Feasibility Study of Mesoporous Silica Particles for Pulmonary Drug Delivery: Therapeutic Treatment with Dexamethasone in a Mouse Model of Airway Inflammation



Abstract
1. Introduction
2. Materials and Methods
2.1. Preparation of the Large (L-MSP) and Small (S-MSP) Mesoporous Silica Particles
2.2. Loading of Drug to the Particles
2.3. Copolymer-Adsorption on the Particles
2.4. Determination of the Loaded DEX Amount
2.5. Animal Models
- Melphalan (MEL)-induced airway inflammation: The mice were briefly anaesthetized with isofluran and melphalan (4-(bis(2-chlorethyl)amino)-l-phenylalanine) (Sigma-Aldrich, St Louis, MO, USA) administered by intratracheal instillation in a volume of 50 µL (1 mg/kg). Melphalan was dissolved in acidic ethanol (30 µL concentrated HCl in 1 mL 99.5% ethanol) to a concentration of 100 mg/mL, and further diluted in phosphate buffered saline (PBS) to a final concentration just before administration [24]. The control mice received only the solvent.
- Lipopolysaccharide (LPS)-induced airway inflammation: The mice were exposed to an aerosol of Lipopolysaccharide (LPS; Escherichia coli O128:B12; Sigma-Aldrich, Merck KGaA, Darmstadt, Germany) for 15 min using a nose-only Battelle exposure chamber. The aerosol was generated by a compressed-air Collison six-jet nebulizer at an airflow of 7 liters/min using a nebulizer concentration of 0.1 mg/mL of LPS dissolved in water [31]. The control mice were exposed to an aerosol of solvent alone.
2.5.1. Treatment
2.5.2. Statistical Analysis of the Animal Data
3. Results and Discussion
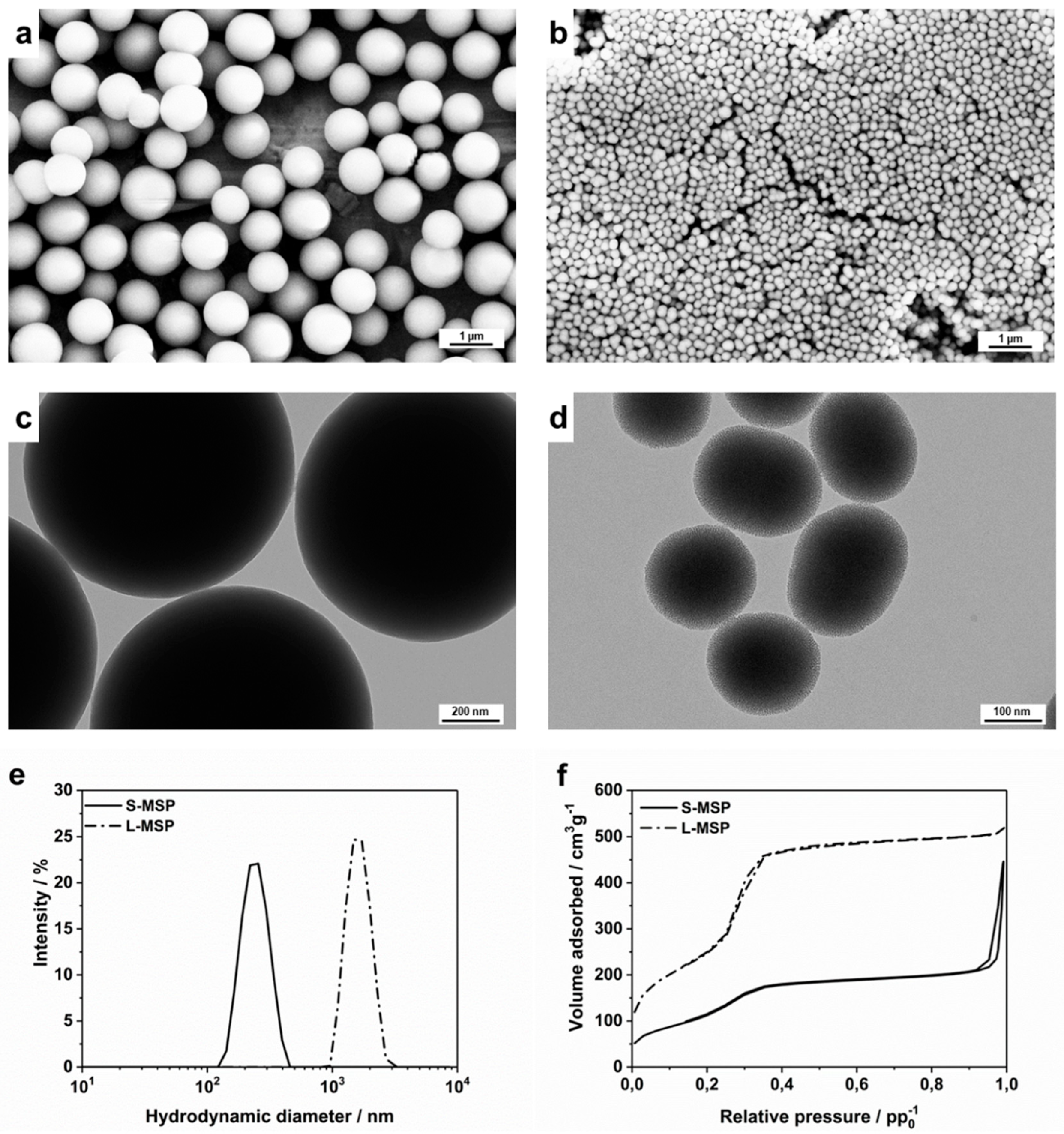
3.1. Design and Characterization of the Carrier Particles for the Anti-Inflammatory Drug
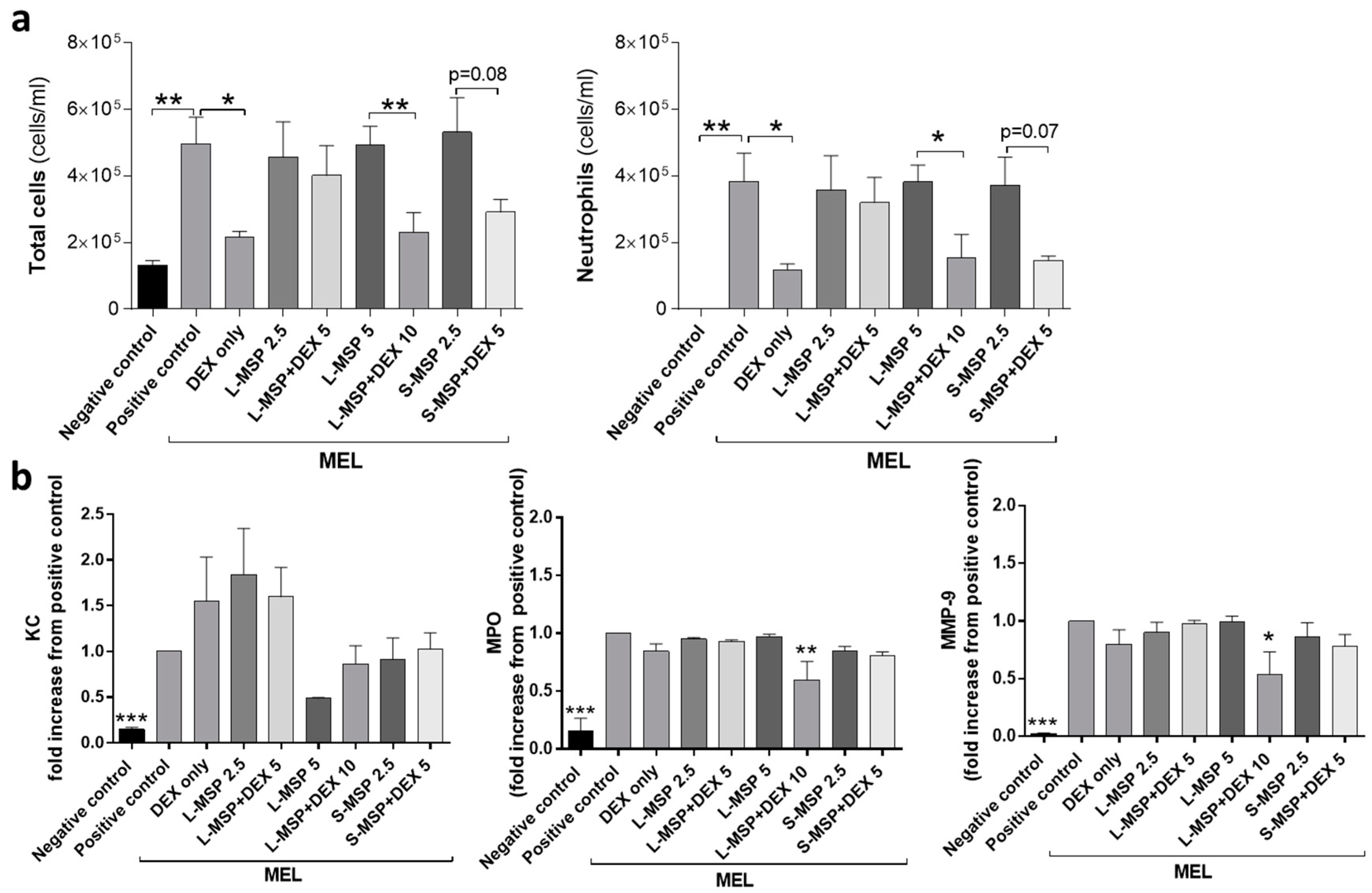
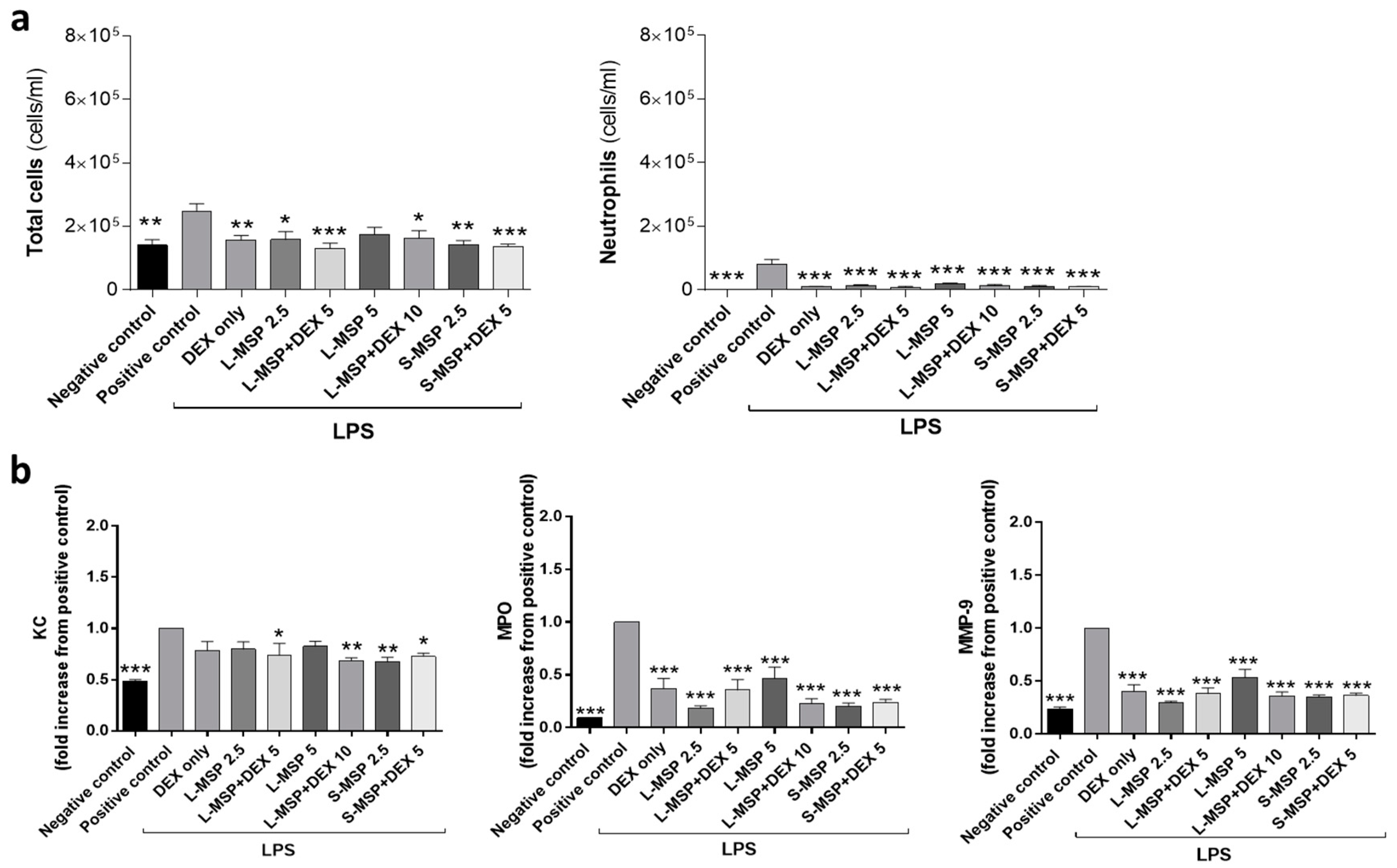
3.2. In Vivo Investigation of the Anti-Inflammatory Response Induced by Drug-Loaded MSPs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sung, J.C.; Pulliam, B.L.; Edwards, D.A. Nanoparticles for drug delivery to the lungs. Trends Biotechnol. 2007, 25, 563–570. [Google Scholar] [CrossRef]
- Babu, A.; Templeton, A.K.; Munshi, A.; Ramesh, R. Nanoparticle-based drug delivery for therapy of lung cancer: Progress and challenges. J. Nanomater. 2013, 2013, 14. [Google Scholar] [CrossRef]
- Yhee, J.Y.; Im, J.; Nho, R.S. Advanced therapeutic strategies for chronic lung disease using nanoparticle-based drug delivery. J. Clin. Med. 2016, 5, 82. [Google Scholar] [CrossRef] [PubMed]
- Global Health Estimates 2016: Deaths by Cause, Age, Sex, by Country and by Region, 2000–2016. Geneva, World Health Organization. 2018. Available online: http://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 2 November 2018).
- Smola, M.; Vandamme, T.; Sokolowski, A. Nanocarriers as pulmonary drug delivery systems to treat and to diagnose respiratory and non-respiratory diseases. Int. J. Nanomed. 2008, 3, 1–19. [Google Scholar]
- Vaughn, J.M.; McConville, J.T.; Burgess, D.; Peters, J.I.; Johnston, K.P.; Talbert, R.L.; Williams III, R.O. Single dose and multiple dose studies of itraconazole nanoparticles. Eur. J. Pharm. Biopharm. 2006, 63, 95–102. [Google Scholar] [CrossRef]
- Zahoor, A.; Sharma, S.; Khuller, G. Inhalable alginate nanoparticles as antitubercular drug carriers against experimental tuberculosis. Int. J. Antimicrob. Agents 2005, 26, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Paranjpe, M.; Müller-Goymann, C.C. Nanoparticle-mediated pulmonary drug delivery: A review. Int. J. Mol. Sci. 2014, 15, 5852–5873. [Google Scholar] [CrossRef]
- Tena, A.F.; Clarà, P.C. Deposition of inhaled particles in the lungs. Archivos de Bronconeumología (Engl. Ed.) 2012, 48, 240–246. [Google Scholar] [CrossRef]
- Barbe, C.; Bartlett, J.; Kong, L.; Finnie, K.; Lin, H.Q.; Larkin, M.; Calleja, S.; Bush, A.; Calleja, G. Silica particles: A novel drug-delivery system. Adv. Mater. 2004, 16, 1959–1966. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, Q.; Han, N.; Bai, L.; Li, J.; Liu, J.; Che, E.; Hu, L.; Zhang, Q.; Jiang, T. Mesoporous silica nanoparticles in drug delivery and biomedical applications. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 313–327. [Google Scholar] [CrossRef]
- Li, X.; Xue, M.; Raabe, O.G.; Aaron, H.L.; Eisen, E.A.; Evans, J.E.; Hayes, F.A.; Inaga, S.; Tagmount, A.; Takeuchi, M. Aerosol droplet delivery of mesoporous silica nanoparticles: A strategy for respiratory-based therapeutics. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 1377–1385. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Rosenholm, J.M. The viability of mesoporous silica nanoparticles for drug delivery. Ther. Deliv. 2015, 6, 891–893. [Google Scholar] [CrossRef]
- Li, Z.; Barnes, J.C.; Bosoy, A.; Stoddart, J.F.; Zink, J.I. Mesoporous silica nanoparticles in biomedical applications. Chem. Soc. Rev. 2012, 41, 2590–2605. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Peters, J.I.; Williams III, R.O. Inhaled nanoparticles—A current review. Int. J. Pharm. 2008, 356, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Wicki, A.; Witzigmann, D.; Balasubramanian, V.; Huwyler, J. Nanomedicine in cancer therapy: Challenges, opportunities, and clinical applications. J. Control. Release 2015, 200, 138–157. [Google Scholar] [CrossRef]
- Desai, D.; Karaman, D.S.; Prabhakar, N.; Tadayon, S.; Duchanoy, A.; Toivola, D.M.; Rajput, S.; Näreoja, T.; Rosenholm, J.M. Design considerations for mesoporous silica nanoparticulate systems in facilitating biomedical applications. Open Mater. Sci. 2014, 1. [Google Scholar] [CrossRef]
- Maleki, A.; Kettiger, H.; Schoubben, A.; Rosenholm, J.M.; Ambrogi, V.; Hamidi, M. Mesoporous silica materials: From physico-chemical properties to enhanced dissolution of poorly water-soluble drugs. J. Control. Release 2017, 262, 329–347. [Google Scholar] [CrossRef]
- Lu, J.; Liong, M.; Zink, J.I.; Tamanoi, F. Mesoporous silica nanoparticles as a delivery system for hydrophobic anticancer drugs. Small 2007, 3, 1341–1346. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, Q.; Hu, Y.; Sun, L.; Bai, L.; Jiang, T.; Wang, S. Ordered nanoporous silica as carriers for improved delivery of water insoluble drugs: A comparative study between three dimensional and two dimensional macroporous silica. Int. J. Nanomed. 2013, 8, 4015–4031. [Google Scholar] [CrossRef]
- Barnes, P.J.; Adcock, I.M. Glucocorticoid resistance in inflammatory diseases. Lancet 2009, 373, 1905–1917. [Google Scholar] [CrossRef]
- de Lange, D.W.; Meulenbelt, J. Do corticosteroids have a role in preventing or reducing acute toxic lung injury caused by inhalation of chemical agents? Clin. Toxicol. 2011, 49, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Jonasson, S.; Wigenstam, E.; Koch, B.; Bucht, A. Early treatment of chlorine-induced airway hyperresponsiveness and inflammation with corticosteroids. Toxicol. Appl. Pharmacol. 2013, 271, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Wigenstam, E.; Rocksén, D.; Ekstrand-Hammarström, B.; Bucht, A. Treatment with dexamethasone or liposome-encapsuled vitamin E provides beneficial effects after chemical-induced lung injury. Inhal. Toxicol. 2009, 21, 958–964. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. PubChem Compound Database. CID 5743. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/5743 (accessed on 2 November 2018).
- Tsapis, N.; Bennett, D.; Jackson, B.; Weitz, D.A.; Edwards, D. Trojan particles: Large porous carriers of nanoparticles for drug delivery. Proc. Natl. Acad. Sci. USA 2002, 99, 12001–12005. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.; Löbenberg, R.; Ku, T.; Azarmi, S.; Roa, W.; Prenner, E.J. Nanoparticles: Characteristics, mechanisms of action, and toxicity in pulmonary drug delivery—A review. J. Biomed. Nanotechnol. 2007, 3, 107–119. [Google Scholar] [CrossRef]
- Wigenstam, E.; Jonasson, S.; Koch, B.; Bucht, A. Corticosteroid treatment inhibits airway hyperresponsiveness and lung injury in a murine model of chemical-induced airway inflammation. Toxicology 2012, 301, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Ekstrand-Hammarström, B.; Wigenstam, E.; Bucht, A. Inhalation of alkylating mustard causes long-term T cell-dependent inflammation in airways and growth of connective tissue. Toxicology 2011, 280, 88–97. [Google Scholar] [CrossRef]
- Rocksén, D.; Ekstrand-Hammarstrom, B.; Johansson, L.; Bucht, A. Vitamin E reduces transendothelial migration of neutrophils and prevents lung injury in endotoxin-induced airway inflammation. Am. J. Respir. Cell Mol. Biol. 2003, 28, 199–207. [Google Scholar] [CrossRef]
- Rocksén, D.; Lilliehöök, B.; Larsson, R.; Johansson, T.; Bucht, A. Differential anti-inflammatory and anti-oxidative effects of dexamethasone and N-acetylcysteine in endotoxin-induced lung inflammation. Clin. Exp. Immunol. 2000, 122, 249–256. [Google Scholar] [CrossRef]
- Kumar, D.; Schumacher, K.; von Hohenesche, C.d.F.; Grün, M.; Unger, K. MCM-41, MCM-48 and related mesoporous adsorbents: Their synthesis and characterisation. Colloids Surf. A Physicochem. Eng. Asp. 2001, 187, 109–116. [Google Scholar] [CrossRef]
- Karaman, D.Ş.; Gulin-Sarfraz, T.; Hedström, G.; Duchanoy, A.; Eklund, P.; Rosenholm, J.M. Rational evaluation of the utilization of PEG-PEI copolymers for the facilitation of silica nanoparticulate systems in biomedical applications. J. Colloid Interface Sci. 2014, 418, 300–310. [Google Scholar] [CrossRef]
- Martín, A.; García, R.; Karaman, D.S.; Rosenholm, J. Polyethyleneimine-functionalized large pore ordered silica materials for poorly water-soluble drug delivery. J. Mater. Sci. 2014, 49, 1437–1447. [Google Scholar] [CrossRef]
- Wigenstam, E.; Koch, B.; Bucht, A.; Jonasson, S. N-acetyl cysteine improves the effects of corticosteroids in a mouse model of chlorine-induced acute lung injury. Toxicology 2015, 328, 40–47. [Google Scholar] [CrossRef] [PubMed]
- De Boer, A.H.; Hagedoorn, P.; Hoppentocht, M.; Buttini, F.; Grasmeijer, F.; Frijlink, H.W. Dry powder inhalation: Past, present and future. Expert Opin. Drug Deliv. 2017, 14, 499–512. [Google Scholar] [CrossRef]
- Muthas, D.; Reznichenko, A.; Balendran, C.A.; Böttcher, G.; Clausen, I.G.; Kärrman Mårdh, C.; Ottosson, T.; Uddin, M.; MacDonald, T.T.; Danese, S.; et al. Neutrophils in ulcerative colitis: A review of selected biomarkers and their potential therapeutic implications. Scand. J. Gastroenterol. 2017, 52, 125–135. [Google Scholar] [CrossRef]
- Sanfins, E.; Correia, A.; Gunnarsson, S.B.; Vilanova, M.; Cedervall, T. Nanoparticle effect on neutrophil produced myeloperoxidase. PLoS ONE 2018, 13, e0191445. [Google Scholar] [CrossRef]
- Sun, L.; Guo, R.F.; Newstead, M.W.; Standiford, T.J.; Macariola, D.R.; Shanley, T.P. Effect of IL-10 on neutrophil recruitment and survival after pseudomonas aeruginosa challenge. Am. J. Respir. Cell Mol. Biol. 2009, 41, 76–84. [Google Scholar] [CrossRef]
- Kroll, A.; Pillukat, M.H.; Hahn, D.; Schnekenburger, J. Interference of engineered nanoparticles with in vitro toxicity assays. Arch. Toxicol. 2012, 86, 1123–1136. [Google Scholar] [CrossRef]
- Veranth, J.M.; Kaser, E.G.; Veranth, M.M.; Koch, M.; Yost, G.S. Cytokine responses of human lung cells (BEAS-2B) treated with micron-sized and nanoparticles of metal oxides compared to soil dusts. Part. Fibre Toxicol. 2007, 4, 2. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.M.; Dickson, C.; Duncan, P.; Al-Attili, F.; Stone, V. Interaction between nanoparticles and cytokine proteins: Impact on protein and particle functionality. Nanotechnology 2010, 21, 215104. [Google Scholar] [CrossRef] [PubMed]
- Shim, K.H.; Hulme, J.; Maeng, E.H.; Kim, M.K.; An, S.S.A. Analysis of zinc oxide nanoparticles binding proteins in rat blood and brain homogenate. Int. J. Nanomed. 2014, 9 (Suppl. 2), 217–224. [Google Scholar]
- Konduru, N.V.; Molina, R.M.; Swami, A.; Damiani, F.; Pyrgiotakis, G.; Lin, P.; Andreozzi, P.; Donaghey, T.C.; Demokritou, P.; Krol, S.; et al. Protein corona: Implications for nanoparticle interactions with pulmonary cells. Part. Fibre Toxicol. 2017, 14, 42. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Exposure | Particle Size | Group | Particle Dose/Vehicle | DEX Dose |
|---|---|---|---|---|
| Solvent | Control | HEPES | - | |
| MEL or LPS exposure | MEL or LPS | HEPES | - | |
| DEX | 5 mg/mL | |||
| L-MSP (1 µm) | L-MSP 2.5 | 2.5 mg/mL | - | |
| L-MSP (1 µm) | L-MSP+DEX 5 | 5 mg/mL | 2.5 mg/mL | |
| L-MSP (1 µm) | L-MSP 5 | 5 mg/mL | - | |
| L-MSP (1 µm) | L-MSP+DEX 10 | 10 mg/mL | 5 mg/mL | |
| S-MSP (200 nm) | S-MSP 2.5 | 2.5 mg/mL | - | |
| S-MSP (200 nm) | S-MSP+DEX 5 | 5 mg/mL | 2.5 mg/mL |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gulin-Sarfraz, T.; Jonasson, S.; Wigenstam, E.; von Haartman, E.; Bucht, A.; Rosenholm, J.M. Feasibility Study of Mesoporous Silica Particles for Pulmonary Drug Delivery: Therapeutic Treatment with Dexamethasone in a Mouse Model of Airway Inflammation. Pharmaceutics 2019, 11, 149. https://doi.org/10.3390/pharmaceutics11040149
Gulin-Sarfraz T, Jonasson S, Wigenstam E, von Haartman E, Bucht A, Rosenholm JM. Feasibility Study of Mesoporous Silica Particles for Pulmonary Drug Delivery: Therapeutic Treatment with Dexamethasone in a Mouse Model of Airway Inflammation. Pharmaceutics. 2019; 11(4):149. https://doi.org/10.3390/pharmaceutics11040149
Chicago/Turabian StyleGulin-Sarfraz, Tina, Sofia Jonasson, Elisabeth Wigenstam, Eva von Haartman, Anders Bucht, and Jessica M. Rosenholm. 2019. "Feasibility Study of Mesoporous Silica Particles for Pulmonary Drug Delivery: Therapeutic Treatment with Dexamethasone in a Mouse Model of Airway Inflammation" Pharmaceutics 11, no. 4: 149. https://doi.org/10.3390/pharmaceutics11040149
APA StyleGulin-Sarfraz, T., Jonasson, S., Wigenstam, E., von Haartman, E., Bucht, A., & Rosenholm, J. M. (2019). Feasibility Study of Mesoporous Silica Particles for Pulmonary Drug Delivery: Therapeutic Treatment with Dexamethasone in a Mouse Model of Airway Inflammation. Pharmaceutics, 11(4), 149. https://doi.org/10.3390/pharmaceutics11040149