Challenges and Recent Progress in Oral Drug Delivery Systems for Biopharmaceuticals



Abstract
1. Introduction
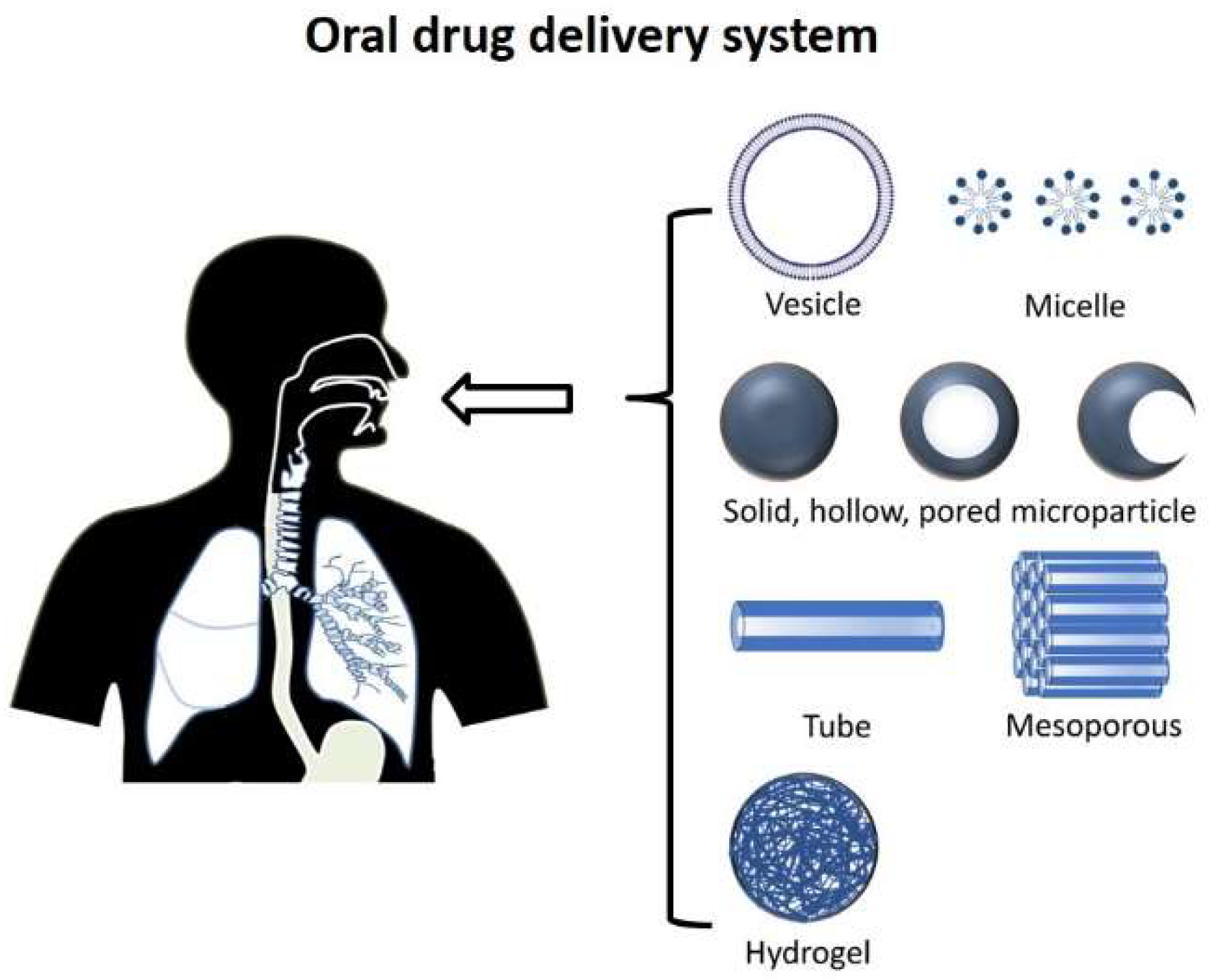
2. Oral Route
3. Challenges Associated with Oral Delivery
3.1. Biological Barriers
3.1.1. Lumen
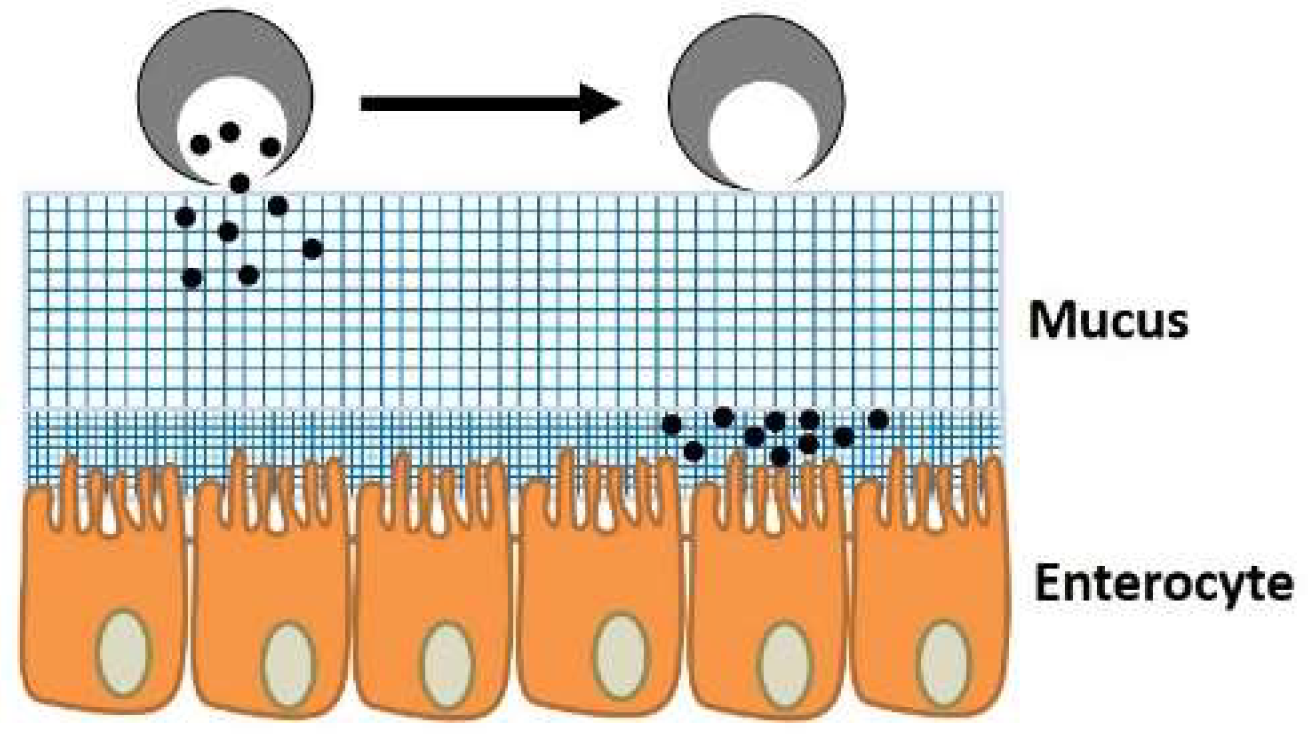
3.1.2. Mucus
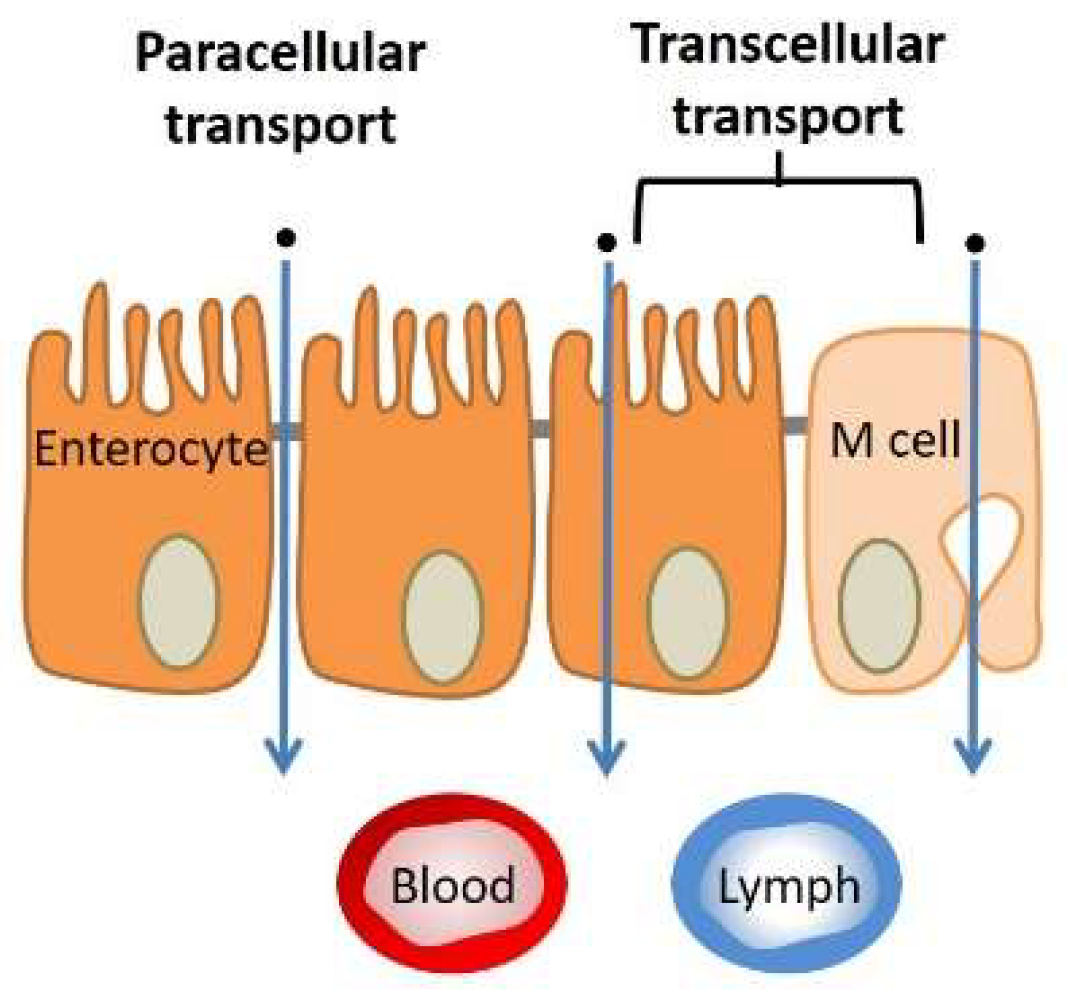
3.1.3. Tissue (Extracellular Barriers)
3.2. Technical Challenges
3.2.1. Oral Delivery Devices and Materials
3.2.2. Sustained Delivery
Mucoadhesive Carriers
Recent Gastric-Resident Architectures
3.3. Solvent-Free Microencapsulation
3.3.1. Multiemulsion Systems
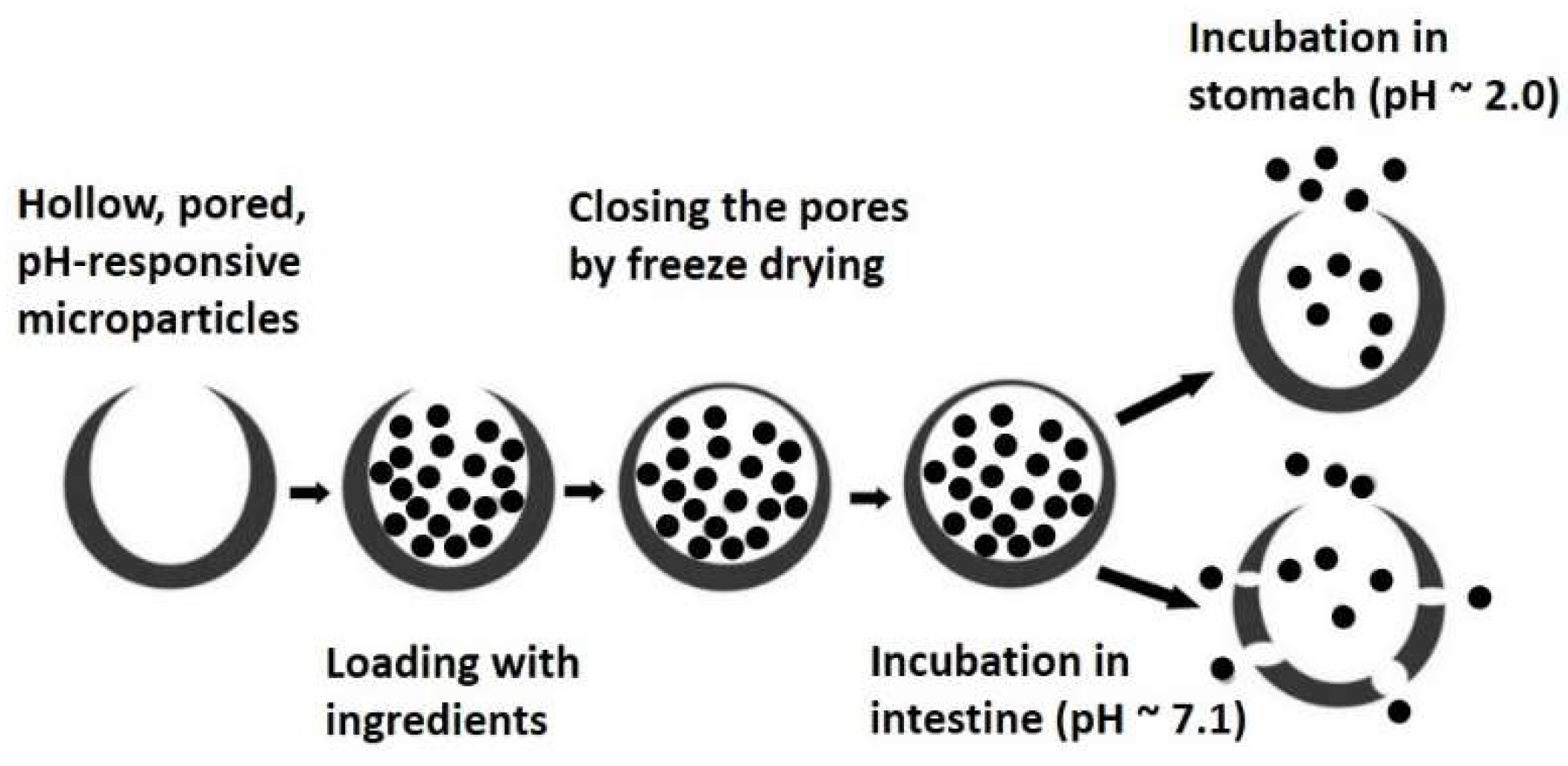
3.3.2. Pored and Hollow Microencapsulation Systems
3.4. Co-Delivery Systems
3.5. Scaling up and Throughput
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siafaka, P.I.; Titopoulou, A.; Koukaras, E.N.; Kostoglou, M.; Koutris, E.; Karavas, E.; Bikiaris, D.N. Chitosan derivatives as effective nanocarriers for ocular release of timolol drug. Int. J. Pharm. 2015, 495, 249–264. [Google Scholar] [CrossRef] [PubMed]
- Ferraiolo, B.L.; Mohler, M.A.; Gloff, C.A. Volume 1: Protein pharmacokinetics and metabolism. In Pharmaeutical Biotechnology.; Borchard, R.T., Ed.; Springer Science+Business Media, LLC: New York, NY, USA, 1992; pp. 78–150. [Google Scholar]
- Römgens, A.M.; Rem-bronneberg, D.; Kassies, R.; Hijlkema, M.; Bader, D.L.; Oomens, C.W.J.; Bruggen, M.P.B. Penetration and delivery characteristics of repetitive microjet injection into the skin. J. Control. Release 2016, 234, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Rodger, M.A.; King, L. Drawing up and administering intramuscular injections: A review of the literature. J. Adv. Nurs. 2000, 31, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Stringer, M.D. Sciatic nerve injury from intramuscular injection: A persistent and global problem. Int. J. Clin. Pract. 2010, 64, 1573–1579. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, L.H.; Hesby, A. Intramuscular injection: An integration research review and guideline for evidence-based practice. Appl. Nurs. Res. 2002, 16, 149–162. [Google Scholar] [CrossRef]
- Liang, F.; Loré, K. Local innate immune responses in the vaccine adjuvant-injected muscle. Clin. Transl. Immunol. 2016, 5, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Herzog, R.W.; Hagstrom, J.N.; Kung, S.H.; Tai, S.J.; Wilson, J.M.; Fisher, K.J.; High, K.A. Stable gene transfer and expression of human blood coagulation factor IX after intramuscular injection of recombinant adeno-associated virus. Proc. Natl. Acad. Sci. USA 1997, 94, 5804–5809. [Google Scholar] [CrossRef] [PubMed]
- Nanaki, S.; Siafaka, P.I.; Zachariadou, D.; Nerantzaki, M.; Giliopoulos, D.J.; Triantafyllidis, K.S.; Kostoglou, M.; Nikolakaki, E.; Bikiaris, D.N. PLGA/SBA-15 mesoporous silica composite microparticles loaded with paclitaxel for local chemotherapy. Eur. J. Pharm. Sci. 2017, 99, 32–44. [Google Scholar] [CrossRef]
- Nanaki, S.; Tseklima, M.; Terzopoulou, Z.; Nerantzaki, M.; Giliopoulos, D.J.; Triantafyllidis, K.; Kostoglou, M.; Bikiaris, D.N. Use of mesoporous cellular foam (MCF) in preparation of polymeric microspheres for long acting injectable release formulations of paliperidone antipsychotic drug. Eur. J. Pharm. Biopharm. 2017, 117, 77–90. [Google Scholar] [CrossRef]
- Fletcher, N.A.; Krebs, M.D. Sustained delivery of anti-VEGF from injectable hydrogel systems provides a prolonged decrease of endothelial cell proliferation and angiogenesis in vitro. RSC Adv. 2018, 8, 8999–9005. [Google Scholar] [CrossRef]
- Moeller, E.H.; Jorgensen, L. Alternative routes of administration for systemic delivery of protein pharmaceuticals. Drug Discov. Today Technol. 2008, 5, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Kale, T.R. Needle free injection technology—An overview. Inov. Pharm. 2014, 5, 1–8. [Google Scholar] [CrossRef]
- Brown, M.B.; Martin, G.P.; Jones, S.A.; Akomeah, F.K.; Brown, M.B.; Martin, G.P.; Jones, S.A.; Akomeah, F.K.; Brown, M.B.; Martin, G.P.; et al. Dermal and transdermal drug delivery systems: Current and future prospects. Drug Deliv. 2006, 13, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Ranade, V.V. Drug delivery systems. 6. Transdermal drug delivery. J. Clin. Pharmacol. 1991, 31, 401–418. [Google Scholar] [CrossRef]
- Nerantzaki, M.; Kehagias, N.; Francone, A.; Ferna, A.; Torres, C.M.S.; Papi, R.; Choli-papadopoulou, T.; Bikiaris, D.N. Design of a multifunctional nanoengineered PLLA surface by maximizing the synergies between biochemical and surface design bactericidal effects. ACS Omega 2018, 3, 1509–1521. [Google Scholar] [CrossRef]
- Kim, H.; Jang, H.; Kim, B.; Kim, M.K.; Wie, D.S.; Lee, H.S.; Kim, D.R.; Lee, C.H.F.; Jaganathan, K.S. Nasal vaccine delivery (Chapter fifteen). Appl. Sci. Eng. 2018, 1, 1–9. [Google Scholar]
- Lee, S.; Mcauliffe, D.J.; Flotte, T.J.; Kollias, N.; Doukas, A.G. Photomechanical transcutaneous delivery of macromolecules. J. Invest. Dermatol. 1998, 111, 925–929. [Google Scholar] [CrossRef] [PubMed]
- Prausnitz, M.R.; Langer, R. Transdermal drug delivery. Nat. Biotechnol. 2008, 26, 1261–1268. [Google Scholar] [CrossRef]
- Chen, Y.; Quan, P.; Liu, X.; Wang, M.; Fang, L. Novel chemical permeation enhancers for transdermal drug delivery. Asian J. Pharm. Sci. 2014, 9, 51–64. [Google Scholar] [CrossRef]
- Karande, P.; Jain, A.; Mitragotri, S. Insights into synergistic interactions in binary mixtures of chemical permeation enhancers for transdermal drug delivery. J. Control. Release 2006, 115, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, L.; Lamb, J.; Elliott, T. Section 2. Practice. In Intravenous Therapy in Nursing Practice; Finlay, T., Lamb, J., Dougherty, L., Quinn, C., Eds.; Blackwell Publishing: Oxford, UK, 2008; pp. 143–225. [Google Scholar]
- Maxwell, M.J.; Wilson, M.J.A. Complications of blood transfusion. Contin. Educ. Anaesth. Crit. Care Pain 2006, 6, 225–229. [Google Scholar] [CrossRef]
- Korttila, K.; Aromaa, U. Venous complications after intravenous injection of diazepam, flunitrazepam, thiopentone and etomidate. Acta Anaesthesiol. Scand. 1980, 24, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Awate, S.; Babiuk, L.A.; Mutwiri, G. Mechanisms of action of adjuvants. Front. Immunol. 2013, 4, 1–10. [Google Scholar] [CrossRef]
- Harshad, P.; Anand, B.; Dushyant, S. Recent techniques in nasal drug delivery: A review. Int. J. Drug Dev. Res. 2010, 2, 565–572. [Google Scholar]
- Nanaki, S.; Tseklima, M.; Christodoulou, E.; Triantafyllidis, K.; Kostoglou, M.; Bikiaris, D.N. Thiolated chitosan masked polymeric microspheres with incorporated mesocellular silica foam (MCF) for intranasal delivery of paliperidone. Polymers 2017, 9, 617. [Google Scholar] [CrossRef]
- Grassin-delyle, S.; Buenestado, A.; Naline, E.; Faisy, C.; Blouquit-laye, S.; Couderc, L.; Le, M.; Fischler, M.; Devillier, P. Intranasal drug delivery: An efficient and non-invasive route for systemic administration Focus on opioids. Pharmacol. Ther. 2012, 134, 366–379. [Google Scholar] [CrossRef]
- Bhise, S.B.; Yadav, A.V.; Avachat, A.M.; Malayandi, R. Bioavailability of intranasal drug delivery system. Asian J. Pharm. 2008, 2, 201–215. [Google Scholar] [CrossRef]
- Ramvikas, M.; Arumugam, M.; Chakrabarti, S.R.; Jaganathan, K.S. Nasal vaccine delivery (Chapter fifteen). In Micro- and Nanotechnology in Vaccine Development; Elsevier Inc.: Amsterdam, The Netherlands, 2017; pp. 279–301. [Google Scholar]
- Bakri, W.; Donovan, M.D.; Cueto, M.; Wu, Y.; Orekie, C.; Yang, Z. Overview of intranasally delivered peptides: Key considerations for pharmaceutical development. Expert Opin. Drug Deliv. 2018, 15, 991–1005. [Google Scholar] [CrossRef]
- Choi, H.J.; Kim, M.C.; Kang, S.M.; Montemagno, C.D. The osmotic stress response of split influenza vaccine particles in an acidic environment. Arch. Pahrmacal Res. 2014, 37, 1607–1616. [Google Scholar] [CrossRef]
- Banerjee, A.; Qi, J.; Gogoi, R.; Wong, J.; Mitragotri, S. Role of nanoparticle size, shape and surface chemistry in oral drug delivery. J. Control. Release 2016, 238, 176–185. [Google Scholar] [CrossRef]
- Araújo, F.; Pedro, J.; Granja, P.L.; Santos, H.A.; Sarmento, B. Functionalized materials for multistage platforms in the oral delivery of biopharmaceuticals. Prog. Mater. Sceince 2017, 89, 306–344. [Google Scholar] [CrossRef]
- Hu, Q.; Luo, Y. Recent advances of polysaccharide-based nanoparticles for oral insulin delivery. Int. J. Biol. Macromol. 2018, 120, 775–782. [Google Scholar] [CrossRef]
- Choi, H.-J.; Ebersbacher, C.F.; Kim, M.C.; Kang, S.M.; Montemagno, C.D. A mechanistic study on the destabilization of whole inactivated influenza virus vaccine in gastric environment. PLoS ONE 2013, 8, 1–14. [Google Scholar] [CrossRef]
- Schenk, M.; Mueller, C. The mucosal immune system at the gastrointestinal barrier. Best Pract. Res. 2008, 22, 391–409. [Google Scholar] [CrossRef]
- Ensign, L.M.; Cone, R.; Hanes, J. Oral drug delivery with polymeric nanoparticles: The gastrointestinal mucus barriers. Adv. Drug Deliv. Rev. 2012, 64, 557–570. [Google Scholar] [CrossRef]
- Leal, J.; Smyth, H.D.C.; Ghosh, D. Physicochemical properties of mucus and their impact on transmucosal drug delivery. Int. J. Pharm. 2017, 532, 555–572. [Google Scholar] [CrossRef]
- Fievez, V.; Garinot, M.; Schneider, Y.; Préat, V. Nanoparticles as potential oral delivery systems of proteins and vaccines: A mechanistic approach. J. Control. Release 2006, 116, 1–27. [Google Scholar]
- Brayden, D.J.; Jepson, M.A.; Baird, A.W. Intestinal Peyer’s patch M cells and oral vaccine targeting. Drug Discov. Today 2005, 10, 1145–1157. [Google Scholar] [CrossRef]
- Kwon, K.; Daniell, H. Oral delivery of protein drugs bioencapsulated in plant cells. Mol. Ther. 2016, 24, 1342–1350. [Google Scholar] [CrossRef]
- Ma, S.; Wang, L.; Huang, X.; Wang, X.; Chen, S.; Shi, W.; Qiao, X.; Jiang, Y. Oral recombinant Lactobacillus vaccine targeting the intestinal microfold cells and dendritic cells for delivering the core neutralizing epitope of porcine epidemic diarrhea virus. Microb. Cell Fact. 2018, 17, 1–12. [Google Scholar] [CrossRef]
- Maharjan, S.; Singh, B.; Jiang, T.; Yoon, S.; Li, H.; Kim, G.; Jeong, M.; Ji, S.; Park, O.; Hyun, S.; et al. Systemic administration of RANKL overcomes the bottleneck of oral vaccine delivery through microfold cells in ileum. Biomaterials 2016, 84, 286–300. [Google Scholar] [CrossRef] [PubMed]
- Varum, F.J.O.; Mcconnell, E.L.; Sousa, J.J.S.; Veiga, F.; Basit, A.W. Mucoadhesion and the gastrointestinal tract. Crit. Rev. Ther. Drug Carrier Syst. 2008, 25, 207–258. [Google Scholar] [CrossRef]
- Dawson, M.; Krauland, E.; Wirtz, D.; Hanes, J. Transport of polymeric nanoparticle gene carriers in gastric mucus. Biotechnol. Prog. 2004, 20, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Hounnou, G.; Destrieux, C.; Desme, J.; Bertrand, P.; Velut, S. Anatomical study of the length of the human intestine. Surg. Radiol. Anat. 2002, 24, 290–294. [Google Scholar]
- Helander, H.F.; Fändriks, L. Surface area of the digestive tract – revisited. Scand. J. Gastroenterol. 2014, 49, 681–689. [Google Scholar] [CrossRef]
- Azizi, A.; Kumar, A.; Diaz-mitoma, F.; Mestecky, J. Enhancing oral vaccine potency by targeting intestinal M cells. PLoS Pathog. 2010, 6, 1001147–1001154. [Google Scholar] [CrossRef] [PubMed]
- Mudie, D.M.; Amidon, G.L.; Amidon, G.E. Physiological parameters for oral delivery and in vitro testing. Mol. Pharm. 2010, 7, 1388–1405. [Google Scholar] [CrossRef]
- Vllasaliu, D.; Thanou, M.; Stolnik, S.; Fowler, R. Recent advances in oral delivery of biologics: Nanomedicine and physical modes of delivery. Expert Opin. Drug Deliv. 2018, 15, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Tao, S.L.; Desai, T.A. Micromachined devices: The impact of controlled geometry from cell-targeting to bioavailability. J. Control. Release 2005, 109, 127–138. [Google Scholar] [CrossRef]
- Rzhevskiy, A.S.; Raghu, T.; Singh, R.; Donnelly, R.F.; Anissimov, Y.G. Microneedles as the technique of drug delivery enhancement in diverse organs and tissues. J. Control. Release 2018, 270, 184–202. [Google Scholar] [CrossRef]
- Dimmitt, R.A.; Sellers, Z.M.; Sibley, E. XIV-Gastrointestinal system-70 Gastrointestinal tract development. In Avery’s Diseases of the Newborn; Elsevier Inc.: Amsterdam, The Netherlands, 2012; pp. 1032–1038. [Google Scholar]
- Treuting, P.M.; Dintzis, S.M.; Montine, K. Upper gastrointestinal tract. In Comparative Anatomy and Histology (Second Edition), A Mouse, Rat, and Human Atlas; Academic Press, Elsevier: London, UK, 2018; pp. 190–211. [Google Scholar]
- Cheng, H. Origin, differentiation and renewal of the four main epithelial cell types in the mouse small intestine. Am. J. Anat. 1974, 141, 481–502. [Google Scholar] [CrossRef]
- Lennernas, H. Human intestinal permeability. Int. J. Pharm. Sci. 1998, 87, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Rubin, D.C.; Langer, J.C. Anatomy and development-small intestine: Anatomy and structural anomalies. In Yamada’s Atlas of Gastroenterology; Podolsky, D.K., Camilleri, M., Shanahan, F., Fitz, J.G., Wang, T.C., Kalloo, A.N., Eds.; Wiley Blackwell: Oxford, UK, 2016; pp. 19–24. [Google Scholar]
- Dressman, J.B.; Berardi, R.R.; Dermentzoglou, L.C.; Russell, T.L.; Schmaltz, S.P.; Barett, J.L.; Jarvenpaa, K.M. Upper gastrointestinal (GI) pH in young, healthy men and women. Pharm. Res. 1990, 7, 756–761. [Google Scholar] [CrossRef] [PubMed]
- Rouge, N.; Buri, P.; Doelker, E. Drug absorption sites in the gastrointestinal tract and dosage forms for site-specific delivery. Int. J. Pharm. 1996, 136, 117–139. [Google Scholar] [CrossRef]
- Moroz, E.; Matoori, S.; Leroux, J. Oral delivery of macromolecular drugs: Where we are after almost 100 years of attempts. Adv. Drug Deliv. Rev. 2016, 101, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Bar-zeev, M.; Assaraf, Y.G.; Livney, Y.D. β-casein nanovehicles for oral delivery of chemotherapeutic drug combinations overcoming P-glycoprotein-mediated multidrug resistance in human gastric cancer cells. Oncotarget 2016, 7, 23322–23335. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Shu, Q.; Wang, L.; Wu, H.; Wang, A.Y.; Mao, H. Layer-by-layer assembled milk protein coated magnetic nanoparticle enabled oral drug delivery with high stability in stomach and enzyme-responsive release in small intestine. Biomaterials 2015, 39, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, G.A.; Opazo-Navarrete, M.; Meurs, M.; Minor, M.; Sala, G.; Van Boekel, M.; Stieger, M.; Janssen, A.E.M. Denaturation and in vitro gastric digestion of heat-treated quinoa protein isolates obtained at various extraction pH. Food Biophys. 2016, 11, 184–197. [Google Scholar] [CrossRef]
- Yamagata, T.; Morishita, M.; Kavimandan, N.J.; Nakamura, K. Characterization of insulin protection properties of complexation hydrogels in gastric and intestinal enzyme fluids. J. Control. Release 2006, 112, 343–349. [Google Scholar] [CrossRef]
- Cerchiara, T.; Abruzzo, A.; Parolin, C.; Vitali, B.; Bigucci, F.; Gallucci, M.C.; Nicoletta, F.P.; Luppi, B. Microparticles based on chitosan/carboxymethylcellulose polyelectrolyte complexes for colon delivery of vancomycin. Carbohydr. Polym. 2016, 143, 124–130. [Google Scholar] [CrossRef]
- O’Neill, M.J.; Bourre, L.; Melgar, S.; O’Driscoll, C.M. Intestinal delivery of non-viral gene therapeutics: Physiological barriers and preclinical models. Drug Discov. Today 2011, 16, 203–218. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Barrett, A.J. Families of serine peptidases. In Methods in Enzymology; Academic Press, Elsevier, Inc.: Amsterdam, The Netherlands, 1994; Volume 244, pp. 19–61. [Google Scholar]
- Davies, M.; Pieber, T.R.; Hartoft-Nielsen, M.L.; Hansen, O.K.H.; Jabbour, S.; Rosenstock, J. Effect of oral semaglutide compared with placebo and subcutaneous semaglutide on glycemic control in patients with type 2 diabetes a randomized clinical trial. J. Am. Med. Assoc. 2017, 318, 1460–1470. [Google Scholar] [CrossRef]
- Layer, P.; Go, V.L.W.; Dimagno, E.P. Fate of pancreatic enzymes aboral transit in humans during small intestinal aboral transit in humans. Am. J. Physiol. 1986, 251, 475–480. [Google Scholar]
- Fallingborg, J.; Christensen, L.A.; Ingeman-Nielsen, M.; Jacobsen, B.A.; Abildgaard, K.; Rasmussen, H.H. pH-profile and regional transit fimes of the normal gut measured by a radiotelemetry device. Aliment. Pharmacol. Ther. 1989, 3, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Lozoya-agullo, I.; Araújo, F.; González-álvarez, I.; Merino-sanjuán, M.; González-álvarez, M.; Bermejo, M.; Sarmento, B. PLGA nanoparticles are effective to control the colonic release and absorption on ibuprofen. Eur. J. Pharm. Sci. 2018, 115, 119–125. [Google Scholar] [CrossRef]
- Valon, L.; Levayer, R. Dying under pressure: Cellular characterisation and in vivo functions of cell death induced by compaction. Biol. Cell 2019, 111, 1–16. [Google Scholar] [CrossRef] [PubMed]
- De France, K.J.; Chan, K.J.W.; Cranston, E.D.; Hoare, T. Enhanced mechanical properties in cellulose nanocrystal−poly(oligoethylene glycol methacrylate) injectable nanocomposite hydrogels through control of physical and chemical cross-linking. Biomacromolecules 2016, 17, 649–660. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhao, J.; Xu, F.; Sun, R. Revealing strong nanocomposite hydrogels reinforced by cellulose nanocrystals: Insight into morphologies and interactions. Appl. Mater. Interfaces 2013, 5, 12960–12967. [Google Scholar] [CrossRef] [PubMed]
- Mert, O.; Lai, S.K.; Ensign, L.; Yang, M.; Wang, Y.; Wood, J.; Hanes, J. A poly(ethylene glycol)-based surfactant for formulation of drug-loaded mucus penetrating particles. J. Control. Release 2012, 157, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, T.; Wei, S.; Zhou, C.; Lan, Y.; Cao, A. Mucus adhesion- and penetration-enhanced liposomes for paclitaxel oral delivery. Int. J. Pharm. 2018, 537, 245–256. [Google Scholar] [CrossRef]
- Shan, W.; Zhu, X.; Liu, M.; Li, L.; Zhong, J.; Sun, W.; Zhang, Z.; Huang, Y. Overcoming the diffusion barrier of mucus and absorption barrier of epithelium by self-assembled nanoparticles for oral delivery of insulin. ACS Nano 2015, 9, 2345–2356. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhang, J.; Zhu, X.; Shan, W.; Li, L.; Zhong, J.; Zhang, Z.; Huang, Y. Efficient mucus permeation and tight junction opening by dissociable “mucus-inert” agent coated trimethyl chitosan nanoparticles for oral insulin delivery. J. Control. Release 2016, 222, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Leal, J.; Dong, T.; Taylor, A.; Siegrist, E.; Gao, F.; Smyth, H.D.C. Mucus-penetrating phage-displayed peptides for improved transport across a mucus-like model. Int. J. Pharm. 2018, 553, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Cheng, H.; Dong, W.; Zhang, M.; Liu, Q.; Wang, X.; Guan, J. Design and intestinal mucus penetration mechanism of core-shell nanocomplex. J. Control. Release 2018, 272, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Navarro, L.A.; French, D.L.; Zauscher, S. Advances in mucin mimic synthesis and applications in surface science. Curr. Opin. Colloid Interface Sci. 2018, 38, 122–134. [Google Scholar] [CrossRef]
- Kufe, D.W. Mucins in cancer: Function, prognosis and therapy Donald. Nat. Rev. Cancer 2009, 9, 874–885. [Google Scholar] [CrossRef] [PubMed]
- Atuma, C.; Strugala, V.; Allen, A.; Holm, L. The adherent gastrointestinal mucus gel layer: Thickness and physical state in vivo. Am. J. Physiol. Liver Physiol. 2001, 280, 922–929. [Google Scholar] [CrossRef]
- Chassaing, B.; Gewirtz, A.T. Identification of inner mucus-associated bacteria by laser capture microdissection. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Bansil, R.; Turner, B.S. The biology of mucus: Composition, synthesis and organization. Adv. Drug Deliv. Rev. 2018, 124, 3–15. [Google Scholar] [CrossRef]
- Boegh, M.; García-díaz, M.; Müllertz, A.; Nielsen, H.M. Steric and interactive barrier properties of intestinal mucus elucidated by particle diffusion and peptide permeation. Eur. J. Pharm. Biopharm. 2015, 95, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.C.; Johansson, M.E. V The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Gut Microbes 2010, 1, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.E.V.; Larsson, J.M.H.; Hansson, G.C. The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host–microbial interactions. PNAS 2011, 108, 4659–4665. [Google Scholar] [CrossRef]
- Bajka, B.H.; Rigby, N.M.; Cross, K.L.; Macierzanka, A.; Mackie, A.R. The influence of small intestinal mucus structure on particle transport ex vivo. Colloids Surf. B Biointerfaces 2015, 135, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, D.; Le, C.; Zhu, C.; Gan, Y.; Hovgaard, L.; Yang, M. Novel mucus-penetrating liposomes as a potential oral drug delivery system: Preparation, in vitro characterization, and enhanced cellular uptake. Int. J. Nanomed. 2011, 6, 3151–3162. [Google Scholar]
- Cu, Y.; Saltzmanr, W.M. Controlled surface modification with poly(ethylene)glycol enhances diffusion of PLGA nanoparticles in human cervical mucus. Mol. Pharm. 2009, 6, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Muller, C.; Leithner, K.; Hauptstein, S.; Hintzen, F.; Salvenmoser, W.; Bernkop-Schnurch, A. Preparation and characterization of mucus-penetrating papain/poly(acrylic acid) nanoparticles for oral drug delivery applications. J. Nanopart. Res. 2013, 15, 1353–1366. [Google Scholar] [CrossRef]
- DeSousa, I.P.; Cattoz, B.; Wilcox, M.D.; Griffiths, P.C.; Dalgliesh, R.; Rogers, S.; Bernkop-schnürch, A. Nanoparticles decorated with proteolytic enzymes, a promising strategy to overcome the mucus barrier. Eur. J. Pharm. Biopharm. 2015, 97, 257–264. [Google Scholar]
- Moreno, J.A.S.; Mendes, A.C.; Stephansen, K.; Engwer, C. Development of electrosprayed mucoadhesive chitosan microparticles. Carbohydr. Polym. 2018, 190, 240–247. [Google Scholar] [CrossRef]
- Park, C.G.; Huh, B.K.; Kim, S.; Lee, S.H.; Hong, H.R.; Choy, Y.B. Nanostructured mucoadhesive microparticles to enhance oral drug bioavailability. J. Ind. Eng. Chem. 2017, 54, 262–269. [Google Scholar] [CrossRef]
- Krauland, A.H.; Guggi, D.; Bernkop-schnurch, A. Thiolated chitosan microparticles: A vehicle for nasal peptide drug delivery. Int. J. Pharm. 2006, 307, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Romero, G.B.; Keck, C.M.; Müller, R.H.; Bou-chacra, N.A. Development of cationic nanocrystals for ocular delivery. Eur. J. Pharm. Biopharm. 2016, 107, 215–222. [Google Scholar] [CrossRef]
- De DeLima, J.A.; Paines, T.C.; Motta, M.H.; Weber, W.B.; Santos, S.S.; Cruz, L.; Silva, C.D.B. Novel Pemulen/Pullulan blended hydrogel containing clotrimazole-loaded cationic nanocapsules: Evaluation of mucoadhesion and vaginal permeation. Mater. Sci. Eng. C 2017, 79, 886–893. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Kim, K.; Hyun, J.; Lee, H. Chitosan-catechol: A polymer with long-lasting mucoadhesive properties. Biomaterials 2015, 52, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Ertl, B.; Heigl, F.; Wirth, M.; Gabor, F. Lectin-mediated bioadhesion: Preparation, stability and Caco-2 binding of wheat germ agglutinin-functionalized poly(D,L-lactic-co-glycolic acid)-microspheres. J. Drug Target. 2000, 8, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Anirudhan, T.S.; Parvathy, J. Novel thiolated chitosan-polyethyleneglycol blend/Montmorillonite composite formulations for the oral delivery of insulin. Bioact. Carbohydr. Diet. Fibre 2018, 16, 22–29. [Google Scholar] [CrossRef]
- Bernkop-schnurch, A.; Hornof, M.; Guggi, D. Thiolated chitosans. Eur. J. Pharm. Biopharm. 2004, 57, 9–17. [Google Scholar] [CrossRef]
- Deutel, B.; Laf, F.; Palmberger, T.; Saxer, A.; Thaler, M.; Bernkop-schnürch, A. In vitro characterization of insulin containing thiomeric microparticles as nasal drug delivery system. Eur. J. Pharm. Sci. 2016, 81, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Sajeesh, S.; Vauthier, C.; Gueutin, C.; Ponchel, G.; Sharma, C.P. Thiol functionalized polymethacrylic acid-based hydrogel microparticles for oral insulin delivery. Acta Biomater. 2010, 6, 3072–3080. [Google Scholar] [CrossRef] [PubMed]
- Farris, E.; Heck, K.; Lampe, A.T.; Brown, D.M.; Ramer-tait, A.E.; Pannier, A.K. Oral non-viral gene delivery for applications in DNA vaccination and gene therapy. Curr. Opin. Biomed. Eng. 2018, 7, 51–57. [Google Scholar] [CrossRef]
- Batista, P.; Castro, P.M.; Raquel, A.; Sarmento, B. Recent insights in the use of nanocarriers for the oral delivery of bioactive proteins and peptides. Peptides 2018, 101, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, X.; Meng, L.; Zhang, Y.; Ai, R.; Qi, N.; He, H.; Xu, H.; Tang, X. Thiolated Eudragit nanoparticles for oral insulin delivery: Preparation, characterization and in vivo evaluation. Int. J. Pharm. 2012, 436, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Cone, R.A. Barrier properties of mucus. Adv. Drug Deliv. Rev. 2009, 61, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Huckaby, J.T.; Lai, S.K. PEGylation for enhancing nanoparticle diffusion in mucus. Adv. Drug Deliv. Rev. 2018, 124, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.; Kamm, W.; Breitenbach, A.; Kaiserling, E.; Xiao, J.X.; Kissel, T. Biodegradable nanoparticles for oral delivery of peptides: Is there a role for polymers to affect mucosal uptake? Eur. J. Pharm. Biopharm. 2000, 50, 147–160. [Google Scholar] [CrossRef]
- Lai, S.K.; Wang, Y.Y.; Hanes, J. Mucus-penetrating nanoparticles for drug and gene delivery to mucosal tissues. Adv. Drug Deliv. Rev. 2009, 61, 158–171. [Google Scholar] [CrossRef] [PubMed]
- Zhaeentana, S.; Amjadib, F.S.; Zandieb, Z.; Joghataei, M.T.; Bakhtiyari, M.; Aflatoonian, R. The effects of hydrocortisone on tight junction genes in an in vitro model of the human fallopian epithelial cells. Eur. J. Obstet. Gynecol. Reprod. Biol. 2018, 229, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Bein, A.; Eventov-friedman, S.; Arbell, D.; Schwartz, B. Intestinal tight junctions are severely altered in NEC preterm neonates. Pediatr. Neonatol. 2018, 59, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Gamboa, J.M.; Leong, K.W. In vitro and in vivo models for the study of oral delivery of nanoparticles. Adv. Drug Deliv. Rev. 2013, 65, 800–810. [Google Scholar] [CrossRef] [PubMed]
- Linnankoski, J.; Makela, J.; Palmgren, J.; Mauriala, T.; Vedin, C.; Ungell, A.-L.; Artursson, P.; Urtti, A.; Yliperttula, M. Paracellular porosity and pore size of the human intestinal epithelium in tissue and cell culture models. J. Pharm. Sci. 2010, 99, 2166–2175. [Google Scholar] [CrossRef]
- Salama, N.N.; Eddington, N.D.; Fasano, A. Tight junction modulation and its relationship to drug delivery. Adv. Drug Deliv. Rev. 2006, 58, 15–28. [Google Scholar] [CrossRef]
- Kim, J.; Yoon, I.; Cho, H.; Kim, D.; Choi, Y.; Kim, D. Emulsion-based colloidal nanosystems for oral delivery of doxorubicin: Improved intestinal paracellular absorption and alleviated cardiotoxicity. Int. J. Pharm. 2014, 464, 117–126. [Google Scholar] [CrossRef]
- Taverner, A.; Dondi, R.; Almansour, K.; Laurent, F.; Owens, S.; Eggleston, I.M.; Fotaki, N.; Mrsny, R.J. Enhanced paracellular transport of insulin can be achieved via transient induction of myosin light chain phosphorylation. J. Control. Release 2015, 210, 189–197. [Google Scholar] [CrossRef]
- Almansour, K.; Taverner, A.; Eggleston, I.M.; Mrsny, R.J. Mechanistic studies of a cell-permeant peptide designed to enhance myosin light chain phosphorylation in polarized intestinal epithelia. J. Control. Release 2018, 279, 208–219. [Google Scholar] [CrossRef]
- Banerjee, A.; Mitragotri, S. Intestinal patch systems for oral drug delivery. Curr. Opin. Pharmacol. 2017, 36, 58–65. [Google Scholar] [CrossRef]
- Banerjee, A.; Lee, J.; Mitragotri, S. Intestinal mucoadhesive devices for oral delivery of insulin. Bioeng. Transl. Med. 2016, 1, 338–346. [Google Scholar] [CrossRef]
- Shen, Z.; Mitragotri, S. Intestinal patches for oral drug delivery. Pharm. Res. 2002, 19, 391–395. [Google Scholar] [CrossRef]
- Toorisaka, E.; Hashida, M.; Kamiya, N.; Ono, H. An enteric-coated dry emulsion formulation for oral insulin delivery. J. Control. Release 2005, 107, 91–96. [Google Scholar] [CrossRef]
- Toorisaka, E.; Watanabe, K.; Ono, H.; Hirata, M.; Kamiya, N. Intestinal patches with an immobilized solid-in-oil formulation for oral protein delivery. Acta Biomater. 2012, 8, 653–658. [Google Scholar] [CrossRef]
- Lee, J.W.; Prausnitz, M.R. Drug delivery using microneedle patches: Not just for skin. Expert Opin. Drug Deliv. 2018, 15, 541–543. [Google Scholar] [CrossRef]
- Ma, Y.; Tao, W.; Krebs, S.J.; Sutton, W.F.; Haigwood, N.L.; Gill, H.S. Vaccine delivery to the oral cavity using coated microneedles induces systemic and mucosal immunity. Pharm. Res. 2014, 31, 2393–2403. [Google Scholar] [CrossRef]
- Traverso, G.; Schoellhammer, C.M.; Schroeder, A.; Maa, R.; Lauwers, G.Y.; Polat, B.E.; Anderson, D.G.; Blankschtein, D.; Langer, R. Microneedles for Drug Delivery via the Gastrointestinal Tract. J. Pharm. Sci. 2015, 104, 362–367. [Google Scholar] [CrossRef]
- Dabholkar, R.D.; Sawant, R.M.; Mongayt, D.A.; Devarajan, P.V.; Torchilin, V.P. Polyethylene glycol–phosphatidylethanolamine conjugate (PEG–PE)-based mixed micelles: Some properties, loading with paclitaxel, and modulation of P-glycoprotein-mediated efflux. Int. J. Pharm. 2006, 315, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Cui, Z.; Yu, P.; Guo, C.; Feng, B.; Jiang, T. pH- and NIR light-responsive micelles with hyperthermia-triggered tumor penetration and cytoplasm drug release to reverse doxorubicin resistance in breast cancer. Adv. Funct. Mater. 2015, 25, 2489–2500. [Google Scholar] [CrossRef]
- Suzuki, H.; Bae, Y.H. Evaluation of drug penetration with cationic micelles and their penetration mechanism using an in vitro tumor model. Biomaterials 2016, 98, 120–130. [Google Scholar] [CrossRef]
- Sosnik, A.; Raskin, M.M. Polymeric micelles in mucosal drug delivery: Challenges towards clinical translation. Biotechnol. Adv. 2015, 33, 1380–1392. [Google Scholar] [CrossRef]
- Torchilin, V.P. Fluorescence microscopy to follow the targeting of liposomes and micelles to cells and their intracellular fate. Adv. Drug Deliv. Rev. 2005, 57, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Byrne, R.S.; Deasy, P.B. Use of commercial porous ceramic particles for sustained drug delivery. Int. J. Pharm. 2002, 246, 61–73. [Google Scholar] [CrossRef]
- Hoffman, A.S. Hydrogels for biomedical applications. Adv. Drug Deliv. Rev. 2012, 64, 18–23. [Google Scholar] [CrossRef]
- Li, J.; Mooney, D.J. Designing hydrogels for controlled drug delivery. Nat. Rev. Mater. 2016, 1, 1–17. [Google Scholar] [CrossRef]
- Chai, Q.; Jiao, Y.; Yu, X. Hydrogels for biomedical applications: Their characteristics and the mechanisms behind them. Gels 2017, 3, 6. [Google Scholar] [CrossRef]
- Caló, E.; Khutoryanskiy, V. Biomedical applications of hydrogels: A review of patents and commercial products. Eur. Polym. J. 2015, 65, 252–267. [Google Scholar] [CrossRef]
- Klouda, L. Thermoresponsive hydrogels in biomedical applications A seven-year update. Eur. J. Pharm. Biopharm. 2015, 97, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Torres-lugo, M.; Peppas, N.A. Molecular design and in vitro studies of novel pH-sensitive hydrogels for the oral delivery of calcitonin. Macromolecules 1999, 32, 6646–6651. [Google Scholar] [CrossRef]
- Simpson, M.J.; Corbett, B.; Arezina, A.; Hoare, T. Narrowly dispersed, degradable, and scalable poly(oligoethylene glycol methacrylate)-based nanogels via thermal self-assembly. Ind. Eng. Chem. Res. 2018, 57, 7495–7506. [Google Scholar] [CrossRef]
- Choi, J.; Moquin, A.; Bomal, E.; Na, L.; Maysinger, D.; Kakkar, A. Telodendrimers for physical encapsulation and covalent linking of individual or combined therapeutics. Mol. Pharm. 2017, 14, 2607–2615. [Google Scholar] [CrossRef]
- DeFrance, K.J.; Xu, F.; Hoare, T. Structured macroporous hydrogels: Progress, challenges, and opportunities. Adv. Healthc. Mater. 2018, 7, 1–17. [Google Scholar]
- Annabi, N.; Nichol, J.W.; Zhong, X.; Ji, C. Controlling the porosity and microarchitecture of hydrogels for tissue engineering. Tissue Eng. Part B 2010, 16, 371–385. [Google Scholar] [CrossRef] [PubMed]
- Kim, U.; Park, J.; Li, C.; Jin, H.; Valluzzi, R.; Kaplan, D.L. Structure and properties of silk hydrogels. Biomacromolecules 2004, 5, 786–792. [Google Scholar] [CrossRef]
- Yokoyama, E.; Masada, I.; Shimamura, K.; Ikawa, T.; Monobe, K. Morphology and structure of highly elastic poly(vinyl alcohol) hydrogel prepared by repeated freezing-and-melting. Colloid Polym. Sci. 1986, 601, 595–601. [Google Scholar] [CrossRef]
- Hermansson, A.M.; Buchheim, W. Characterization of protein gels by scanning and transmission electron microscopy. J. Colloid Interface Sci. 1981, 81, 510–530. [Google Scholar] [CrossRef]
- Hyuk Im, S.; Jeong, U.; Xia, Y. Polymer hollow particles with controllable holes in their surfaces. Nat. Mater. 2005, 4, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Staruch, R.M.T.; Glass, G.E.; Rickard, R.; Hettiaratchy, S.; Butler, P.E.M. Injectable pore-forming hydrogel scaffolds for complex wound tissue engineering: Designing and controlling their porosity and mechanical properties. Tissue Eng. Part B 2017, 23, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Loh, Q.L.; Choong, C. Three-dimensional scaffolds for tissue engineering applications: Role of porosity and pore size. Tissue Eng. Part B 2013, 19, 485–502. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, G.; Zhang, X.; Wang, L.; Du, Y.; Jian, T.; Xu, F. Engineering cell alignment in vitro. Biotechnol. Adv. 2014, 32, 347–365. [Google Scholar] [CrossRef] [PubMed]
- Vlierberghe, S.; Cnudde, V.; Dubruel, P.; Masschaele, B.; Cosijns, A.; De Paepe, I.; Jacobs, P.J.S.; Hoorebeke, L.; Remon, J.P.; Schacht, E. Porous gelatin hydrogels: 1. Cryogenic formation and structure analysis. Biomacromolecules 2007, 8, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.; Li, Y. Functional assessment of cross-linked porous gelatin hydrogels for bioengineered cell sheet carriers. Biomacromolecules 2010, 11, 1387–1397. [Google Scholar] [CrossRef]
- Lewus, R.K.; Carta, G. Protein transport in constrained anionic hydrogels: Diffusion and boundary-layer mass transfer. Ind. Eng. Chem. Res. 2001, 40, 1548–1558. [Google Scholar] [CrossRef]
- Pundir, S.; Badola, A.; Sharma, D. Sustained release matrix technology and recent advance in matrix drug delivery system: A review. Int. J. Drug Res. Technol. 2013, 3, 12–20. [Google Scholar]
- Thedrattanawong, C.; Manaspon, C.; Nasongkla, N. Controlling the burst release of doxorubicin from polymeric depots via adjusting hydrophobic/hydrophilic properties. J. Drug Deliv. Sci. Technol. 2018, 46, 446–451. [Google Scholar] [CrossRef]
- Li, R.; Deng, L.; Cai, Z.; Zhang, S.; Wang, K.; Li, L.; Ding, S.; Zhou, C. Liposomes coated with thiolated chitosan as drug carriers of curcumin. Mater. Sci. Eng. C 2017, 80, 156–164. [Google Scholar] [CrossRef]
- Martins, A.L.L.; Oliveira, A.C.; Nascimento, C.M.O.L.; Silva, D.; Gaeti, M.P.N.; Lima, E.M.; Taveira, S.F.; Fernandes, K.F.; Marreto, R.N. Mucoadhesive properties of thiolated pectin-based pellets prepared by extrusion-spheronization technique. J. Pharm. Sci. 2017, 106, 1363–1370. [Google Scholar] [CrossRef]
- Lichtenberger, L.M. The hydrophobic barrier properties of gastrointestinal mucus. Annu. Rev. Physiol. 1995, 57, 565–583. [Google Scholar] [CrossRef] [PubMed]
- Traverso, G.; Langer, R. Special delivery for the gut. Nature 2015, 519, S19. [Google Scholar] [CrossRef] [PubMed]
- Lam, P.L.; Gambari, R. Advanced progress of microencapsulation technologies: In vivo and in vitro models for studying oral and transdermal drug deliveries. J. Control. Release 2014, 178, 25–45. [Google Scholar] [CrossRef] [PubMed]
- Salessiotis, N. Measurement of the diameter of the Pylorus in man: Part I. Experimental project for clinical application. Am. J. Surg. 1972, 124, 331–333. [Google Scholar] [CrossRef]
- Sultan, M.; Norton, R.A. Esophageal diameter and the treatment of achalasia. Am. J. Dig. Dis. 1969, 14, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Bellinger, A.M.; Glettig, D.L.; Barman, R.; Lee, Y.A.; Zhu, J.; Cleveland, C.; Montgomery, V.A.; Gu, L.; Nash, L.D.; et al. A pH-responsive supramolecular polymer gel as an enteric elastomer for use in gastric devices. Nat. Mater. 2015, 14, 1065–1071. [Google Scholar] [CrossRef]
- Soudry-kochavi, L.; Naraykin, N.; DiPaola, R.; Gugliandolo, E.; Peritore, A.; Cuzzocrea, S.; Ziv, E.; Nassar, T.; Benita, S. Pharmacodynamical effects of orally administered exenatide nanoparticles embedded in gastro-resistant microparticles. Eur. J. Pharm. Biopharm. 2018, 133, 214–223. [Google Scholar] [CrossRef]
- Agüero, L.; Zaldivar-silva, D.; Pena, L.; Dias, M.L. Alginate microparticles as oral colon drug delivery device: A review. Carbohydr. Polym. 2017, 168, 32–43. [Google Scholar] [CrossRef]
- Chen, Q.; Gou, S.; Huang, Y.; Zhou, X.; Li, Q. Facile fabrication of bowl-shaped microparticles for oral curcumin delivery to ulcerative colitis tissue. Colloids Surf. B Biointerfaces 2018, 169, 92–98. [Google Scholar] [CrossRef]
- Nadal, J.M.; Gomes, M.L.S.; Borsato, D.M.; Almeida, M.A.; Barboza, F.M.; Zawadzki, S.F.; Kanunfre, C.C.; Farago, P.V.; Zanin, S.M.W. Spray-dried Eudragit® L100 microparticles containing ferulic acid: Formulation, in vitro cytoprotection and in vivo anti-platelet effect. Mater. Sci. Eng. C 2016, 64, 318–328. [Google Scholar] [CrossRef]
- Ratzinger, G.; Agrawal, P.; Korner, W.; Lonkai, J.; Sanders, H.M.H.F.; Terreno, E.; Wirth, M.; Strijkers, G.J.; Nicolay, K.; Gabor, F. Surface modification of PLGA nanospheres with Gd-DTPA and Gd-DOTA for high-relaxivity MRI contrast agents. Biomaterials 2010, 31, 8716–8723. [Google Scholar] [CrossRef] [PubMed]
- Koch, B.; Rubino, I.; Quan, F.-S.; Yoo, B.; Choi, H.-J. Microfabrication for drug delivery. Materials 2016, 9, 646. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, H.; Takasu, M. Fusion pathways of vesicles: A Brownian dynamics simulation. J. Chem. Phys. 2001, 115, 9547–9551. [Google Scholar] [CrossRef]
- Pekarek, K.J.; Jacob, J.S.; Mathiowitz, E. Double-walled polymer microspheres for controlled drug release. Nature 1994, 367, 258–260. [Google Scholar] [CrossRef]
- Zolnik, B.S.; Burgess, D.J. Effect of acidic pH on PLGA microsphere degradation and release. J. Control. Release 2007, 122, 338–344. [Google Scholar] [CrossRef]
- Wong, M.S.; Cha, J.N.; Choi, K.S.; Deming, T.J.; Stucky, G.D. Assembly of nanoparticles into hollow spheres using block copolypeptides. Nano Lett. 2002, 2, 583–587. [Google Scholar] [CrossRef]
- Du, J.; O’Reilly, R.K. Advances and challenges in smart and functional polymer vesicles. Soft Matter 2009, 5, 3544–3561. [Google Scholar] [CrossRef]
- Yokoyama, M.; Inoue, S.; Kataoka, K.; Yui, N.; Okano, T.; Sakurai, Y. Molecular design for missile drug: Synthesis of adriamycin conjugated with immunoglobulin G using poly(ethylene glycol)-block-poly(aspartic acid) as intermediate carrier. Die Makromol. Chemie 1989, 190, 2041–2054. [Google Scholar] [CrossRef]
- Batycky, R.P.; Hanes, J.; Langer, R.; Edwards, D.A. A theoretical model of erosion and macromolecular drug release from biodegrading microspheres. J. Pharm. Sci. 1997, 86, 1464–1477. [Google Scholar] [CrossRef]
- Steijn, V.; Korczyk, P.M.; Derzsi, L.; Abate, A.R.; Weitz, D.A.; Garstecki, P. Block-and-break generation of microdroplets with fixed volume. Biomicrofluidics 2013, 7, 24108. [Google Scholar] [CrossRef] [PubMed]
- Abbaspourrad, A.; Carroll, N.J.; Kim, S.; Weitz, D.A. Polymer microcapsules with programmable active release. J. Am. Chem. Soc. 2013, 135, 7744–7750. [Google Scholar] [CrossRef] [PubMed]
- Ostafe, R.; Prodanovic, R.; Ung, W.L.; Weitz, D.A.; Fischer, R. A high-throughput cellulase screening system based on droplet microfluidics. Biomicrofluidics 2014, 8, 041102. [Google Scholar] [CrossRef] [PubMed]
- Bitar, C.; Markwick, K.E.; Hoesli, C.A. Encapsulation of Pancreatic Islet Cells for Type 1 Diabetes Treatment. In Proceedings of the XXV International Conference on Bioencapsulation, Nantes, France, 3–6 July 2017; Available online: http://bioencapsulation.net/221_newsletters/Bioencap_innov_2017_11/Bioencap_innov_2017_11.pdf (accessed on 18 March 2019).
- Deng, N.-N.; Wang, W.; Ju, X.-J.; Xie, R.; Weitz, D.A.; Chu, L.-Y. Reply to the ‘Comment on “Wetting-induced formation of controllable monodisperse multiple emulsions in microfluidics”’ by J. Guzowski and P.; Garstecki, Lab Chip, 2014, 14, DOI:10.1039/ C3LC51229K. Lab Chip 2014, 14, 1479–1480. [Google Scholar] [CrossRef] [PubMed]
- Massenburg, S.S.; Amstad, E.; Weitz, D.A. Clogging in parallelized tapered microfluidic channels. Microfluid. Nanofluidics 2016, 20, 1–5. [Google Scholar] [CrossRef]
- Wyss, H.M.; Blair, D.L.; Morris, J.F.; Stone, H.A.; Weitz, D.A. Mechanism for clogging of microchannels. Phys. Rev. E 2006, 74, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Montemagno, C.; Choi, H.-j. Smart microparticles with a pH-responsive macropore for targeted oral drug delivery. Sci. Rep. 2017, 7, 061402. [Google Scholar] [CrossRef] [PubMed]
- Caruso, F. Nanoengineering of particle surfaces. Adv. Mater. 2001, 13, 11–22. [Google Scholar] [CrossRef]
- Cai, Y.; Chen, Y.; Hong, X.; Liu, Z.; Yuan, W. Porous microsphere and its applications. Int. J. Nanomed. 2013, 8, 1111–1120. [Google Scholar]
- Kim, K.K.; Pack, D.W. Volume I: Biological and biomedical nanotechnology-Microspheres for drug delivery. In BioMEMS and Biomedical Nanotechnology; Ferrari, M., Lee, A., Lee, J., Eds.; Springer: Boston, MA, USA, 2006; pp. 19–50. [Google Scholar]
- Homayun, B.; Sun, C.; Kumar, A.; Montemagno, C.; Choi, H.-J. Facile fabrication of microparticles with pH-responsive macropores for small intestine targeted drug formulation. Eur. J. Pharm. Biopharm. 2018, 128, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Homayun, B.; Kumar, A.; Nascimento, P.T.H.; Choi, H.-J. Macropored microparticles with a core–shell architecture for oral delivery of biopharmaceuticals. Arch. Pharm. Res. 2018, 41, 848–860. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.M.; Zhang, M.; Wei, D.; Stueber, D.; Taratula, O.; Minko, T.; He, H. Co-delivery of doxorubicin and Bcl-2 siRNA by mesoporous silica nanoparticles enhances the efficacy of chemotherapy in multidrug-resistant cancer cells. Small 2009, 5, 2673–2677. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, Q.; Han, N.; Bai, L.; Li, J.; Liu, J.; Che, E.; Hu, L.; Zhang, Q.; Jiang, T.; et al. Mesoporous silica nanoparticles in drug delivery and biomedical applications. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 313–327. [Google Scholar] [CrossRef]
- Lai, C.Y.; Trewyn, B.G.; Jeftinija, D.M.; Jeftinija, K.; Xu, S.; Jeftinija, S.; Lin, V.S.Y. A mesoporous silica nanosphere-based carrier system with chemically removable CdS nanoparticle caps for stimuli-responsive controlled release of neurotransmitters and drug molecules. J. Am. Chem. Soc. 2003, 125, 4451–4459. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, H.; Ma, M.; Chen, F.; Guo, L.; Zhang, L.; Shi, J. Double mesoporous silica shelled spherical/ellipsoidal nanostructures: Synthesis and hydrophilic/hydrophobic anticancer drug delivery. J. Mater. Chem. 2011, 21, 5290–5298. [Google Scholar] [CrossRef]
- Zhao, Y.; Trewyn, B.G.; Slowing, I.I.; Lin, S.-Y. Mesoporous silica nanoparticle-based double drug delivery system for glucose responsive controlled release of insulin and cyclic AMP. J. Am. Chem. Soc. 2009, 131, 8398–8400. [Google Scholar] [CrossRef] [PubMed]
- Teo, P.Y.; Cheng, W.; Hedrick, J.L.; Yang, Y.Y. Co-delivery of drugs and plasmid DNA for cancer therapy. Adv. Drug Deliv. Rev. 2016, 98, 41–63. [Google Scholar] [CrossRef]
- Kang, S.H.; Cho, H.; Shim, G.; Lee, S.; Kim, S.; Choi, H.; Kim, C.; Oh, Y.-K. Cationic liposomal co-delivery of small interfering RNA and a MEK inhibitor for enhanced anticancer efficacy. Pharm. Res. 2011, 28, 3069–3078. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Zeng, Q.; Xu, C.; Shi, S.; Zhang, Z.; Sun, X. Enhanced antitumor response mediated by the codelivery of paclitaxel and adenoviral vector expressing IL-12. Mol. Pharm. 2013, 10, 1804–1814. [Google Scholar] [CrossRef]
- Mamaeva, V.; Sahlgren, C.; Lindén, M. Mesoporous silica nanoparticles in medicine-Recent advances. Adv. Drug Deliv. Rev. 2013, 65, 689–702. [Google Scholar] [CrossRef]
- Santos, A.C.; Ferreira, C.; Veiga, F.; Ribeiro, A.J.; Panchal, A.; Lvov, Y.; Agarwal, A. Halloysite clay nanotubes for life sciences applications: From drug encapsulation to bioscaffold. Adv. Colloid Interface Sci. 2018, 257, 58–70. [Google Scholar] [CrossRef] [PubMed]
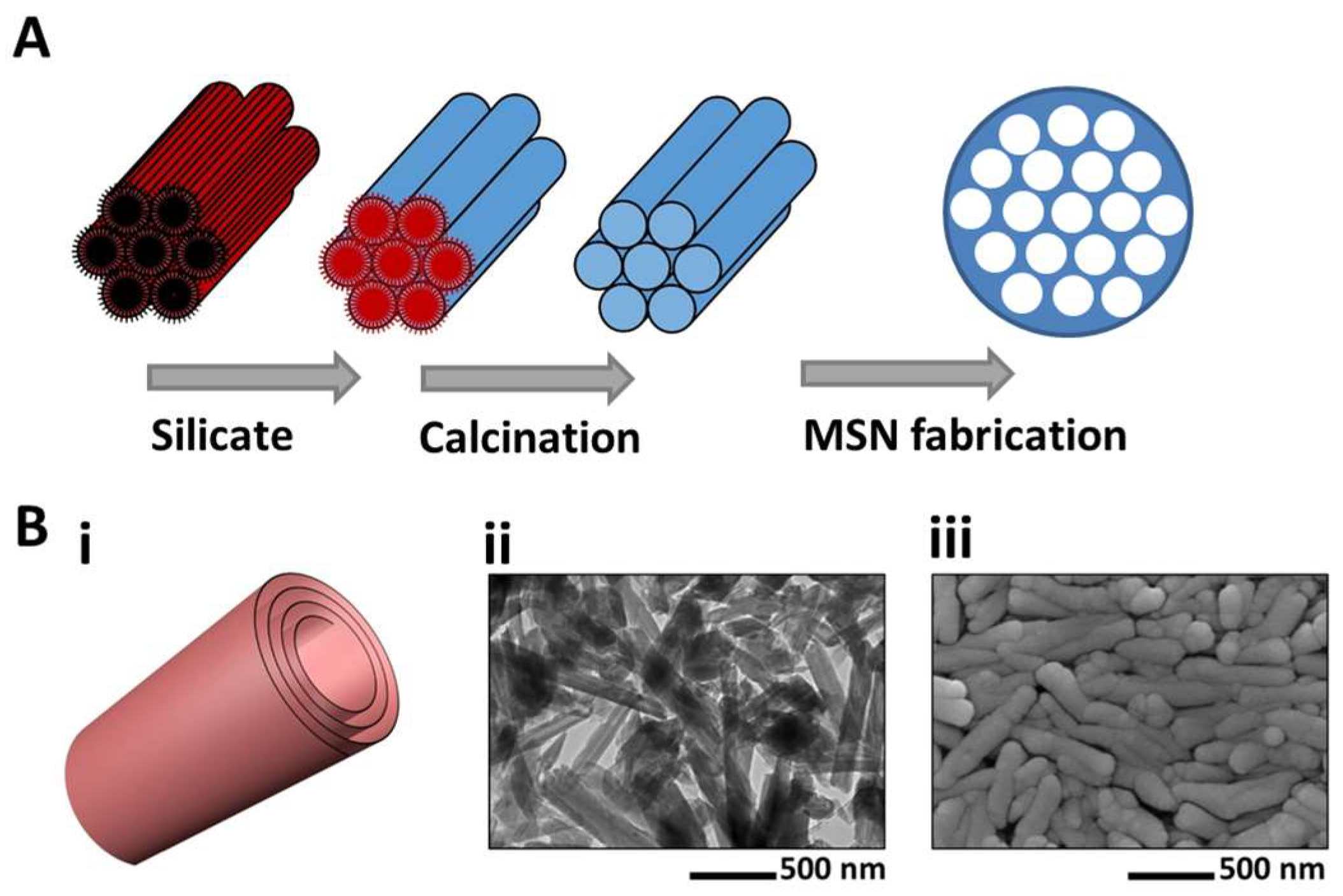
- Slowing, I.I.; Vivero-escoto, J.L.; Wu, C.; Lin, V.S. Mesoporous silica nanoparticles as controlled release drug delivery and gene transfection carriers. Adv. Drug Deliv. Rev. 2008, 60, 1278–1288. [Google Scholar] [CrossRef] [PubMed]
- Kresge, C.T.; Leonowicz, M.E.; Roth, W.J.; Vartuli, J.C.; Beck, J.S. Ordered mesoporous molecular sieves synthesized by a liquid crystal template mechanism. Nature 1992, 359, 710–712. [Google Scholar] [CrossRef]
- Inagaki, S.; Fukushima, Y.; Kuroda, K. Synthesis of highly ordered mesoporous materials from a layered polysilicate. J. Chem. Soc. Chem. Commun. 1993, 8, 680–682. [Google Scholar] [CrossRef]
- Gaaz, T.S.; Sulong, A.B.; Akhtar, M.N.; Kadhum, A.A.H.; Mohamad, A.B.; Al-Amiery, A.A. Properties and applications of polyvinyl alcohol, halloysite nanotubes and their nanocomposites. Molecules 2015, 20, 22833–22847. [Google Scholar] [CrossRef] [PubMed]
- Lvov, Y.M.; Shchukin, D.G.; Mohwald, H.; Price, R.R. Halloysite clay nanotubes for controlled release of protective agents. ACS Nano 2008, 2, 814–820. [Google Scholar] [CrossRef]
- Lvov, Y.M.; Devilliers, M.M.; Fakhrullin, R.F. The application of halloysite tubule nanoclay in drug delivery. Expert Opin. Drug Deliv. 2016, 13, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Fizir, M.; Dramou, P.; Zhang, K.; Sun, C.; Pham-Huy, C.; He, H. Polymer grafted-magnetic halloysite nanotube for controlled and sustained release of cationic drug. J. Colloid Interface Sci. 2017, 505, 476–488. [Google Scholar] [CrossRef]
- Lvov, Y.; Wang, W.; Zhang, L.; Fakhrullin, R. Halloysite clay nanotubes for loading and sustained release of functional compounds. Adv. Mater. 2016, 28, 1227–1250. [Google Scholar] [CrossRef]
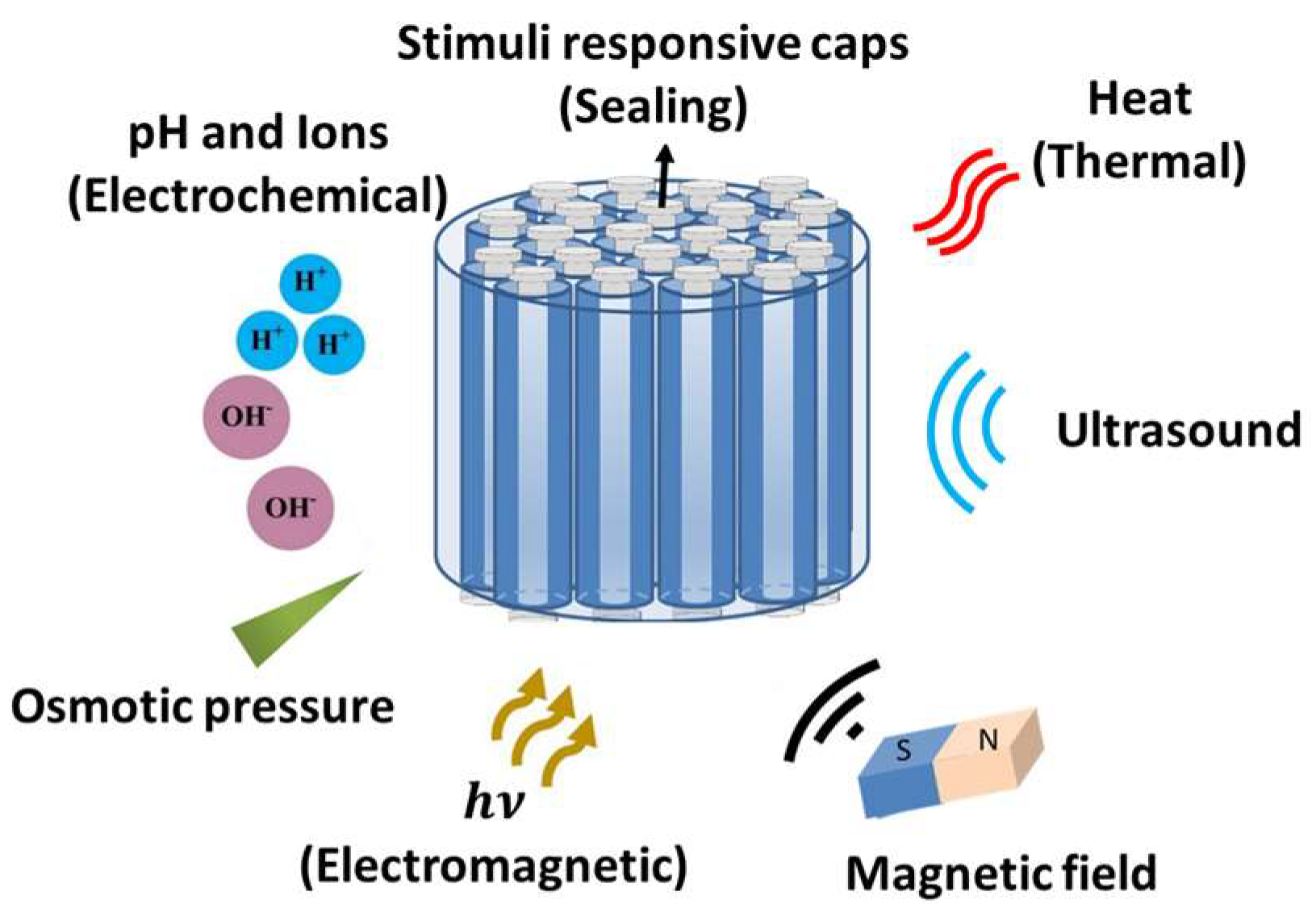
- Baeza, A.; Colilla, M.; Vallet-regí, M. Advances in mesoporous silica nanoparticles for targeted stimuli-responsive drug delivery. Expert Opin. Drug Deliv. 2015, 12, 319–337. [Google Scholar] [CrossRef]
- Croissant, J.G.; Zhang, D.; Alsaiari, S.; Lu, J.; Deng, L.; Tamanoi, F.; Almalik, A.M.; Zink, J.I.; Khashab, N.M. Protein-gold clusters-capped mesoporous silica nanoparticles for high drug loading, autonomous gemcitabine/doxorubicin co-delivery, and in-vivo tumor imaging. J. Control. Release 2016, 229, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ai, K.; Liu, J.; Sun, G.; Yin, Q.; Lu, L. Multifunctional envelope-type mesoporous silica nanoparticles for pH-responsive drug delivery and magnetic resonance imaging. Biomaterials 2015, 60, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Niedermayer, S.; Weiss, V.; Herrmann, A.; Schmidt, A.; Datz, S.; Muller, K.; Wagner, E.; Bein, T.; Bräuchle, C. Multifunctional polymer-capped mesoporous silica nanoparticles for pH-responsive targeted drug delivery. Nanoscale 2015, 7, 7953–7964. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Niu, X.; Ma, K.; Huang, P.; Grothe, J.; Kaskel, S.; Zhu, Y. Graphene quantum dots-capped magnetic mesoporous silica nanoparticles as a multifunctional platform for controlled drug delivery, magnetic hyperthermia, and photothermal therapy. Small 2017, 13, 1–11. [Google Scholar] [CrossRef]
- Lee, C.H.; Cheng, S.H.; Huang, I.P.; Souris, J.S.; Yang, C.S.; Mou, C.Y.; Lo, L.W. Intracellular pH-responsive mesoporous silica nanoparticles for the controlled release of anticancer chemotherapeutics. Angew. Chem. Int. Ed. 2010, 49, 8214–8219. [Google Scholar] [CrossRef] [PubMed]
- Mekaru, H.; Lu, J.; Tamanoi, F. Development of mesoporous silica-based nanoparticles with controlled release capability for cancer therapy. Adv. Drug Deliv. Rev. 2015, 95, 40–49. [Google Scholar] [CrossRef]
- Chen, P.J.; Hu, S.H.; Hsiao, C.S.; Chen, Y.Y.; Liu, D.M.; Chen, S.Y. Multifunctional magnetically removable nanogated lids of Fe3O4-capped mesoporous silica nanoparticles for intracellular controlled release and MR imaging. Chem. J. Mater. 2011, 21, 2535–2543. [Google Scholar] [CrossRef]
- Saint-Crizq, P.; Deshayes, S.; Zink, J.I.; Kasko, A.M. Magnetic field activated drug delivery using thermodegradable azo-functionalised PEG-coated core–shell mesoporous silica nanoparticles. Nanoscale 2015, 7, 13168–13172. [Google Scholar] [CrossRef]
- Gil, M.; Vicente, J.; Gaspar, F. Scale-up methodology for pharmaceutical spray drying. Chem. Today 2010, 28, 18–23. [Google Scholar]
- Ofner, A.; Moore, D.G.; Rühs, P.A.; Schwendimann, P.; Eggersdorfer, M.; Amstad, E.; Weitz, D.A.; Studart, A.R. High-throughput step emulsification for the production of functional materials using a glass microfluidic device. Macromol. Chem. Phys. 2017, 218, 1–10. [Google Scholar] [CrossRef]
- Stolovicki, E.; Ziblat, R.; Weitz, D.A. Throuput enhancement of parallel step emulsifier devices by shear-free and efficient nozzle clearance. Lab Chip 2018, 18, 132–138. [Google Scholar] [CrossRef]
- Jeong, H.; Issadore, D.; Lee, D. Recent developments in scale-up of microfluidic emulsion generation via parallelization. Korean J. Chem. Eng. 2016, 33, 1757–1766. [Google Scholar] [CrossRef]
- Holtze, C. Large-scale droplet production in microfluidic devices—An industrial perspective. J. Phys. D. Appl. Phys. 2013, 46, 1–10. [Google Scholar] [CrossRef]
- Tendulkar, S.; Mirmalek-Sani, S.-H.; Childers, C.; Saul, J.; Opara, E.C.; Ramasubramanian, M.K. A three-dimensional microfluidic approach to scaling up microencapsulation of cells. Biomed. Microdevices 2012, 14, 9623–9626. [Google Scholar] [CrossRef]
- Nisisako, T.; Ando, T.; Hatsuzawa, T. High-volume production of single and compound emulsions in a microfluidic parallelization arrangement coupled with coaxial annular world-to-chip interfaces. Lab Chip 2012, 12, 3426–3435. [Google Scholar] [CrossRef]
- Conchouso, D.; Castro, D.; Khan, S.A.; Foulds, I.G. Three-dimensional parallelization of microfluidic droplet generators for a litre per hour volume production of single emulsions. Lab Chip 2014, 14, 3011–3020. [Google Scholar] [CrossRef]
- Amstad, E.; Chemama, M.; Eggersdorfer, M.; Arriaga, L.R.; Brenner, M.P.; Weitz, D.A. Robust scalable high throughput production of monodisperse drops. Lab Chip 2016, 16, 4163–4172. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Segment | pH | Length (cm) | Mean Diameter (cm) | Mucus Average Thickness (µm) | Mucus Turnover (hour) |
|---|---|---|---|---|---|
| Stomach | 0.8–5 [45] | 20 [45] | NA | 245 ± 200 [38] | 24–48 [46] |
| Duodenum | ~7 [38] | 17–56 [47] | 4 [48] | 15.5 [45] | 24–48 [46] |
| Jejunum | ≥ 7 [38] | 280–1000 [47] | 2–2.5 [48] | 15.5 [45] | |
| Ileum | ≥ 7 [38] | 3 [48] | 15.5 [45] | ||
| Colon | 7–8 [38] | 80–313 [47] | 4–4.8 [48] | 135 ± 25 [38] | 24–48 [46] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Homayun, B.; Lin, X.; Choi, H.-J. Challenges and Recent Progress in Oral Drug Delivery Systems for Biopharmaceuticals. Pharmaceutics 2019, 11, 129. https://doi.org/10.3390/pharmaceutics11030129
Homayun B, Lin X, Choi H-J. Challenges and Recent Progress in Oral Drug Delivery Systems for Biopharmaceuticals. Pharmaceutics. 2019; 11(3):129. https://doi.org/10.3390/pharmaceutics11030129
Chicago/Turabian StyleHomayun, Bahman, Xueting Lin, and Hyo-Jick Choi. 2019. "Challenges and Recent Progress in Oral Drug Delivery Systems for Biopharmaceuticals" Pharmaceutics 11, no. 3: 129. https://doi.org/10.3390/pharmaceutics11030129
APA StyleHomayun, B., Lin, X., & Choi, H.-J. (2019). Challenges and Recent Progress in Oral Drug Delivery Systems for Biopharmaceuticals. Pharmaceutics, 11(3), 129. https://doi.org/10.3390/pharmaceutics11030129