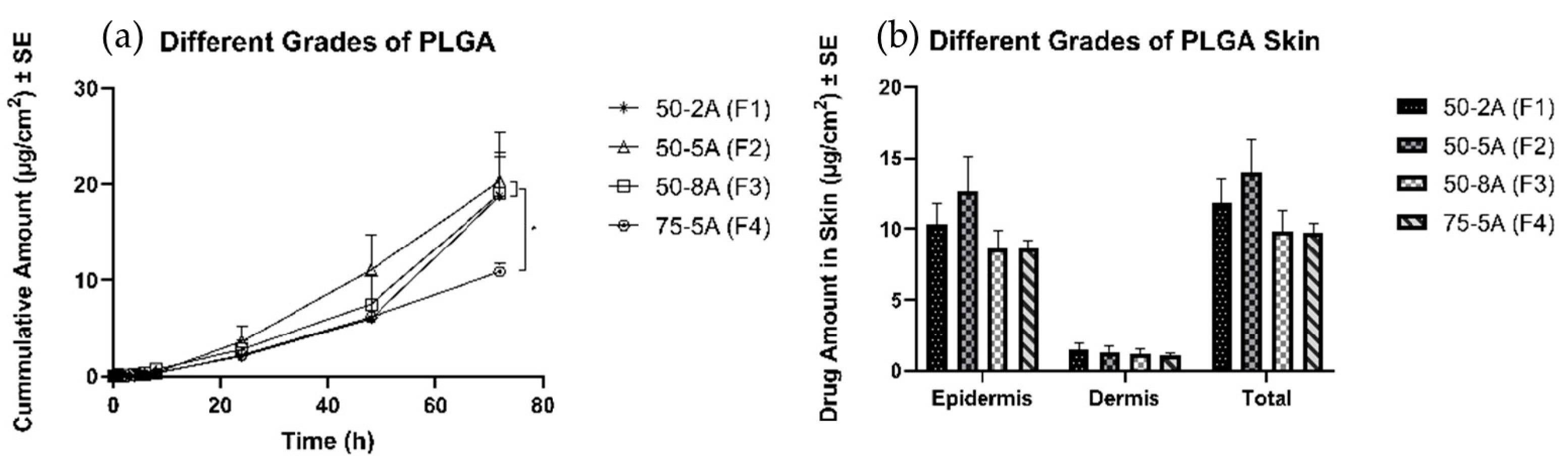
3.4.2. Effects of Different Types of PLGA
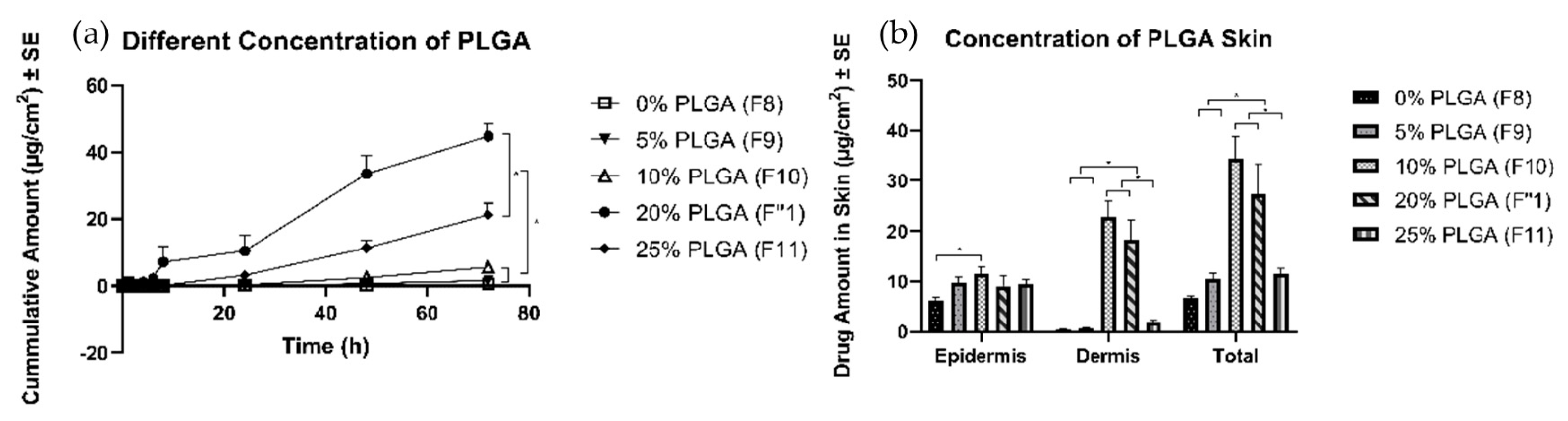
3.4.3. Effects of Concentration of PLGA
4. Discussion
4.1. Parameters for the Development of Film-Forming Polymeric Solutions
4.2. Characterization Methods for Film-Forming Polymeric Solutions
4.3. In Vitro Release Study for Different Types of PLGA
4.4. In Vitro Permeation Study
4.4.1. Effects of Plasticizers
4.4.2. Effects of Different Types of PLGA
4.4.3. Effects of Different Amounts of PLGA
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Singh, G. Recent considerations in nonsteroidal anti-inflammatory drug gastropathy. Am. J. Med. 1998, 105, 31S–38S. [Google Scholar] [CrossRef]
- Banga, A.K. Transdermal and Intradermal Delivery of Therapeutic Agents: Application of Physical Technologies; CRC Press: Boca Raton, FL, USA, 2011. [Google Scholar]
- Trottet, L.; Maibach, H. Dermal Drug Selection and Development; Springer: Berlin, Germany, 2017. [Google Scholar]
- Cross, S.E.; Anderson, C.; Roberts, M.S. Topical penetration of commercial salicylate esters and salts using human isolated skin and clinical microdialysis studies. Br. J. Clin. Pharmacol. 1998, 46, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Barkin, R.L. The pharmacology of topical analgesics. Postgrad. Med. 2013, 125, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Alkilani, A.Z.; McCrudden, M.T.; Donnelly, R.F. Transdermal drug delivery: Innovative pharmaceutical developments based on disruption of the barrier properties of the stratum corneum. Pharmaceutics 2015, 7, 438–470. [Google Scholar] [CrossRef] [PubMed]
- Wiedersberg, S.; Guy, R.H. Transdermal drug delivery: 30+ years of war and still fighting! J. Control. Release 2014, 190, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Haroutiunian, S.; Drennan, D.A.; Lipman, A.G. Topical NSAID therapy for musculoskeletal pain. Pain Med. 2010, 11, 535–549. [Google Scholar] [CrossRef] [PubMed]
- Cagnie, B.; Vinck, E.; Rimbaut, S.; Vanderstraeten, G. Phonophoresis versus topical application of ketoprofen: comparison between tissue and plasma levels. Phys. Ther. 2003, 83, 707–712. [Google Scholar] [PubMed]
- Schroeder, I.Z.; Franke, P.; Schaefer, U.F.; Lehr, C.-M. Development and characterization of film forming polymeric solutions for skin drug delivery. Eur. J. Pharm. Biopharm. 2007, 65, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Lü, J.-M.; Wang, X.; Marin-Muller, C.; Wang, H.; Lin, P.H.; Yao, Q.; Chen, C. Current advances in research and clinical applications of PLGA-based nanotechnology. Expert Rev. Mol. Diagn. 2009, 9, 325–341. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, F.; van der Walle, C.F. Engineering biodegradable polyester particles with specific drug targeting and drug release properties. J. Pharm. Sci. 2008, 97, 71–87. [Google Scholar] [CrossRef]
- Allison, S.D. Effect of structural relaxation on the preparation and drug release behavior of poly (lactic-co-glycolic) acid microparticle drug delivery systems. J. Pharm. Sci. 2008, 97, 2022–2035. [Google Scholar] [CrossRef] [PubMed]
- Mundargi, R.C.; Babu, V.R.; Rangaswamy, V.; Patel, P.; Aminabhavi, T.M. Nano/micro technologies for delivering macromolecular therapeutics using poly (d, l-lactide-co-glycolide) and its derivatives. J. Control. Release 2008, 125, 193–209. [Google Scholar] [CrossRef] [PubMed]
- Makadia, H.K.; Siegel, S.J. Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef] [PubMed]
- Bakshi, P.; Jiang, Y.; Nakata, T.; Akaki, J.; Matsuoka, N.; Banga, A.K. Formulation development and characterization of nanoemulsion-based formulation for topical delivery of heparinoid. J. Pharm. Sci. 2018, 107, 2883–2890. [Google Scholar] [CrossRef] [PubMed]
- Kathe, K.; Kathpalia, H. Film forming systems for topical and transdermal drug delivery. Asian J. Pharm. Sci. 2017, 12, 487–497. [Google Scholar] [CrossRef]
- Amarante, M.; Constantinescu, M.A.; O’Connor, D.; Yaremchuk, M.J. Cyanoacrylate fixation of the craniofacial skeleton: an experimental study. Plast. Reconstr. Surg. 1995, 95, 639–646. [Google Scholar] [CrossRef]
- Bonutti, P.M.; Weiker, G.G.; Andrish, J.T. Isobutyl cyanoacrylate as a soft tissue adhesive. An in vitro study in the rabbit Achilles tendon. Clin. Orthop. Relat. Res. 1988, 229, 241–248. [Google Scholar]
- Ambrose, C.G.; Clyburn, T.A.; Louden, K.; Joseph, J.; Wright, J.; Gulati, P.; Gogola, G.R.; Mikos, A.G. Effective treatment of osteomyelitis with biodegradable microspheres in a rabbit model. Clin. Orthop. Relat. Res. 2004, 421, 293–299. [Google Scholar] [CrossRef]
- Cheski, P.J.; Matthews, T.W. Endoscopic reduction and internal cyanoacrylate fixation of the zygoma. J. Otolaryngol. 1997, 26, 2. [Google Scholar]
- Meskin, S.W.; Ritterband, D.C.; Shapiro, D.E.; Kusmierczyk, J.; Schneider, S.S.; Seedor, J.A.; Koplin, R.S. Liquid bandage (2-octyl cyanoacrylate) as a temporary wound barrier in clear corneal cataract surgery. Ophthalmology 2005, 112, 2015–2021. [Google Scholar] [CrossRef]
- Shermak, M.A.; Wong, L.; Inoue, N.; Crain, B.J.; Im, M.J.; Chao, E.; Manson, P.N. Fixation of the craniofacial skeleton with butyl-2-cyanoacrylate and its effects on histotoxicity and healing. Plast. Reconstr. Surg. 1998, 102, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Trott, A.T. Cyanoacrylate tissue adhesives: an advance in wound care. JAMA 1997, 277, 1559–1560. [Google Scholar] [CrossRef] [PubMed]
- Eskandari, M.M.; Ozturk, O.G.; Eskandari, H.G.; Balli, E.; Yilmaz, C. Cyanoacrylate adhesive provides efficient local drug delivery. Clin. Orthop. Relat. Res. 2006, 451, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Williams, A. Chemical penetration enhancement, possibilities and problems. Dermal Absorpt. Toxic. Assess. 1998, 12, 645–651. [Google Scholar]
- US Food and Drug Administration. Inactive Ingredient Search for Approved Drug Products; FDA Database: Silver Spring, MD, USA, 2017. [Google Scholar]
- Hansen, H.; Wilbur, S.B. Toxicological Profile for Acetone; Agency for Toxic Substances and Disease Registry: Atlanta, GA, USA, 1994. [Google Scholar]
- Gal, A.; Nussinovitch, A. Plasticizers in the manufacture of novel skin-bioadhesive patches. Int. J. Pharm. 2009, 370, 103–109. [Google Scholar] [CrossRef]
- Bharkatiya, M.; Nema, R.; Bhatnagar, M. Designing and characterization of drug free patches for transdermal application. Int. J. Pharm. Sci. Drug Res. 2010, 2, 35–39. [Google Scholar]
- Jain, P.; Banga, A.K. Induction and inhibition of crystallization in drug-in-adhesive-type transdermal patches. Pharm. Res. 2013, 30, 562–571. [Google Scholar] [CrossRef]
- Liechty, W.B.; Kryscio, D.R.; Slaughter, B.V.; Peppas, N.A. Polymers for drug delivery systems. Annu. Rev. Chem. Biomol. Eng. 2010, 1, 149–173. [Google Scholar] [CrossRef]
- Duncan, R. Polymer conjugates as anticancer nanomedicines. Nat. Rev. Cancer 2006, 6, 688. [Google Scholar] [CrossRef]
- Naves, L.; Dhand, C.; Almeida, L.; Rajamani, L.; Ramakrishna, S.; Soares, G. Poly (lactic-co-glycolic) acid drug delivery systems through transdermal pathway: An overview. Prog. Biomater. 2017, 6, 1–11. [Google Scholar] [CrossRef]
- Marsac, P.J.; Shamblin, S.L.; Taylor, L.S. Theoretical and practical approaches for prediction of drug–polymer miscibility and solubility. Pharm. Res. 2006, 23, 2417. [Google Scholar] [CrossRef]
- Mahieu, A.l.; Willart, J.-F.o.; Dudognon, E.; Danède, F.; Descamps, M. A new protocol to determine the solubility of drugs into polymer matrixes. Mol. Pharm. 2013, 10, 560–566. [Google Scholar] [CrossRef]
- Kim, J.-H.; Choi, H.-K. Effect of additives on the crystallization and the permeation of ketoprofen from adhesive matrix. Int. J. Pharm. 2002, 236, 81–85. [Google Scholar] [CrossRef]
- Guideline, I.H.T. Impurities: Guideline for residual solvents Q3C (R5). Curr. Step 2005, 4, 1–25. [Google Scholar]
- Baino, F. Biomaterials and implants for orbital floor repair. Acta Biomater. 2011, 7, 3248–3266. [Google Scholar] [CrossRef]
- D’Souza, S.S.; Faraj, J.A.; DeLuca, P.P. A model-dependent approach to correlate accelerated with real-time release from biodegradable microspheres. Aaps Pharmscitech 2005, 6, E553–E564. [Google Scholar] [CrossRef]
- Fredenberg, S.; Wahlgren, M.; Reslow, M.; Axelsson, A. The mechanisms of drug release in poly (lactic-co-glycolic acid)-based drug delivery systems—A review. Int. J. Pharm. 2011, 415, 34–52. [Google Scholar] [CrossRef]
- Salamanca, C.; Barrera-Ocampo, A.; Lasso, J.; Camacho, N.; Yarce, C. Franz diffusion cell approach for pre-formulation characterisation of ketoprofen semi-solid dosage forms. Pharmaceutics 2018, 10, 148. [Google Scholar] [CrossRef]
- Abd, E.; Yousef, S.A.; Pastore, M.N.; Telaprolu, K.; Mohammed, Y.H.; Namjoshi, S.; Grice, J.E.; Roberts, M.S. Skin models for the testing of transdermal drugs. Clin. Pharmacol. Adv. Appl. 2016, 8, 163. [Google Scholar] [CrossRef]
- Miranda, M.; Sousa, J.J.; Veiga, F.; Cardoso, C.; Vitorino, C. Bioequivalence of topical generic products. Part 1: Where are we now? Eur. J. Pharm. Sci. 2018, 123, 260–267. [Google Scholar] [CrossRef]
- Roberts, M. Correlation of Physicochemical Characteristics and In Vitro Permeation Test (IVPT) Results for Acyclovir and Metronidazole Topical Products. Available online: https://www.fda.gov/media/110256/download (accessed on 31 July 2019).
- U.S. Food and Drug Administration. FY2016 Regulatory Science Report: Topical Dermatological Drug Products. Available online: https://www.fda.gov/industry/generic-drug-user-fee-amendments/fy2016-regulatory-science-report-topical-dermatological-drug-products (accessed on 31 July 2019).
- Wypych, G. Handbook of Plasticizers; ChemTec Publishing: Toronto, ON, Canada, 2004. [Google Scholar]
- Barhate, S.D.; Patel, M.; Sharma, A.S.; Nerkar, P.; Shankhpal, G. Formulation and evaluation of transdermal drug delivery system of carvedilol. J. Pharm. Res. 2009, 2, 663–665. [Google Scholar]
- Chereddy, K.K.; Payen, V.L.; Préat, V. PLGA: From a classic drug carrier to a novel therapeutic activity contributor. J. Control. Release 2018, 289, 10–13. [Google Scholar] [CrossRef]
- Martanto, W.; Davis, S.P.; Holiday, N.R.; Wang, J.; Gill, H.S.; Prausnitz, M.R. Transdermal delivery of insulin using microneedles in vivo. Pharm. Res. 2004, 21, 947–952. [Google Scholar] [CrossRef]
- Farahani, T.D.; Entezami, A.A.; Mobedi, H.; Abtahi, M. Degradation of poly (d, l-lactide-co-glycolide) 50: 50 implant in aqueous medium. Iran. Polym. J. 2005, 14, 753–763. [Google Scholar]
- Bré, L.P.; Zheng, Y.; Pêgo, A.P.; Wang, W. Taking tissue adhesives to the future: from traditional synthetic to new biomimetic approaches. Biomater. Sci. 2013, 1, 239–253. [Google Scholar] [CrossRef]
- Leonard, F.; Kulkarni, R.; Brandes, G.; Nelson, J.; Cameron, J.J. Synthesis and degradation of poly (alkyl α-cyanoacrylates). J. Appl. Polym. Sci. 1966, 10, 259–272. [Google Scholar] [CrossRef]
- Petersen, B.; Barkun, A.; Carpenter, S.; Chotiprasidhi, P.; Chuttani, R.; Silverman, W.; Hussain, N.; Liu, J.; Taitelbaum, G.; Ginsberg, G.G. Tissue adhesives and fibrin glues: November 2003. Gastrointest. Endosc. 2004, 60, 327–333. [Google Scholar] [CrossRef]
- Kull, S.; Martinelli, I.; Briganti, E.; Losi, P.; Spiller, D.; Tonlorenzi, S.; Soldani, G. Glubran2 surgical glue: in vitro evaluation of adhesive and mechanical properties. J. Surg. Res. 2009, 157, e15–e21. [Google Scholar] [CrossRef]
- Bhagat, V.; Becker, M.L. Degradable adhesives for surgery and tissue engineering. Biomacromolecules 2017, 18, 3009–3039. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PLGA Polymer | Lactide/Glycolide Molar Ratio | End Group | Molecular Weight (Indicative Range, kDa) |
|---|---|---|---|
| EXPANSORB® DLG 50-2A | 1:1 | –COOH | 5–20 |
| EXPANSORB® DLG 50-5A | 1:1 | –COOH | 42–65 |
| EXPANSORB® DLG 50-8A | 1:1 | –COOH | 80–130 |
| EXPANSORB® DLG 75-5A | 3:1 | –COOH | 37–84 |
| Different Types of PLGA | Different Amounts of PEG 400 | Different Concentrations of PLGA | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F1 | F2 | F3 | F4 | F1′ | F5 | F6 | F7 | F1″ | F8 | F9 | F10 | F11 | |
| Code | 50-2A | 50-5A | 50-8A | 75-5A | 4:1:1 | 4:1:0 | 4:1:2 | 4:1:3 | 20% | 0% | 5% | 10% | 25% |
| Polymer | 50-2A | 50-5A | 50-8A | 75-5A | 50-2A | 50-2A | 50-2A | 50-2A | 50-2A | 50-2A | 50-2A | 50-2A | 50-2A |
| PLGA | 20 | 20 | 20 | 20 | 20 | 24 | 17 | 15 | 20 | 0 | 5 | 10 | 25 |
| Cyanoacrylate | 5 | 5 | 5 | 5 | 5 | 6 | 4.25 | 3.75 | 5 | 15 | 12.5 | 10 | 2.5 |
| Plasticizer | |||||||||||||
| PEG 400 | 5 | 5 | 5 | 5 | 5 | 0 | 8.75 | 11.25 | 5 | 15 | 12.5 | 10 | 2.5 |
| Solvent | |||||||||||||
| Acetone | 69 | 69 | 69 | 69 | 69 | 69 | 69 | 69 | 69 | 69 | 69 | 69 | 69 |
| Drug | |||||||||||||
| Trolamine Salicylate | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Groups | Epidermis (μg/cm2) | Dermis (μg/cm2) | Total (μg/cm2) | |
|---|---|---|---|---|
| Different types of PLGA | 50-2A (F1) | 10.4 ± 1.4 | 1.5 ± 0.4 | 11.9 ± 1.7 |
| 50-5A (F2) | 12.6 ± 2.5 | 1.3 ± 0.4 | 13.9 ± 2.4 | |
| 50-8A (F3) | 8.6 ± 1.3 | 1.1 ± 0.4 | 9.8 ± 1.5 | |
| 75-5A (F4) | 8.7 ± 0.5 | 1.0 ± 0.2 | 9.7 ± 0.7 | |
| Effect of plasticizer | 4:1:1 (F1’) | 8.6 ± 0.6 | 1.6 ± 0.3 | 10.1 ± 0.6 |
| 4:1:0 (F5) | 2.5 ± 0.2 | 0.3 ± 0.1 | 2.7 ± 0.3 | |
| 4:1:2 (F6) | 14.3 ± 2.5 | 1.9 ± 0.5 | 16.2 ± 2.8 | |
| 4:1:3 (F7) | 14.2 ± 0.7 | 3.7 ± 0.6 | 17.9 ± 1.0 | |
| Different amount of PLGA | 20% (F1″) | 9.1 ± 2.0 | 18.2 ± 4.0 | 27.4 ± 6.0 |
| 0% (F8) | 6.3 ± 0.6 | 0.40 ± 0.1 | 6.7 ± 0.5 | |
| 5% (F9) | 9.9 ± 1.0 | 0.62 ± 0.2 | 10.5 ± 1.2 | |
| 10% (F10) | 11.5 ± 1.5 | 22.9 ± 3.0 | 34.4 ± 4.5 | |
| 25% (F11) | 9.5 ± 0.8 | 1.9 ± 0.4 | 11.4 ± 1.1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.; Beck-Broichsitter, M.; Banga, A.K. Design and Evaluation of a Poly(Lactide-co-Glycolide)-Based In Situ Film-Forming System for Topical Delivery of Trolamine Salicylate. Pharmaceutics 2019, 11, 409. https://doi.org/10.3390/pharmaceutics11080409
Kim Y, Beck-Broichsitter M, Banga AK. Design and Evaluation of a Poly(Lactide-co-Glycolide)-Based In Situ Film-Forming System for Topical Delivery of Trolamine Salicylate. Pharmaceutics. 2019; 11(8):409. https://doi.org/10.3390/pharmaceutics11080409
Chicago/Turabian StyleKim, Yujin, Moritz Beck-Broichsitter, and Ajay K. Banga. 2019. "Design and Evaluation of a Poly(Lactide-co-Glycolide)-Based In Situ Film-Forming System for Topical Delivery of Trolamine Salicylate" Pharmaceutics 11, no. 8: 409. https://doi.org/10.3390/pharmaceutics11080409
APA StyleKim, Y., Beck-Broichsitter, M., & Banga, A. K. (2019). Design and Evaluation of a Poly(Lactide-co-Glycolide)-Based In Situ Film-Forming System for Topical Delivery of Trolamine Salicylate. Pharmaceutics, 11(8), 409. https://doi.org/10.3390/pharmaceutics11080409