Mucosal Vaccination via the Respiratory Tract


Abstract
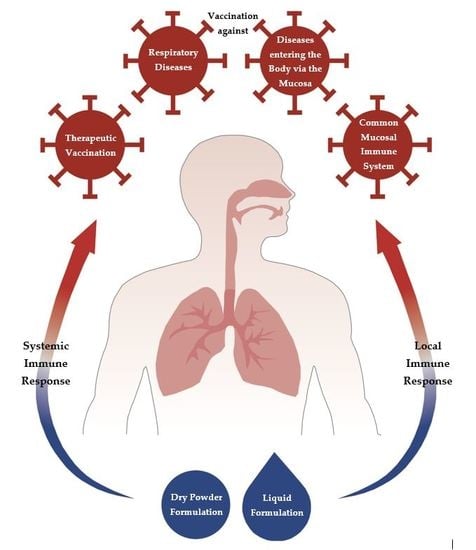
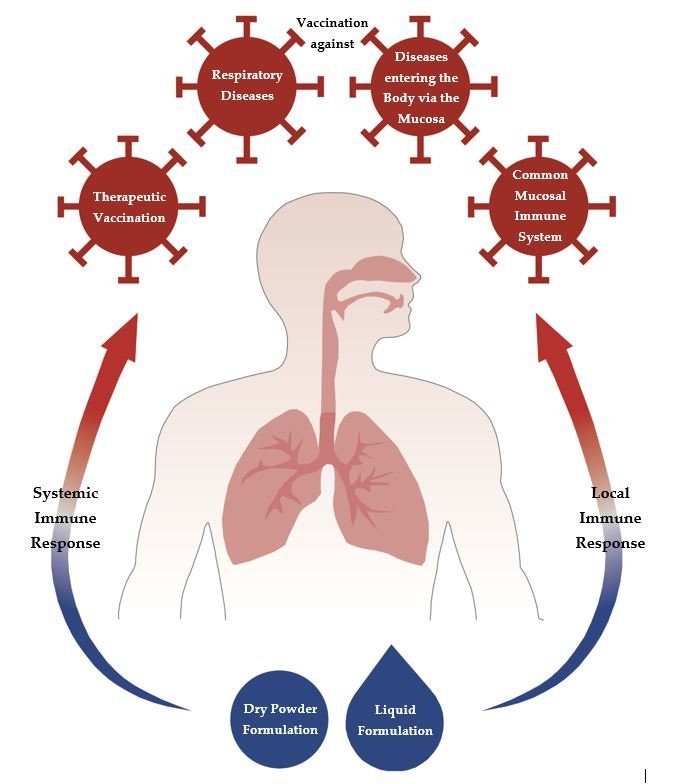
1. Introduction
2. The Immune System of the Respiratory Tract
- Protect the mucosal surface against invasion by microbial pathogens;
- Prevent internalisation of commensal bacteria or antigens as non-degraded proteins;
- Induct tolerance against innocuous soluble substances.
2.1. Immune System in the Nose
2.2. Immune System in the Lung
3. Advantages of Mucosal Immunisation
- Easily accessible;
- Highly vascularised;
- After intranasal immunisation, both mucosal and systemic immune responses are induced;
- Immune response can be induced at distant mucosal sites owing to the dissemination of effector immune cells in the common mucosal immune system;
- Can be used for the immunisation of large population groups;
- Does not require needles and syringes, which are potential sources of infection.
- Rapidly immunisation of the population;
- Avoid the risk of transmitting hepatitis B, HIV and other blood-borne diseases;
- Highly responsive immune system.
4. Prerequisites for Mucosal Vaccination
4.1. Particle Uptake in Immune Cells
4.2. Particle Size
4.3. Particle Shape
4.4. Charge and Functionalisation
5. History of Respiratory Vaccination
6. Formulations for Respiratory Vaccination: General Considerations
7. Adjuvants for Respiratory Vaccines
8. Therapeutic Vaccination
9. Respiratory Vaccine Formulations
9.1. Nasal Vaccine Formulations
9.2. Pulmonary Vaccine Formulations
9.3. Respiratory Vaccines in Research and Development
10. Pulmonary and Nasal Administration Devices
10.1. Nasal Administration
10.1.1. Liquid Preparations
10.1.2. Dry Powder Devices
10.2. Pulmonary Administration
10.2.1. Liquid Preparations
10.2.2. Dry Powder Inhalers
11. Conclusions
12. Patents
Funding
Conflicts of Interest
References
- World Health Organization. 2018 Assessment Report of the Global Vaccine Action Plan. Strategic Advisory Group of Experts on Immunization; WHO/IVB/18.11; World Health Organization: Geneva, Switzerland, 2018; p. 4. [Google Scholar]
- Orenstein, W.; Hinman, A.; Nkowane, B.; Olive, J.; Reingold, A. Measles and Rubella Global Strategic Plan 2012–2020 midterm review. Vaccine 2018, 36, A1–A34. [Google Scholar] [CrossRef] [PubMed]
- Thiel, B. Measles cases in Europe tripled in 2018, WHO says. Infect. Dis. Child. 2019, 3, 5. [Google Scholar]
- Niewiesk, S. Maternal Antibodies: Clinical Significance, Mechanism of Interference with Immune Responses, and Possible Vaccination Strategies. Front. Immunol. 2014, 5, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Mitragotri, S. Immunization without needles. Nat. Rev. Immunol. 2005, 5, 905–916. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.S. Nasal vaccines. Adv. Drug Deliv. Rev. 2001, 51, 21–42. [Google Scholar] [CrossRef]
- Pavot, V.; Rochereau, N.; Genin, C.; Verrier, B.; Paul, S. New insights in mucosal vaccine development. Vaccine 2012, 30, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Cesta, M.F. Normal Structure, Function, and Histology of Mucosa-Associated Lymphoid Tissue. Toxicol. Pathol. 2006, 34, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Gebert, A.; Pabst, R. M cells at locations outside the gut. Semin. Immunol. 1999, 11, 165–170. [Google Scholar] [CrossRef]
- Hathaway, L.J.; Kraehenbuhl, J.-P. The role of M cells in mucosal immunity. Cell. Mol. Life Sci. 2000, 57, 323–332. [Google Scholar] [CrossRef]
- Haneberg, B.; Holst, J. Can non-living nasal vaccines be made to work? Expert Rev. Vaccines 2002, 1, 227–232. [Google Scholar] [CrossRef]
- Neutra, M.R.; Kozlowski, P.A. Mucosal vaccines: The promise and the challenge. Nat. Rev. Immunol. 2006, 6, 148–158. [Google Scholar] [CrossRef]
- Park, S.-K.; Kim, G.-Y.; Lim, J.-Y.; Kwak, J.-Y.; Bae, Y.-S.; Lee, J.-D.; Oh, Y.-H.; Ahn, S.-C.; Park, Y.-M. Acidic polysaccharides isolated from Phellinus linteus induce phenotypic and functional maturation of murine dendritic cells. Biochem. Biophys. Res. Commun. 2003, 312, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Kiyono, H.; Fukuyama, S. NALT-versus Peyer‘s-patch-mediated mucosal immunity. Nat. Rev. Immunol. 2004, 4, 699–710. [Google Scholar] [CrossRef]
- Blank, F.; Wehrli, M.; Lehmann, A.; Baum, O.; Gehr, P.; Von Garnier, C.; Rothen-Rutishauser, B.M. Macrophages and dendritic cells express tight junction proteins and exchange particles in an in vitro model of the human airway wall. Immunobiology 2011, 216, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Hickey, A.J. Pulmonary vaccine delivery. Vaccines 2007, 6, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Vujanic, A.; Sutton, P.; Snibson, K.J.; Yen, H.H.; Scheerlinck, J.P.Y. Mucosal vaccination: Lung versus nose. Veter-Immunol. Immunopathol. 2012, 148, 172–177. [Google Scholar] [CrossRef]
- De Magistris, M.T. Mucosal delivery of vaccine antigens and its advantages in pediatrics. Adv. Drug Deliv. Rev. 2006, 58, 52–67. [Google Scholar] [CrossRef]
- Illum, L. Nasal drug delivery—Possibilities, problems and solutions. J. Control. Release 2003, 87, 187–198. [Google Scholar] [CrossRef]
- Partidos, C.D. Intranasal vaccines: Forthcoming challenges. Pharm. Sci. Technol. Today 2000, 3, 273–281. [Google Scholar] [CrossRef]
- Pilcer, G.; Amighi, K. Formulation strategy and use of excipients in pulmonary drug delivery. Int. J. Pharm. 2010, 392, 1–19. [Google Scholar] [CrossRef]
- Türker, S.; Onur, E.; Ózer, Y. Nasal route and drug delivery systems. Pharm. World Sci. 2004, 26, 137–142. [Google Scholar] [CrossRef] [PubMed]
- De Temmerman, M.-L.; Rejman, J.; Demeester, J.; Irvine, D.J.; Gander, B.; De Smedt, S.C. Particulate vaccines: On the quest for optimal delivery and immune response. Drug Discov. Today 2011, 16, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, L.; Berman, M.; Kong, Y.-Y.; Dorf, M.E. Mapping a Dynamic Innate Immunity Protein Interaction Network Regulating Type I Interferon Production. Immunity 2011, 35, 647–648. [Google Scholar] [CrossRef][Green Version]
- Rietscher, R.; Schröder, M.; Janke, J.; Czaplewska, J.; Gottschaldt, M.; Scherließ, R.; Hanefeld, A.; Schubert, U.S.; Schneider, M.; Knolle, P.; et al. Antigen delivery via hydrophilic PEG-b-PAGE-b-PLGA nanoparticles boosts vaccination induced T cell immunity. Eur. J. Pharm. Biopharm. 2016, 102, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.F.; Shakir, E.; Carter, K.C.; Mullen, A.B.; Alexander, J.; Ferro, V.A. Lipid vesicle size of an oral influenza vaccine delivery vehicle influences the Th1/Th2 bias in the immune response and protection against infection. Vaccine 2009, 27, 3643–3649. [Google Scholar] [CrossRef] [PubMed]
- Slütter, B.; Hagenaars, N.; Jiskoot, W. Rational design of nasal vaccines. J. Drug Target. 2008, 16, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Tafaghodi, M.; Tabassi, S.S.; Jaafari, M.-R.; Zakavi, S.R.; Momen-Nejad, M. Evaluation of the clearance characteristics of various microspheres in the human nose by gamma-scintigraphy. Int. J. Pharm. 2004, 280, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Rieux, A.D.; Fievez, V.; Garinot, M.; Schneider, Y.-J.; Préat, V. Nanoparticles as potential oral delivery systems of proteins and vaccines: A mechanistic approach. J. Control. Release 2006, 116, 1–27. [Google Scholar] [CrossRef]
- Fifis, T.; Gamvrellis, A.; Crimeen-Irwin, B.; Pietersz, G.A.; Li, J.; Mottram, P.L.; McKenzie, I.F.C.; Plebanski, M. Size-Dependent Immunogenicity: Therapeutic and Protective Properties of Nano-Vaccines against Tumors. J. Immunol. 2004, 173, 3148–3154. [Google Scholar] [CrossRef]
- Singh, M.; Chakrapani, A.; O’Hagan, D. Nanoparticles and microparticles as vaccine-delivery systems. Expert Rev. Vaccines 2007, 6, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K.; Terada, H. Endocytosis of Particle Formulations by Macrophages and Its Application to Clinical Treatment. In Molecular Regulation of Endocytosis; Ceresa, B., Ed.; IntechOpen: London, UK, 2012; pp. 413–428. [Google Scholar]
- Gratton, S.E.A.; Ropp, P.A.; Pohlhaus, P.D.; Luft, J.C.; Madden, V.J.; Napier, M.E.; DeSimone, J.M. The effect of particle design on cellular internalization pathways. Proc. Natl. Acad. Sci. USA 2008, 105, 11613–11618. [Google Scholar] [CrossRef] [PubMed]
- Champion, J.A.; Katare, Y.K.; Mitragotri, S. Particle Shape: A New Design Parameter for Micro- and Nanoscale Drug Delivery Carriers. J. Control. Release 2007, 121, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Champion, J.A.; Walker, A.M.; Matragotri, S. Role of Particle Size in Phagocytosis of Polymeric Microspheres. Pharm. Res. 2008, 25, 1815–1821. [Google Scholar] [CrossRef]
- Sharma, S.; Mukkur, T.; Benson, H.A.; Chen, Y. Pharmaceutical Aspects of Intranasal Delivery of Vaccines Using Particulate Systems. J. Pharm. Sci. 2009, 98, 812–843. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int. J. Nanomedicine 2012, 7, 5577–5591. [Google Scholar] [CrossRef] [PubMed]
- Foged, C.; Brodin, B.; Frokjaer, S.; Sundblad, A. Particle size and surface charge affect particle uptake by human dendritic cells in and in vitro model. Int. J. Pharm. 2005, 298, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Slütter, B.; Bal, S.M.; Que, I.; Kaijzel, E.; Löwik, C.; Bouwstra, J.; Jiskoot, W. Antigen−Adjuvant Nanoconjugates for Nasal Vaccination: An Improvement over the Use of Nanoparticles? Mol. Pharm. 2010, 7, 2207–2215. [Google Scholar] [CrossRef]
- Vyas, S.P.; Gupta, P.N. Implication of nanoparticles/microparticles in mucosal vaccine delivery. Expert Rev. Vaccines 2007, 6, 401–418. [Google Scholar] [CrossRef]
- Illum, L. Nanoparticulate Systems for Nasal Delivery of Drugs: A Real Improvement over Simple Systems? J. Pharm. Sci. 2007, 96, 473–483. [Google Scholar] [CrossRef]
- Cruz, L.J.; Tacken, P.J.; Fokkink, R.; Joosten, B.; Stuart, M.C.; Albericio, F.; Torensma, R.; Figdor, C.G. Targeted PLGA nano- but not microparticles specifically deliver antigen to human dendritic cells via DC-SIGN in vitro. J. Control. Release 2010, 144, 118–126. [Google Scholar] [CrossRef]
- Chadwick, S.; Kriegel, C.; Amiji, M. Delivery strategies to enhance mucosal vaccination. Expert Opin. Biol. Ther. 2009, 9, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Thiele, L.; Rothen-Rutishauser, B.; Jilek, S.; Wunderli-Allenspach, H.; Merkle, H.P.; Walter, E. Evaluation of particle uptake in human blood monocyte-derived cells in vitro. Does phagocytosis activity of dendritic cells measure up with macrophages? J. Control. Release 2001, 76, 59–71. [Google Scholar] [CrossRef]
- Hickey, A.J.; Garmise, R.J. Dry Powder Nasal Vaccines as an Alternative to Needle-Based Delivery. Crit. Rev. Ther. Drug Carr. Syst. 2009, 26, 1–27. [Google Scholar] [CrossRef]
- Roth, Y.; Chapnik, J.S.; Cole, P. Feasibility of Aerosol Vaccination in Humans. Ann. Otol. Rhinol. Laryngol. 2003, 112, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.V.; De Castro, J.F.; Valdespino-Gomez, J.L.; Garcia-Garcia, M.D.L.; Islas-Romero, R.; Echaniz-Aviles, G.; Jimenez-Corona, A.; Sepulveda-Amor, J. Aerosolized measles and measles-rubella vaccines induce better measles antibody booster responses than injected vaccines: Randomized trials in Mexican schoolchildren. Bull. World Heal. Organ. 2002, 80, 806–812. [Google Scholar]
- Hasija, M.; Li, L.; Rahman, N.; Ausar, S.F. Forced degradation studies: An essential tool for the formulation development of vaccines. Vaccine Dev. Ther. 2013, 3, 11. [Google Scholar]
- Brandau, D.T.; Jones, L.S.; Wiethoff, C.M.; Rexroad, J.; Middaugh, C.R. Thermal Stability of Vaccines. J. Pharm. Sci. 2003, 92, 218–231. [Google Scholar] [CrossRef] [PubMed]
- Taneja, S.; Ahmad, F. Increased thermal stability of proteins in the presence of amino acids. Biochem. J. 1994, 303, 147–153. [Google Scholar] [CrossRef]
- Rajapaksa, T.E.; Bennett, K.M.; Hamer, M.; Lytle, C.; Rodgers, V.G.J.; Lo, D.D. Intranasal M Cell Uptake of Nanoparticles Is Independently Influenced by Targeting Ligands and Buffer Ionic Strength. J. Biol. Chem. 2010, 285, 23739–23746. [Google Scholar] [CrossRef]
- Wang, W. Lyophilization and development of solid protein pharmaceuticals. Int. J. Pharm. 2000, 203, 1–60. [Google Scholar] [CrossRef]
- Amorij, J.-P.; Huckriede, A.; Wilschut, J.; Frijlink, H.W.; Hinrichs, W.L.J. Development of Stable Influenza Vaccine Powder Formulations: Challenges and Possibilities. Pharm. Res. 2008, 25, 1256–1273. [Google Scholar] [CrossRef] [PubMed]
- Amorij, J.-P.; Meulenaar, J.; Hinrichs, W.; Stegmann, T.; Huckriede, A.; Coenen, F.; Frijlink, H. Rational design of an influenza subunit vaccine powder with sugar glass technology: Preventing conformational changes of haemagglutinin during freezing and freeze-drying. Vaccine 2007, 25, 6447–6457. [Google Scholar] [CrossRef] [PubMed]
- Hansen, L.; Daoussi, R.; Vervaet, C.; Remon, J.-P.; De Beer, T. Freeze-drying of live virus vaccines: A review. Vaccine 2015, 33, 5507–5519. [Google Scholar] [CrossRef] [PubMed]
- Tonnis, W.; Amorij, J.-P.; Vreeman, M.; Frijlink, H.; Kersten, G.; Hinrichs, W. Improved storage stability and immunogenicity of hepatitis B vaccine after spray-freeze drying in presence of sugars. Eur. J. Pharm. Sci. 2014, 55, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Garmise, R.J.; Staats, H.F.; Hickey, A.J. Novel dry powder preparations of whole inactivated influenza virus for nasal vaccination. J. Am. Assoc. Pharm. Sci. 2007, 8, 2–10. [Google Scholar] [CrossRef]
- Saluja, V.; Amorij, J.-P.; Kapteyn, J.C.; De Boer, A.; Frijlink, H.; Hinrichs, W. A comparison between spray drying and spray freeze drying to produce an influenza subunit vaccine powder for inhalation. J. Control. Release 2010, 144, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Minne, A.; Boireau, H.; Horta, M.J.; Vanbever, R. Optimization of the aerosolization properties of an inhalation dry powder based on selection of excipients. Eur. J. Pharm. Biopharm. 2008, 70, 839–844. [Google Scholar] [CrossRef]
- Raula, J.; Thielmann, F.; Naderi, M.; Lehto, V.-P.; Kauppinen, E.I. Investigations on particle surface characteristics vs. dispersion behaviour of l-leucine coated carrier-free inhalable powders. Int. J. Pharm. 2010, 385, 79–85. [Google Scholar] [CrossRef]
- Weiler, C.; Egen, M.; Trunk, M.; Langguth, P. Force control and powder dispersibility of spray dried particles for inhalation. J. Pharm. Sci. 2010, 99, 303–316. [Google Scholar] [CrossRef]
- Westmeier, R.; Steckel, H. In-Situ Fine Particle Excipient as Dispersion Modifier for a Dry Powder Inhalation Product; DDL 19; Aerosol Society: Edinburgh, UK, 2008. [Google Scholar]
- Trows, S.; Scherließ, R. Preparation and Characterization of Dry Powder Agarose Nano-in-Microparticles for Nasal Vaccination. In Respiratory Drug Delivery; Davies Healthcare International Publishing: Phoenix, AZ, USA, 2012; pp. 491–496. [Google Scholar]
- Azad, N.; Rojanasakul, Y. Nanobiotechnology in drug delivery. Am. J. Drug Deliv. 2006, 4, 79–88. [Google Scholar] [CrossRef]
- Mestecky, J.; Russell, M.W.; Elson, C.O. Perspectives on Mucosal Vaccines: Is Mucosal Tolerance a Barrier? J. Immunol. 2007, 179, 5633–5638. [Google Scholar] [CrossRef] [PubMed]
- Bijker, M.S.; Van den Eeden, S.J.F.; Franken, K.L.; Melief, C.J.M.; Offringa, R.; Van der Burg, S.H. CD8+ CTL Priming by Exact Peptide Epitopes in Incomplete Freund’s Adjuvant Induces a Vanishing CTL Response, whereas Long Peptides Induce Sustained CTL Reactivity. J. Immunol. 2007, 179, 5033–5040. [Google Scholar] [CrossRef] [PubMed]
- Foged, C.; Hansen, J.; Agger, E.M. License to kill: Formulation requirements for optimal priming of CD8+ CTL responses with particulate vaccine delivery systems. Eur. J. Pharm. Sci. 2012, 45, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Chadwick, S.; Kriegel, C.; Amiji, M. Nanotechnology solutions for mucosal immunization. Adv. Drug Deliv. Rev. 2010, 62, 394–407. [Google Scholar] [CrossRef] [PubMed]
- Zuercher, A.W. Upper Respiratory Tract Immunity. Viral Immunol. 2003, 16, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Yanagita, M.; Hiroi, T.; Kitagaki, N.; Hamada, S.; Ito, H.; Shimauchi, H.; Murakami, S.; Okada, H.; Kiyono, H. Nasopharyngeal-Associated Lymphoreticular Tissue (NALT) Immunity: Fimbriae-Specific Th1 and Th2 Cell-Regulated IgA Responses for the Inhibition of Bacterial Attachment to Epithelial Cells and Subsequent Inflammatory Cytokine Production. J. Immunol. 1999, 162, 3559–3565. [Google Scholar]
- Boukhvalova, M.S.; Prince, G.A.; Soroush, L.; Harrigan, D.C.; Vogel, S.N.; Blanco, J.C. The TLR4 agonist, monophosphoryl lipid A, attenuates the cytokine storm associated with respiratory syncytial virus vaccine-enhanced disease. Vaccine 2006, 24, 5027–5035. [Google Scholar] [CrossRef]
- Childers, N.K.; Miller, K.L.; Tong, G.; Llarena, J.C.; Greenway, T.; Ulrich, J.T.; Michalek, S.M. Adjuvant Activity of Monophosphoryl Lipid A for Nasal and Oral Immunization with Soluble or Liposome-Associated Antigen. Infect. Immun. 2000, 68, 5509–5516. [Google Scholar] [CrossRef]
- Wang, C.; Muttil, P.; Lu, D.; Beltran-Torres, A.A.; Garcia-Contreras, L.; Hickey, A.J. Screening for Potential Adjuvants Administered by the Pulmonary Route for Tuberculosis Vaccines. AAPS J. 2009, 11, 139–147. [Google Scholar] [CrossRef]
- Shafique, M.; Meijerhof, T.; Wilschut, J.; De Haan, A. Evaluation of an Intranasal Virosomal Vaccine against Respiratory Syncytial Virus in Mice: Effect of TLR2 and NOD2 Ligands on Induction of Systemic and Mucosal Immune Responses. PLoS ONE 2013, 8, e61287. [Google Scholar] [CrossRef] [PubMed]
- Alignani, D.; Maletto, B.; Liscovsky, M.; Rópolo, A.; Morón, G.; Pistoresi-Palencia, M.C. Orally administered OVA/CpG-ODN induces specific mucosal and systemic immune response in young and aged mice. J. Leukoc. Biol. 2005, 77, 898–905. [Google Scholar] [CrossRef] [PubMed]
- Todoroff, J.; Ucakar, B.; Inglese, M.; Vandermarliere, S.; Fillee, C.; Renauld, J.-C.; Huygen, K.; Vanbever, R. Targeting the deep lungs, Poloxamer 407 and a CpG oligonucletide optimize immune responses to Mycobacterium tuberculosis antigen 85A following pulmonary delivery. Eur. J. Pharm. Biopharm. 2013, 84, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Ogra, P.L.; Faden, H.; Welliver, R.C. Vaccination Strategies for Mucosal Immune Responses. Clin. Microbiol. Rev. 2001, 14, 430–445. [Google Scholar] [CrossRef] [PubMed]
- Giri, P.K.; Khuller, G.K. Is intranasal vaccination a feasible solution for tuberculosis? Expert Rev. Vaccines 2008, 7, 1341–1356. [Google Scholar] [CrossRef]
- Johnson, A.G. Molecular adjuvants and immunomodulators: New approaches to immunization. Clin. Microbiol. Rev. 1994, 7, 277–289. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wolvers, D.A.; Bakker, J.M.; Bagchus, W.M.; Kraal, G. The steroid hormone dehydroepiandrosterone (DHEA) breaks intranasally induced tolerance, when administered at time of systemic immunization. J. Neuroimmunol. 1998, 89, 19–25. [Google Scholar] [CrossRef]
- Boyaka, P.N.; McGhee, J.R. Cytokines as adjuvants for the induction of mucosal immunity. Adv. Drug Deliv. Rev. 2001, 51, 71–79. [Google Scholar] [CrossRef]
- Lycke, N. Recent progress in mucosal vaccine development: Potential and limitations. Nat. Rev. Immunol. 2012, 12, 592–605. [Google Scholar] [CrossRef]
- Jabbal-Gill, I. Nasal vaccine innovation. J. Drug Target. 2010, 18, 771–786. [Google Scholar] [CrossRef]
- Melief, C.J.M.; Van Hall, T.; Arens, R.; Ossendorp, F.; Van der Burg, S.H. Therapeutic cancer vaccines. J. Clin. Investig. 2015, 125, 3401–3412. [Google Scholar] [CrossRef] [PubMed]
- Pannemans, K.; Hellings, N.; Stinissen, P. Therapeutic vaccines for autoimmune diseases. Drug Discov. Today Ther. Strat. 2009, 6, 39–44. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Palucka, K.; Ueno, H.; Banchereau, J. Recent Developments in Cancer Vaccines. J. Immunol. 2011, 186, 1325–1331. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, G.A.; Gabrilovich, D.; Sotomayor, E.M. Immunosuppressive Strategies that are Mediated by Tumor Cells. Annu. Rev. Immunol. 2007, 25, 267–296. [Google Scholar] [CrossRef] [PubMed]
- FDA. Guidance for Industry: Nasal Spray and Inhalation Solution, Suspension, and Spray Drug Products-Chemistry, Manufacturing, and Controls Documentation; USA Department of Health and Human Services, FDA, Center for Drug Evaluation and Research (CDER): Rockville, MD, USA, 2002; pp. 1–49.
- Marple, B.; Roland, P.; Benninger, M. Safety Review of Benzalkonium Chloride Used as a Preservative in Intranasal Solutions: An Overview of Conflicting Data and Opinions. Otolaryngol. Neck Surg. 2004, 130, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Merkus, P.; Romeijn, S.G.; Verhoef, J.C.; Merkus, F.W.H.M.; Schouwenburg, P.F. Classification of Cilio-Inhibiting Effects of Nasal Drugs. Laryngoscope 2001, 111, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Yuki, Y.; Nochi, T.; Harada, N.; Katakai, Y.; Shibata, H.; Mejima, M.; Kohda, T.; Tokuhara, D.; Kurokawa, S.; Takahashi, Y.; et al. In Vivo Molecular Imaging Analysis of a Nasal Vaccine That Induces Protective Immunity against Botulism in Nonhuman Primates. J. Immunol. 2010, 185, 5436–5443. [Google Scholar] [CrossRef] [PubMed]
- Perrie, Y.; Mohammed, A.R.; Kirby, D.J.; McNeil, S.E.; Bramwell, V.W.; Mohammed, A.-U.-R. Vaccine adjuvant systems: Enhancing the efficacy of sub-unit protein antigens. Int. J. Pharm. 2008, 364, 272–280. [Google Scholar] [CrossRef]
- Carter, N.J.; Curran, M.P. Live Attenuated Influenza Vaccine. A Review of its Use in the Prevention of Seasonal Influenza in Children and Adults. Adis Drug Eval. 2011, 71, 1591–1622. [Google Scholar]
- Black, S.V.; Hultquist, M.; Connor, E.M.; Belshe, R.B.; Edwards, K.M.; Vesikari, T.; Walker, R.E.; Kemble, G. Live Attenuated versus Inactivated Influenza Vaccine in Infants and Young Children. N. Engl. J. Med. 2007, 356, 685–696. [Google Scholar]
- Bosquillon, C.; Lombry, C.; Préat, V.; Vanbever, R. Influence of formulation excipients and physical characteristics of inhalation dry powders on their aerosolization performance. J. Control. Release 2001, 70, 329–339. [Google Scholar] [CrossRef]
- Carvalho, T.C.; Peters, J.I.; Williams III, R.O. Influence of particle size on regional lung deposition—What evidence is there? Int. J. Pharm. 2011, 406, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Steckel, H.; Bolzen, N. Alternative sugars as potential carriers for dry powder inhalations. Int. J. Pharm. 2004, 270, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Telko, M.J.; Hickey, A.J. Dry Powder Inhaler Formulation. Respir. Care 2005, 50, 1209–1227. [Google Scholar] [PubMed]
- Newman, S.; Busse, W. Evolution of dry powder inhaler design, formulation, and performance. Respir. Med. 2002, 96, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Baldrick, P. Pharmaceutical Excipient Development: The Need for Preclinical Guidance. Regul. Toxicol. Pharmacol. 2000, 32, 210–218. [Google Scholar] [CrossRef]
- Steinberg, M.; Borzelleca, J.F.; Enters, E.K.; Kinoshita, F.K.; Loper, A.; Mitchell, D.B.; Tamulinas, C.B.; Weiner, M.L. A New Approach to the Safety Assessment of Pharmaceutical Excipients: The Safety Committee of the International Pharmaceutical Excipient Council. Regul. Toxic. Pharmacol. 1996, 24, 149–154. [Google Scholar] [CrossRef]
- VanDevanter, D.R.; Geller, D.E. Tobramycin administered by the TOBI® Podhaler® for persons with cystic fibrosis: A review. Med. Devices Evid. Res. 2011, 4, 179–188. [Google Scholar] [CrossRef]
- Rice-Ficht, A.C.; Arenas-Gamboa, A.M.; Kahl-McDonagh, M.M.; Ficht, T.A. Polymeric particles in vaccine delivery. Curr. Opin. Microbiol. 2010, 13, 106–112. [Google Scholar] [CrossRef]
- Smith, J.D.; Morton, L.D.; Ulery, B.D. Nanoparticles as synthetic vaccines. Curr. Opin. Biotechnol. 2015, 34, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Zhao, D.; Li, D.; Wang, X.; Jin, Z.; Zhao, K. Polymer-Based Nanomaterials and Applications for Vaccines and Drugs. Polymers 2018, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, K.S.; Vyas, S.P. Strong systemic and mucosal immune responses to surface-modified PLGA microspheres containing recombinant Hepatitis B antigen administered intranasally. Vaccine 2006, 24, 4201–4211. [Google Scholar] [CrossRef] [PubMed]
- Pawar, D.; Mangal, S.; Goswami, R.; Jaganathan, K. Development and characterization of surface modified PLGA nanoparticles for nasal vaccine delivery: Effect of mucoadhesive coating on antigen uptake and immune adjuvant activity. Eur. J. Pharm. Biopharm. 2013, 85, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Illum, L.; Jabbal-Gill, I.; Hinchcliffe, M.; Fisher, A.; Davis, S. Chitosan as a novel nasal delivery system for vaccines. Adv. Drug Deliv. Rev. 2001, 51, 81–96. [Google Scholar] [CrossRef]
- Heidland, J.; Scherließ, R. NiM powders for nasal vaccination—Insights into particle forming process. In Proceedings of the 10th World Meeting on Pharmaceutics, Biopharmaceutics and Pharmaceutical Technology, Glasgow, UK, 4–7 April 2016. [Google Scholar]
- Smith, A.; Perelman, M.; Hinchcliffe, M. Chitosan—A promising safe and immune-enhancing adjuvant for intranasal vaccines. Hum. Vaccines Immunother. 2014, 10, 797–807. [Google Scholar] [CrossRef]
- Heidland, J.; Scherließ, R. Nano-in-Microparticle Powders for Mucosal Vaccination—Understanding the Particle Forming Process; DDL 27; Aerosol Society: Edinburgh, UK, 2016. [Google Scholar]
- Heidland, J.; Scherließ, R. Nano-in-Microparticles for Dry Powder Vaccination—Possible or Nasal Application? DDL 2017; Aerosol Society: Edinburgh, UK, 2017. [Google Scholar]
- Buske, S.D.L. Chitosan as Adjuvant and Particle Forming Excipient in a Nano-in-Microparticulate Dry Powder for Nasal and Pulmonary Vaccine Delivery. Ph.D. Thesis, Kiel University , Kiel, Germany, 2014. [Google Scholar]
- Diedrich, A. Entwicklung Einer Nanopartikulären Formulierung zur Vakzinierung über den Respirationstrakt. Ph.D. Thesis, Kiel University, Kiel, Germany, 2015. [Google Scholar]
- Scherließ, R.; Diedrich, A.; Ebensen, T.; Guzmán, C.A.; Wolf, M.; Hanefeld, A. Chitosan Nanoparticulate Formulation for Pulmonary Vaccination—Formulation and in Vivo Proof of Concept. In Respiratory Drug Delivery Europe; Davies Healthcare International Publishing: Antibes, France, 2015; pp. 269–274. [Google Scholar]
- Trows, S. Pulverformulierungen für die Nasale Vakzinierung. Ph.D. Thesis, Kiel University, Kiel, Germany, 2013. [Google Scholar]
- Scherließ, R.; Mönckedieck, M.; Young, K.; Trows, S.; Buske, S.; Hook, S. First in vivo evaluation of particulate nasal dry powder vaccine formulations containing ovalbumin in mice. Int. J. Pharm. 2015, 479, 408–415. [Google Scholar] [CrossRef]
- Tonnis, W.F.; Kersten, G.F.; Frijlink, H.W.; Hinrichs, W.L.; De Boer, A.H.; Amorij, J.-P. Pulmonary Vaccine Delivery: A Realistic Approach? J. Aerosol Med. Pulm. Drug Deliv. 2012, 25, 249–260. [Google Scholar] [CrossRef]
- Sou, T.; Meeusen, E.N.; De Veer, M.; Morton, D.A.; Kaminskas, L.M.; McIntosh, M.P. New developments in dry powder pulmonary vaccine delivery. Trends Biotechnol. 2011, 29, 191–198. [Google Scholar] [CrossRef]
- Kisich, K.O.; Higgins, M.P.; Park, I.; Cape, S.P.; Lindsay, L.; Bennett, D.J.; Winston, S.; Searles, J.; Sievers, R.E. Dry powder measles vaccine: Particle deposition, virus replication, and immune response in cotton rats following inhalation. Vaccine 2011, 29, 905–912. [Google Scholar] [CrossRef]
- Lin, W.-H.; Griffin, D.E.; Rota, P.A.; Papania, M.; Cape, S.P.; Bennett, D.; Quinn, B.; Sievers, R.E.; Shermer, C.; Powell, K.; et al. Successful respiratory immunization with dry powder live-attenuated measles virus vaccine in rhesus macaques. Proc. Natl. Acad. Sci. USA 2011, 108, 2987–2992. [Google Scholar] [CrossRef] [PubMed]
- Sievers, R.E.; Cape, S.P.; McAdams, D.H.; Manion, J.R.; Shah, N.K.; Chen, D.J.; Winston, S.E. Optimizing the Effectiveness of Live-Attenuated Measles Vaccine Aerosols: Effects of Different Delivery Modes. In Respiratory Drug Delivery Europe; Davies Healthcare International Publishing: Phoenix, AZ, USA, 2012; pp. 101–110. [Google Scholar]
- Andrews, N.J.; Shaikh, N.; Jadi, R.S.; Rajagopal, A.; Brown, K.E.; Riveros-Balta, A.X.; Greco, M.; Low, N.; Bavdekar, A.; Jeyaseelan, L.; et al. A Randomized, Controlled Trial of an Aerosolized Vaccine against Measles. New Engl. J. Med. 2015, 372, 1519–1529. [Google Scholar]
- Dhere, R.; Yeolekar, L.; Kulkarni, P.; Menon, R.; Vaidya, V.; Ganguly, M.; Tyagi, P.; Barde, P.; Jadhav, S. A pandemic influenza vaccine in India: From strain to sale within 12 months. Vaccine 2011, 29, A16–A21. [Google Scholar] [CrossRef] [PubMed]
- Audouy, S.A.; Van Der Schaaf, G.; Hinrichs, W.L.; Frijlink, H.W.; Wilschut, J.; Huckriede, A. Development of a dried influenza whole inactivated virus vaccine for pulmonary immunization. Vaccine 2011, 29, 4345–4352. [Google Scholar] [CrossRef] [PubMed]
- Källenius, G.; Pawlowski, A.; Brandtzaeg, P.; Svenson, S. Should a new tuberculosis vaccine be administered intranasally? Tuberculosis 2007, 87, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Fourie, P.; Germishuizen, W.; Wong, Y.-L.; Edwards, D. Spray drying TB vaccines for pulmonary administration. Expert Opin. Biol. Ther. 2008, 8, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Contreras, L.; Wong, Y.-L.; Muttil, P.; Padilla, D.; Sadoff, J.; DeRousse, J.; Germishuizen, W.A.; Goonesekera, S.; Elbert, K.; Bloom, B.R.; et al. Immunization by a bacterial aerosol. Proc. Natl. Acad. Sci. USA 2008, 105, 4656–4660. [Google Scholar] [CrossRef] [PubMed]
- Muttil, P.; Prego, C.; Garcia-Contreras, L.; Pulliam, B.; Fallon, J.K.; Wang, C.; Hickey, A.J.; Edwards, D. Immunization of Guinea Pigs with Novel Hepatitis B Antigen as Nanoparticle Aggregate Powders Administered by the Pulmonary Route. AAPS J. 2010, 12, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Djupesland, P.G. Nasal drug delivery devices: Characteristics and performance in a clinical perspective—A review. Drug Deliv. Transl. Res. 2013, 3, 42–62. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Garmise, R.J.; Crowder, T.M.; Mar, K.; Hwang, C.R.; Hickey, A.J.; Mikszta, J.A.; Sullivan, V.J. A novel dry powder influenza vaccine and intranasal delivery technology: Induction of systemic and mucosal immune responses in rats. Vaccine 2004, 23, 794–801. [Google Scholar] [CrossRef]
- Friebel, C.; Steckel, H.; Müller, B.W. Rational design of a dry powder inhaler: Device design and optimisation. J. Pharm. Pharmacol. 2012, 64, 1303–1315. [Google Scholar] [CrossRef] [PubMed]
- Friebel, C.; Steckel, H. Single-use disposable dry powder inhalers for pulmonary drug delivery. Expert Opin. Drug Deliv. 2010, 7, 1359–1372. [Google Scholar] [CrossRef] [PubMed]
- De Boer, A.H.; Hagedoorn, P.; Westerman, E.M.; Le Brun, P.P.; Heijerman, H.G.; Frijlink, H.W. Design and in vitro performance testing of multiple air classifier technology in a new disposable inhaler concept (Twincer®) for high powder doses. Eur. J. Pharm. Sci. 2006, 28, 171–178. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Cutaneous Immunisation | Mucosal Immunisation |
|---|---|
| Epidermal powder immunisation | Ocular immunisation |
| Nasal immunisation | |
| Liquid-jet immunisation | Pulmonary immunisation |
| Oral immunisation | |
| Topical application | Vaginal immunisation |
| Rectal immunisation |
| Parts of the Immune System | Location | Cells/Structures |
|---|---|---|
| Epithelial compartments with immunocompetent cells | Nose/lung | Macrophages |
| Dendritic cells (DCs) | ||
| M cells | ||
| T and B lymphocytes | ||
| Lymphoid structures of the nose and the bronchus | Nose: nasopharynx and tonsils | Nose-associated lymphoid tissue (NALT) |
| Larynx | Larynx-associated lymphoid tissue (LALT) | |
| Lung: upper and lower airways (branching site) | Bronchus-associated lymphoid tissue (BALT) | |
| Lymph nodes | T and B cells |
| Vaccine | Method | Stabiliser |
|---|---|---|
| Influenza | Spray drying | DPPC/HES (dipalmitoyl phosphatidylcholine/hydroxyethyl starch) |
| Inulin [59] | ||
| Spray-freeze drying | Arginine | |
| Dextran | ||
| Lactose | ||
| Mannitol | ||
| Trehalose | ||
| Freeze drying | Dextran | |
| HYAFF (esterified hyaluronic acid) microspheres | ||
| Inulin | ||
| Sorbitol | ||
| Trehalose | ||
| Air drying | d-xylose | |
| Smallpox | Freeze drying | Mannitol |
| Measles, mumps and rubella | Freeze drying | Sorbitol |
| Sucrose | ||
| Hepatitis B | Spray-freeze drying | Inulin |
| Dextran | ||
| Trehalose |
| Mucosal Adjuvant | Mechanism | Immune Response in the Respiratory Tract |
|---|---|---|
Lipopolysaccharide (LPS)–protein complexes (endotoxins):
| Enhance antigen-specific mucosal IgA and systemic IgG responses to administered proteins [71] | Yes [16] |
| Monophosphoryl lipid A (MPL) | Activate cells via Toll-like Receptor 4 (TLR4) [72] | Yes [73] |
| Muramyl dipeptide (MDP) | Enhance the cell-mediated immune response [74] | Yes [75] |
| Oligonucleotids (CpG) | Stimulate B cells to proliferate and secrete immunoglobulins, activate APCs and stimulate cytokine production [76] | Yes [77] |
| Saponins like QuilA (e.g., ISCOMS) | Improve T cell responses and antigen uptake by APC [78,79] | Yes [79] |
| Non-ionic block polymers (Poloxamers) | Enhance antigen presentation by binding protein antigens to the surface of the oil droplets [80] | Yes [76] |
| Dehydroepiandrosterone (DHEA) | Increase cell-mediated immunity [80] | Yes [81] |
Cytokines
| Enhance B cell growth (Il-12) and influence the differentiation of Th cells (Il-1) [82] | Yes [82] |
| Devices for Liquid Preparations | Dry Powder Devices | ||
|---|---|---|---|
| Device | Company | Device | Company |
| Nasal pressurised metered-dose inhaler | 3M, USA | Unit dose powder device | Aptar, France |
| Unitdose System | Aptar, France | Turbohaler | Astra Zeneca, Sweden |
| Unidose Xtra | Bespak, UK | OptiNose | Optinose UK Ltd. |
| Nebuliser, e.g., ViaNase | Kurve Technology, USA | Fit-lizer | SNBL Pharma, Japan |
| OptiNose | Optinose UK Ltd. | ||
Single-dose spray devices
| Teleflex, USA | ||
| Metered-dose spray pumps | |||
| Devices for Liquid Preparations | Dry Powder Inhalers | ||
|---|---|---|---|
| Device | Company | Device | Company |
| Pressurised metered-dose inhaler | Aptar, France 3M, USA Bespak, UK | ResQhaler | Aespira, Israel |
| Nebuliser | Twister | Aptar, France | |
| Philips N.V., The Netherlands | TwinCaps | Hovione, Portugal |
| Twincer | Stichting Groningen Centre for Drug Research, The Netherlands | ||
| Flores Medical GmbH, Germany | Cyclohaler | |
| PuffHaler | |||
| Pari GmbH Germany | Unihaler | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hellfritzsch, M.; Scherließ, R. Mucosal Vaccination via the Respiratory Tract. Pharmaceutics 2019, 11, 375. https://doi.org/10.3390/pharmaceutics11080375
Hellfritzsch M, Scherließ R. Mucosal Vaccination via the Respiratory Tract. Pharmaceutics. 2019; 11(8):375. https://doi.org/10.3390/pharmaceutics11080375
Chicago/Turabian StyleHellfritzsch, Marie, and Regina Scherließ. 2019. "Mucosal Vaccination via the Respiratory Tract" Pharmaceutics 11, no. 8: 375. https://doi.org/10.3390/pharmaceutics11080375
APA StyleHellfritzsch, M., & Scherließ, R. (2019). Mucosal Vaccination via the Respiratory Tract. Pharmaceutics, 11(8), 375. https://doi.org/10.3390/pharmaceutics11080375