3D Printing of a Multi-Layered Polypill Containing Six Drugs Using a Novel Stereolithographic Method



 , , , and
, , , and 
Abstract
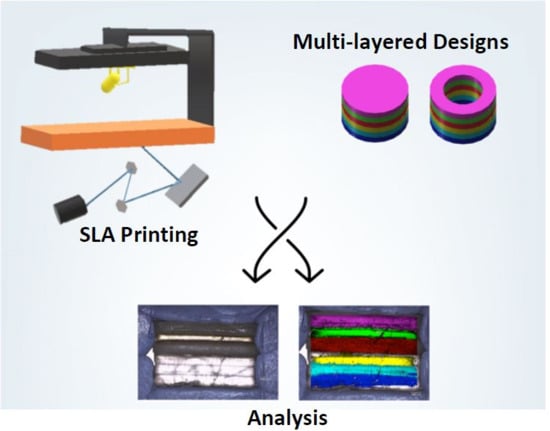
1. Introduction
2. Materials and Methods
2.1. 3D Printing
- Type I: Cylinder shape
- Type II: Ring shape
- Type III: Ring shape with a soluble filler (PEG 300)
2.2. Printlet Dimensions
2.3. Raman Spectroscopy and Mapping
2.4. X-ray Powder Diffraction (XRPD)
2.5. Determination of Drug Concentration in the Polypills
2.6. Dynamic Drug Dissolution Testing Conditions
2.7. Determination of Swelling Ratio (SR) for Individual Layers
3. Results and Discussion
3.1. 3D Printing Process
3.2. Physical Characteristics
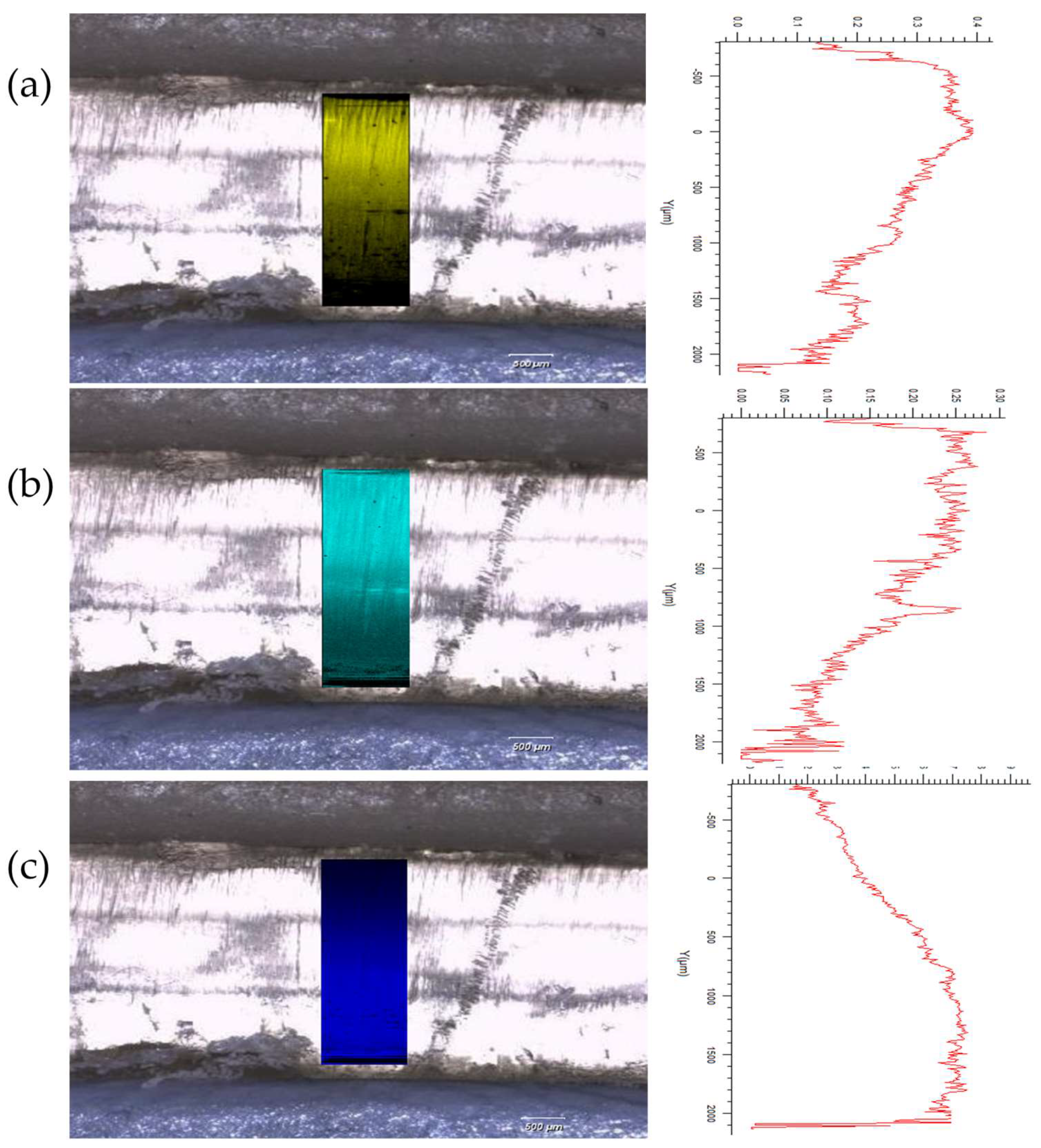
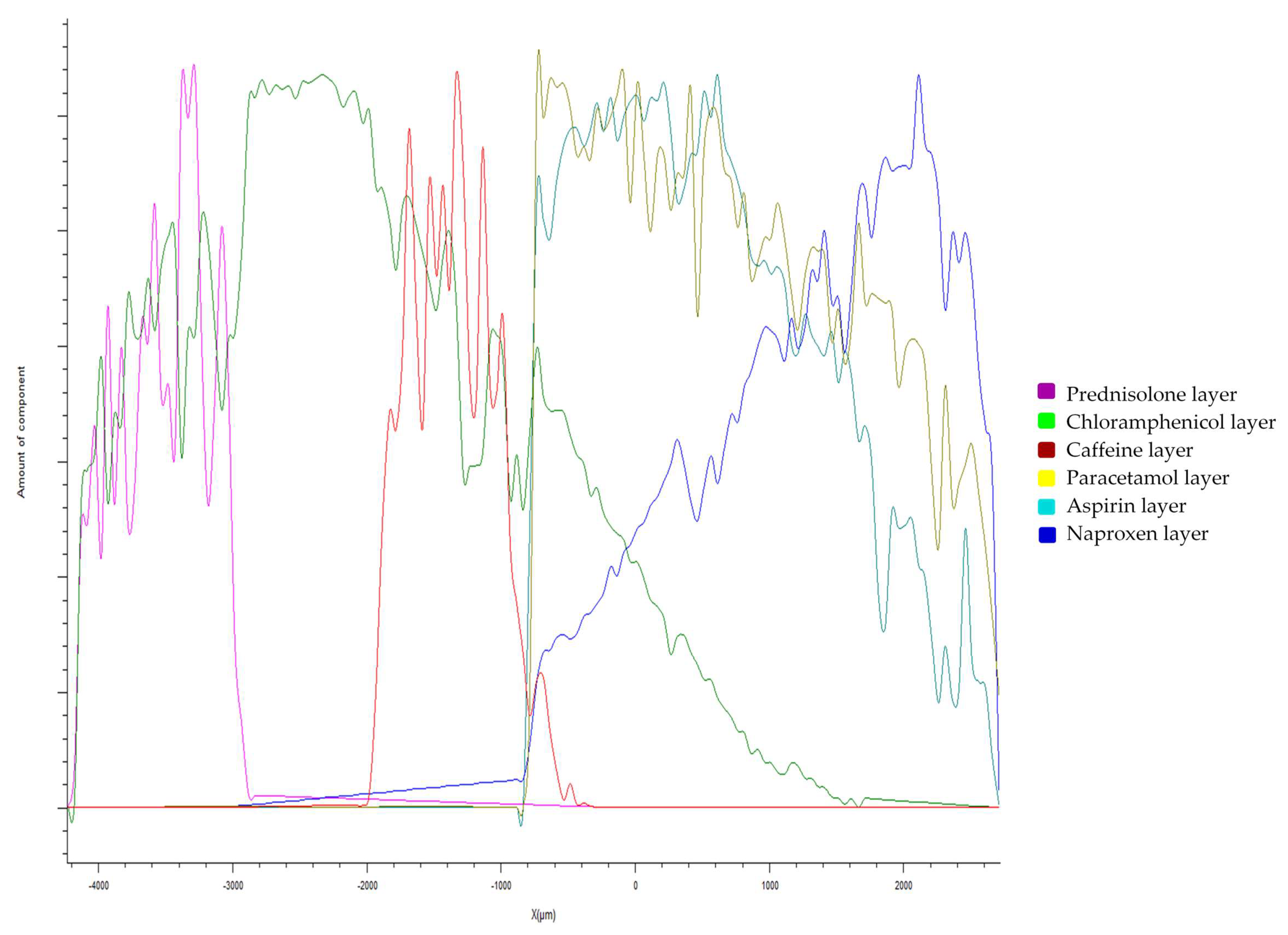
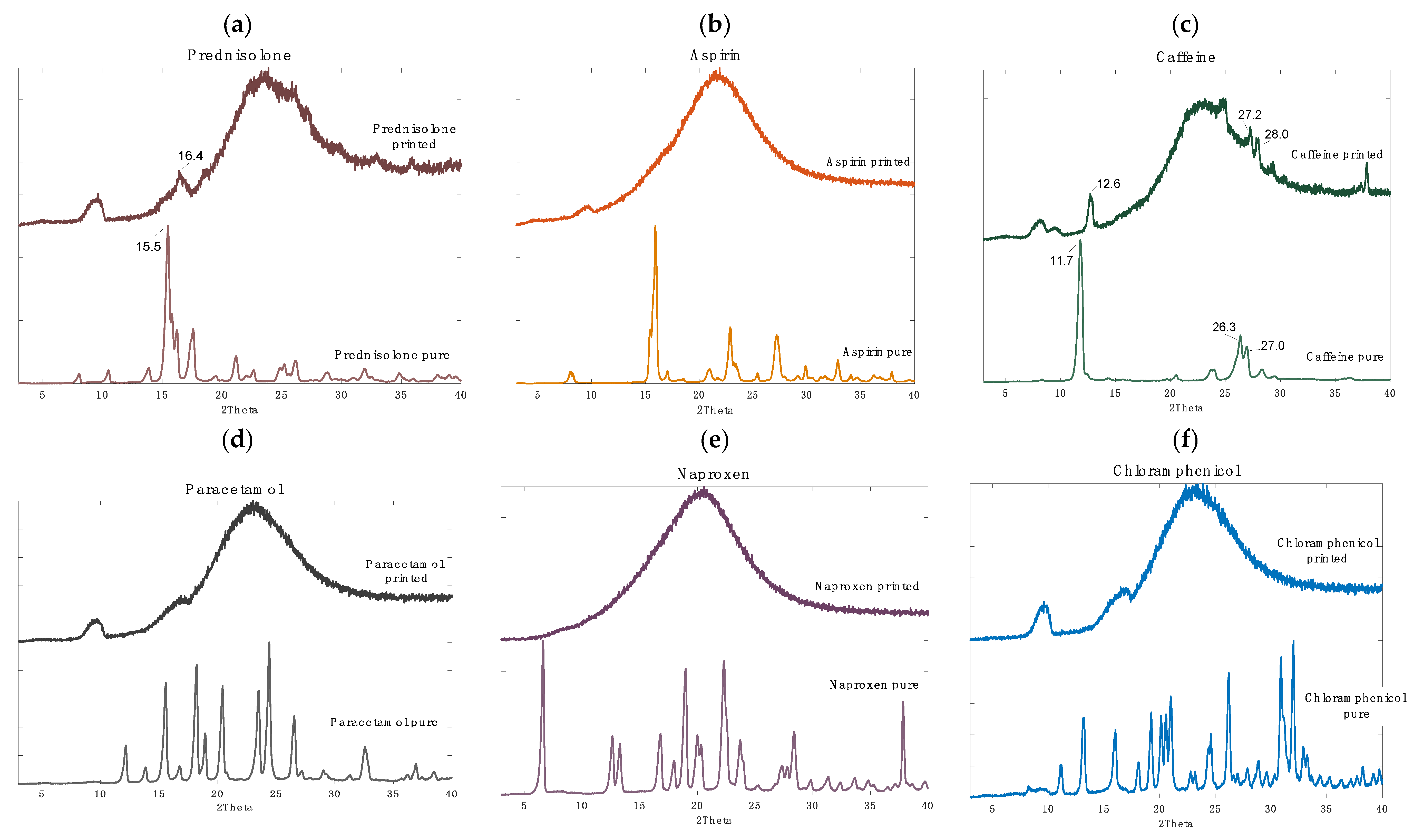
3.2.1. Drug Distribution and Solid-State Characteristics
3.2.2. Printlet Dimensions and Weight Variation
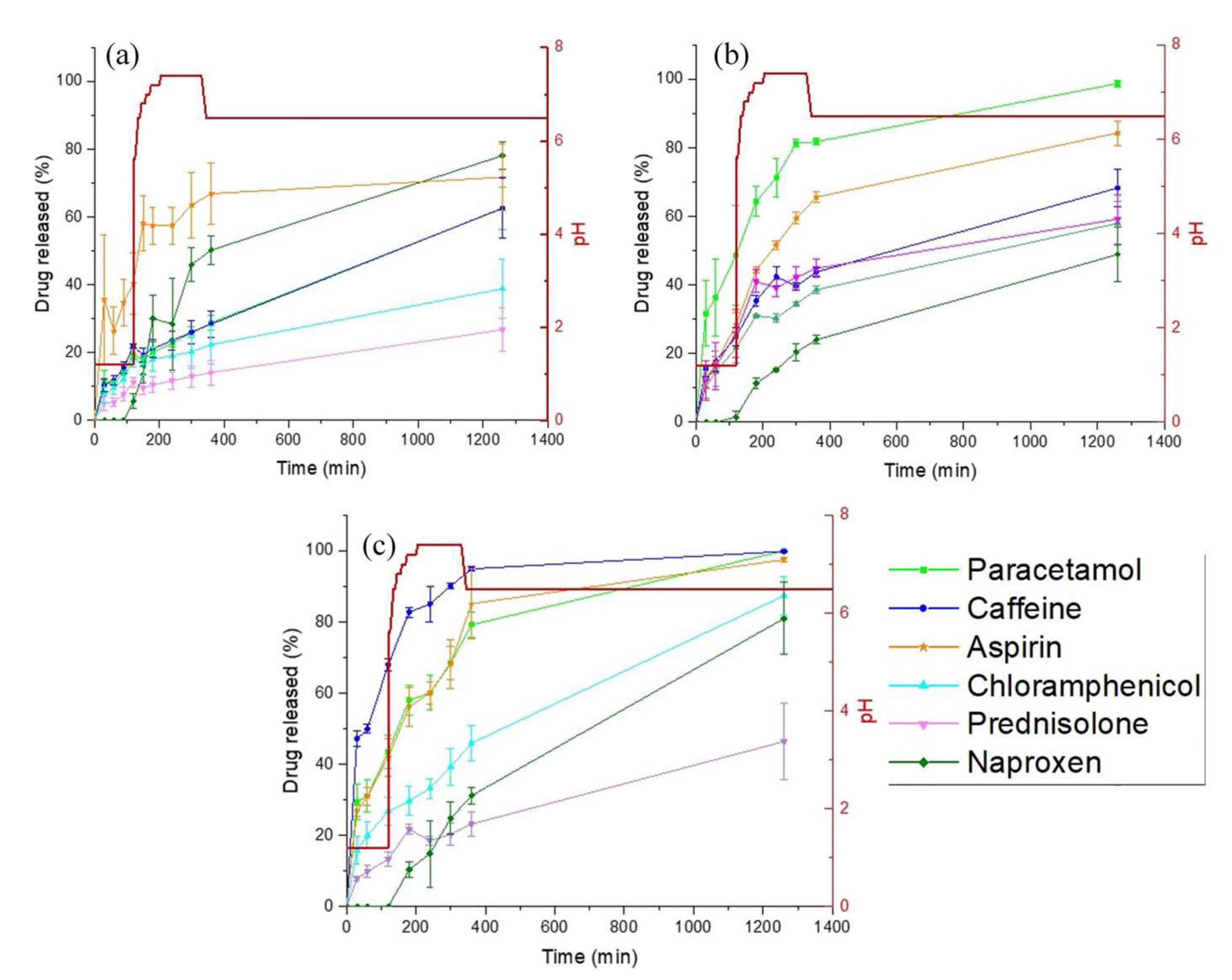
3.3. Drug Release Studies
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fonseca, V.; Rosenstock, J.; Patwardhan, R.; Salzman, A. Effect of Metformin and Rosiglitazone Combination Therapy in Patients With Type 2 Diabetes Mellitus. JAMA 2000, 283, 1695–1702. [Google Scholar] [CrossRef] [PubMed]
- Gradman, A.H.; Basile, J.N.; Carter, B.L.; Bakris, G.L. Combination therapy in hypertension. J. Am. Soc. Hypertens. 2010, 4, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Cruz, A.T.; Garcia-Prats, A.J.; Furin, J.; Seddon, J.A. Treatment of Multidrug-resistant Tuberculosis Infection in Children. Pediatr. Infect. Dis. J. 2018, 37, 831–834. [Google Scholar] [CrossRef] [PubMed]
- Weverling, G.J.; Lange, J.M.; Jurriaans, S.; Prins, J.M.; Lukashov, V.V.; Notermans, D.W.; Roos, M.; Schuitemaker, H.; Hoetelmans, R.M.; Danner, S.A.; et al. Alternative multidrug regimen provides improved suppression of HIV-1 replication over triple therapy. AIDS 1998, 12, F117–F122. [Google Scholar] [CrossRef] [PubMed]
- Payne, R.A.; Avery, A.J. Polypharmacy: One of the greatest prescribing challenges in general practice. Br. J. Gen. Pract. 2011, 61, 83. [Google Scholar] [CrossRef] [PubMed]
- Bangalore, S.; Kamalakkannan, G.; Parkar, S.; Messerli, F.H. Fixed-dose combinations improve medication compliance: A meta-analysis. Am. J. Med. 2007, 120, 713–719. [Google Scholar] [CrossRef]
- Castellano, J.M.; Sanz, G.; Penalvo, J.L.; Bansilal, S.; Fernandez-Ortiz, A.; Alvarez, L.; Guzman, L.; Linares, J.C.; Garcia, F.; D’Aniello, F.; et al. A polypill strategy to improve adherence: Results from the FOCUS project. J. Am. Coll. Cardiol. 2014, 64, 2071–2082. [Google Scholar] [CrossRef]
- Connor, J.; Rafter, N.; Rodgers, A. Do fixed-dose combination pills or unit-of-use packaging improve adherence? A systematic review. Bull. World Health Organ. 2004, 82, 935–939. [Google Scholar]
- Yusuf, S.; Pais, P.; Afzal, R.; Xavier, D.; Teo, K.; Eikelboom, J.; Sigamani, A.; Mohan, V.; Gupta, R.; Thomas, N. Effects of a polypill (Polycap) on risk factors in middle-aged individuals without cardiovascular disease (TIPS): A phase II, double-blind, randomised trial. Lancet 2009, 373, 1341–1351. [Google Scholar]
- Patel, A.; Shah, T.; Shah, G.; Jha, V.; Ghosh, C.; Desai, J.; Khamar, B.; Chakraborty, B.S. Preservation of Bioavailability of Ingredients and Lack of Drug-Drug Interactions in a Novel Five-Ingredient Polypill (Polycap™). Am. J. Cardiovasc. Drugs 2010, 10, 95–103. [Google Scholar] [CrossRef]
- Webster, R.; Castellano, J.M.; Onuma, O.K. Putting polypills into practice: Challenges and lessons learned. Lancet 2017, 389, 1066–1074. [Google Scholar] [CrossRef]
- Alomari, M.; Vuddanda, P.R.; Trenfield, S.J.; Dodoo, C.C.; Velaga, S.; Basit, A.W.; Gaisford, S. Printing T3 and T4 oral drug combinations as a novel strategy for hypothyroidism. Int. J. Pharm. 2018, 549, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Tian, P.; Yang, F.; Xu, Y.; Lin, M.-M.; Yu, L.-P.; Lin, W.; Lin, Q.-F.; Lv, Z.-F.; Huang, S.-Y.; Chen, Y.-Z. Oral disintegrating patient-tailored tablets of warfarin sodium produced by 3D printing. Drug Dev. Ind. Pharm. 2018, 44, 1918–1923. [Google Scholar] [CrossRef] [PubMed]
- Vuddanda, P.R.; Alomari, M.; Dodoo, C.C.; Trenfield, S.J.; Velaga, S.; Basit, A.W.; Gaisford, S. Personalisation of warfarin therapy using thermal ink-jet printing. Eur. J. Pharm. Sci. 2018, 117, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Goyanes, A.; Chang, H.; Sedough, D.; Hatton, G.B.; Wang, J.; Buanz, A.; Gaisford, S.; Basit, A.W. Fabrication of controlled-release budesonide tablets via desktop (FDM) 3D printing. Int. J. Pharm. 2015, 496, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Alomari, M.; Mohamed, F.H.; Basit, A.W.; Gaisford, S. Personalised dosing: Printing a dose of one’s own medicine. Int. J. Pharm. 2015, 494, 568–577. [Google Scholar] [CrossRef] [PubMed]
- El Aita, I.; Breitkreutz, J.; Quodbach, J. On-demand manufacturing of immediate release levetiracetam tablets using pressure-assisted microsyringe printing. Eur. J. Pharm. Biopharm. 2019, 134, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Basit, A.W.; Gaisford, S. 3D Printing of Pharmaceuticals; Springer International Publishing: Cham, Switzerland, 2018. [Google Scholar]
- Awad, A.; Fina, F.; Trenfield, S.J.; Patel, P.; Goyanes, A.; Gaisford, S.; Basit, A.W. 3D Printed Pellets (Miniprintlets): A Novel, Multi-Drug, Controlled Release Platform Technology. Pharmaceutics 2019, 11, 148. [Google Scholar] [CrossRef]
- Vithani, K.; Goyanes, A.; Jannin, V.; Basit, A.W.; Gaisford, S.; Boyd, B.J. A Proof of Concept for 3D Printing of Solid Lipid-Based Formulations of Poorly Water-Soluble Drugs to Control Formulation Dispersion Kinetics. Pharm. Res. 2019, 36, 102. [Google Scholar] [CrossRef]
- Vithani, K.; Goyanes, A.; Jannin, V.; Basit, A.W.; Gaisford, S.; Boyd, B.J. An Overview of 3D Printing Technologies for Soft Materials and Potential Opportunities for Lipid-based Drug Delivery Systems. Pharm. Res. 2018, 36, 4–24. [Google Scholar] [CrossRef]
- Linares, V.; Casas, M.; Caraballo, I. Printfills: 3D printed systems combining fused deposition modeling and injection volume filling. Application to colon-specific drug delivery. Eur. J. Pharm. Biopharm. 2019, 134, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, W.-K.; Lorber, B.; Reitsamer, H.; Khinast, J. 3D printing of oral drugs: A new reality or hype? Expert Opin. Drug Deliv. 2017, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Awad, A.; Trenfield, S.J.; Goyanes, A.; Gaisford, S.; Basit, A.W. Reshaping drug development using 3D printing. Drug Discov. Today 2018, 23, 1547–1555. [Google Scholar] [CrossRef] [PubMed]
- Fina, F.; Madla, C.M.; Goyanes, A.; Zhang, J.; Gaisford, S.; Basit, A.W. Fabricating 3D printed orally disintegrating printlets using selective laser sintering. Int. J. Pharm. 2018, 541, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Fina, F.; Goyanes, A.; Gaisford, S.; Basit, A.W. Selective laser sintering (SLS) 3D printing of medicines. Int. J. Pharm. 2017, 529, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Goyanes, A.; Det-Amornrat, U.; Wang, J.; Basit, A.W.; Gaisford, S. 3D scanning and 3D printing as innovative technologies for fabricating personalized topical drug delivery systems. J. Control. Release 2016, 234, 41–48. [Google Scholar] [CrossRef]
- Kadry, H.; Wadnap, S.; Xu, C.; Ahsan, F. Digital light processing (DLP) 3D-printing technology and photoreactive polymers in fabrication of modified-release tablets. Eur. J. Pharm. Sci. 2019, 135, 60–67. [Google Scholar] [CrossRef]
- Fina, F.; Goyanes, A.; Madla, C.M.; Awad, A.; Trenfield, S.J.; Kuek, J.M.; Patel, P.; Gaisford, S.; Basit, A.W. 3D printing of drug-loaded gyroid lattices using selective laser sintering. Int. J. Pharm. 2018, 547, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Sadia, M.; Arafat, B.; Ahmed, W.; Forbes, R.T.; Alhnan, M.A. Channelled tablets: An innovative approach to accelerating drug release from 3D printed tablets. J. Control. Release 2018, 269, 355–363. [Google Scholar] [CrossRef]
- Isreb, A.; Baj, K.; Wojsz, M.; Isreb, M.; Peak, M.; Alhnan, M.A. 3D printed oral theophylline doses with innovative ‘radiator-like’ design: Impact of polyethylene oxide (PEO) molecular weight. Int. J. Pharm. 2019, 564, 98–105. [Google Scholar] [CrossRef]
- Siyawamwaya, M.; du Toit, L.C.; Kumar, P.; Choonara, Y.E.; Kondiah, P.P.P.D.; Pillay, V. 3D printed, controlled release, tritherapeutic tablet matrix for advanced anti-HIV-1 drug delivery. Eur. J. Pharm. Biopharm. 2019, 138, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, O.N.; Albadarin, A.B.; Croker, D.M.; Healy, A.M.; Walker, G.M. Maximising success in multidrug formulation development: A review. J. Control. Release 2018, 283, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Genina, N.; Boetker, J.P.; Colombo, S.; Harmankaya, N.; Rantanen, J.; Bohr, A. Anti-tuberculosis drug combination for controlled oral delivery using 3D printed compartmental dosage forms: From drug product design to in vivo testing. J. Control. Release 2017, 268, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.C.; Isreb, A.; Forbes, R.T.; Dores, F.; Habashy, R.; Petit, J.-B.; Alhnan, M.A.; Oga, E.F. ‘Temporary Plasticiser’: A novel solution to fabricate 3D printed patient-centred cardiovascular ‘Polypill’ architectures. Eur. J. Pharm. Biopharm. 2019, 135, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Sadia, M.; Isreb, A.; Abbadi, I.; Isreb, M.; Aziz, D.; Selo, A.; Timmins, P.; Alhnan, M.A. From ‘fixed dose combinations’ to ‘a dynamic dose combiner’: 3D printed bi-layer antihypertensive tablets. Eur. J. Pharm. Sci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Trenfield, S.J.; Awad, A.; Goyanes, A.; Gaisford, S.; Basit, A.W. 3D Printing Pharmaceuticals: Drug Development to Frontline Care. Trends Pharmacol. Sci. 2018, 39, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Gioumouxouzis, C.I.; Baklavaridis, A.; Katsamenis, O.L.; Markopoulou, C.K.; Bouropoulos, N.; Tzetzis, D.; Fatouros, D.G. A 3D printed bilayer oral solid dosage form combining metformin for prolonged and glimepiride for immediate drug delivery. Eur. J. Pharm. Sci. 2018, 120, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Goyanes, A.; Wang, J.; Buanz, A.; Martinez-Pacheco, R.; Telford, R.; Gaisford, S.; Basit, A.W. 3D Printing of Medicines: Engineering Novel Oral Devices with Unique Design and Drug Release Characteristics. Mol. Pharm. 2015, 12, 4077–4084. [Google Scholar] [CrossRef] [PubMed]
- Khaled, S.A.; Burley, J.C.; Alexander, M.R.; Yang, J.; Roberts, C.J. 3D printing of five-in-one dose combination polypill with defined immediate and sustained release profiles. J. Control. Release 2015, 217, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Khaled, S.A.; Burley, J.C.; Alexander, M.R.; Yang, J.; Roberts, C.J. 3D printing of tablets containing multiple drugs with defined release profiles. Int. J. Pharm. 2015, 494, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Awad, A.; Trenfield, S.J.; Gaisford, S.; Basit, A.W. 3D printed medicines: A new branch of digital healthcare. Int. J. Pharm. 2018, 548, 586–596. [Google Scholar] [CrossRef] [PubMed]
- Beck, R.C.R.; Chaves, P.S.; Goyanes, A.; Vukosavljevic, B.; Buanz, A.; Windbergs, M.; Basit, A.W.; Gaisford, S. 3D printed tablets loaded with polymeric nanocapsules: An innovative approach to produce customized drug delivery systems. Int. J. Pharm. 2017, 528, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Goyanes, A.; Fernández-Ferreiro, A.; Majeed, A.; Gomez-Lado, N.; Awad, A.; Luaces-Rodríguez, A.; Gaisford, S.; Aguiar, P.; Basit, A.W. PET/CT imaging of 3D printed devices in the gastrointestinal tract of rodents. Int. J. Pharm. 2018, 536, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Feng, X.; Patil, H.; Tiwari, R.V.; Repka, M.A. Coupling 3D printing with hot-melt extrusion to produce controlled-release tablets. Int. J. Pharm. 2017, 519, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Kollamaram, G.; Croker, D.M.; Walker, G.M.; Goyanes, A.; Basit, A.W.; Gaisford, S. Low temperature fused deposition modeling (FDM) 3D printing of thermolabile drugs. Int J Pharm 2018, 545, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Pietrzak, K.; Isreb, A.; Alhnan, M.A. A flexible-dose dispenser for immediate and extended release 3D printed tablets. Eur. J. Pharm. Biopharm. 2015, 96, 380–387. [Google Scholar] [CrossRef]
- Alhnan, M.A.; Okwuosa, T.C.; Sadia, M.; Wan, K.W.; Ahmed, W.; Arafat, B. Emergence of 3D Printed Dosage Forms: Opportunities and Challenges. Pharm. Res. 2016, 33, 1817–1832. [Google Scholar] [CrossRef]
- Goyanes, A.; Buanz, A.B.M.; Hatton, G.B.; Gaisford, S.; Basit, A.W. 3D printing of modified-release aminosalicylate (4-ASA and 5-ASA) tablets. Eur. J. Pharm. Biopharm. 2015, 89, 157–162. [Google Scholar] [CrossRef]
- Gardan, J. Additive manufacturing technologies: State of the art and trends. Int. J. Prod. Res. 2016, 54, 3118–3132. [Google Scholar] [CrossRef]
- Vitale, A.; Cabral, J. Frontal Conversion and Uniformity in 3D Printing by Photopolymerisation. Materials 2016, 9, 760. [Google Scholar] [CrossRef]
- Wang, J.; Goyanes, A.; Gaisford, S.; Basit, A.W. Stereolithographic (SLA) 3D printing of oral modified-release dosage forms. Int. J. Pharm. 2016, 503, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Martinez, P.R.; Goyanes, A.; Basit, A.W.; Gaisford, S. Fabrication of drug-loaded hydrogels with stereolithographic 3D printing. Int. J. Pharm. 2017, 532, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Martinez, P.R.; Goyanes, A.; Basit, A.W.; Gaisford, S. Influence of Geometry on the Drug Release Profiles of Stereolithographic (SLA) 3D-Printed Tablets. AAPS PharmSciTech 2018. [Google Scholar] [CrossRef] [PubMed]
- Fadda, H.M.; Merchant, H.A.; Arafat, B.T.; Basit, A.W. Physiological bicarbonate buffers: Stabilisation and use as dissolution media for modified release systems. Int. J. Pharm. 2009, 382, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Merchant, H.A.; Kulkarni, R.P.; Alkademi, M.; Basit, A.W. Evolution of a physiological pH6.8 bicarbonate buffer system: Application to the dissolution testing of enteric coated products. Eur. J. Pharm. Biopharm. 2011, 78, 151–157. [Google Scholar] [CrossRef]
- Goyanes, A.; Hatton, G.B.; Basit, A.W. A dynamic in vitro model to evaluate the intestinal release behaviour of modified-release corticosteroid products. J. Drug Deliv. Sci. Technol. 2015, 25, 36–42. [Google Scholar] [CrossRef]
- Goyanes, A.; Hatton, G.B.; Merchant, H.A.; Basit, A.W. Gastrointestinal release behaviour of modified-release drug products: Dynamic dissolution testing of mesalazine formulations. Int. J. Pharm. 2015, 484, 103–108. [Google Scholar] [CrossRef]
- Merchant, H.A.; Frost, J.A.; Basit, A.W. Apparatus and Method for Testing Medicaments. U.S. Patent 15/392,113, 20 April 2014. [Google Scholar]
- Merchant, H.A.; Goyanes, A.; Parashar, N.; Basit, A.W. Predicting the gastrointestinal behaviour of modified-release products: Utility of a novel dynamic dissolution test apparatus involving the use of bicarbonate buffers. Int. J. Pharm. 2014, 475, 585–591. [Google Scholar] [CrossRef]
- Trenfield, S.J.; Goyanes, A.; Telford, R.; Wilsdon, D.; Rowland, M.; Gaisford, S.; Basit, A.W. 3D printed drug products: Non-destructive dose verification using a rapid point-and-shoot approach. Int. J. Pharm. 2018, 549, 283–292. [Google Scholar] [CrossRef]
- Yonemochi, E.; Sano, S.; Yoshihashi, Y.; Terada, K. Diffusivity of amorphous drug in solid dispersion. J. Therm. Anal. Calorim. 2013, 113, 1505–1510. [Google Scholar] [CrossRef]
- Corcione, C. Development and Characterization of Novel Photopolymerizable Formulations for Stereolithography. J. Polym. Eng. 2014, 34, 85–93. [Google Scholar] [CrossRef]
- Rao, V.M.; Haslam, J.L.; Stella, V.J. Controlled and complete release of a model poorly water-soluble drug, prednisolone, from hydroxypropyl methylcellulose matrix tablets using (SBE)7m-β-cyclodextrin as a solubilizing agent. J. Pharm. Sci. 2001, 90, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Di Colo, G.; Baggiani, A.; Zambito, Y.; Mollica, G.; Geppi, M.; Serafini, M.F. A new hydrogel for the extended and complete prednisolone release in the GI tract. Int. J. Pharm. 2006, 310, 154–161. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Type I (% w/w) | Type II (% w/w) | Type III (% w/w) | |
|---|---|---|---|---|
| Material | ||||
| PEGda | 89 | 89 | 44.5 | |
| PEG300 | - | - | 44.5 | |
| TPO | 1 | 1 | 1 | |
| Drug | 10 | 10 | 10 | |
| Type I | ||
| Width (mm) ± %CV | Height ± SD (mm) | Weight ± SD (mg) |
| 10.99 ± 1.0 | 2.81 ± 9.8 | 329 ± 13.6 |
| Type II | ||
| Width (mm) ± %CV | Height ± SD (mm) | Weight ± SD (mg) |
| 11.07 ± 0.1 | 6.12 ± 0.03 | 501.13 ± 6.3 |
| Type III | ||
| Width (mm) ± %CV | Height ± SD (mm) | Weight ± SD (mg) |
| 10.73 ± 0.18 | 6.12 ± 0.02 | 553 ± 8.9 |
| Drug | Swelling Ratio |
|---|---|
| Paracetamol | 1.21 ± 0.07 |
| Aspirin | 1.15 ± 0.03 |
| Naproxen | 1.18 ± 0.03 |
| Prednisolone * | 1.11 ± 0.05 |
| Chloramphenicol * | 1.05 ± 0.02 |
| Caffeine * | 1.17 ± 0.02 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robles-Martinez, P.; Xu, X.; Trenfield, S.J.; Awad, A.; Goyanes, A.; Telford, R.; Basit, A.W.; Gaisford, S. 3D Printing of a Multi-Layered Polypill Containing Six Drugs Using a Novel Stereolithographic Method. Pharmaceutics 2019, 11, 274. https://doi.org/10.3390/pharmaceutics11060274
Robles-Martinez P, Xu X, Trenfield SJ, Awad A, Goyanes A, Telford R, Basit AW, Gaisford S. 3D Printing of a Multi-Layered Polypill Containing Six Drugs Using a Novel Stereolithographic Method. Pharmaceutics. 2019; 11(6):274. https://doi.org/10.3390/pharmaceutics11060274
Chicago/Turabian StyleRobles-Martinez, Pamela, Xiaoyan Xu, Sarah J. Trenfield, Atheer Awad, Alvaro Goyanes, Richard Telford, Abdul W. Basit, and Simon Gaisford. 2019. "3D Printing of a Multi-Layered Polypill Containing Six Drugs Using a Novel Stereolithographic Method" Pharmaceutics 11, no. 6: 274. https://doi.org/10.3390/pharmaceutics11060274
APA StyleRobles-Martinez, P., Xu, X., Trenfield, S. J., Awad, A., Goyanes, A., Telford, R., Basit, A. W., & Gaisford, S. (2019). 3D Printing of a Multi-Layered Polypill Containing Six Drugs Using a Novel Stereolithographic Method. Pharmaceutics, 11(6), 274. https://doi.org/10.3390/pharmaceutics11060274