Electrostatically Driven Encapsulation of Hydrophilic, Non-Conformational Peptide Epitopes into Liposomes

 ,
,
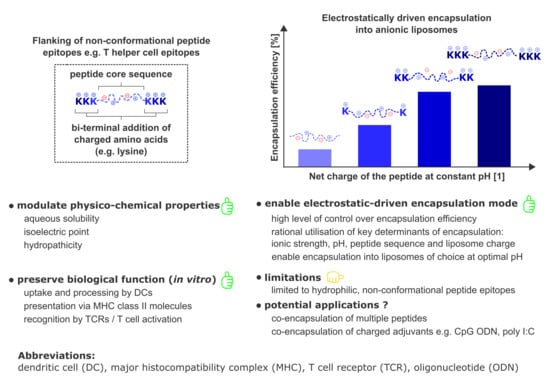
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Buffers
2.3. Liposome Preparation
2.3.1. Liposome Composition
2.3.2. Thin-Film Hydration
2.3.3. Microfluidic Mixing
2.3.4. Tangential Flow Filtration (TFF)
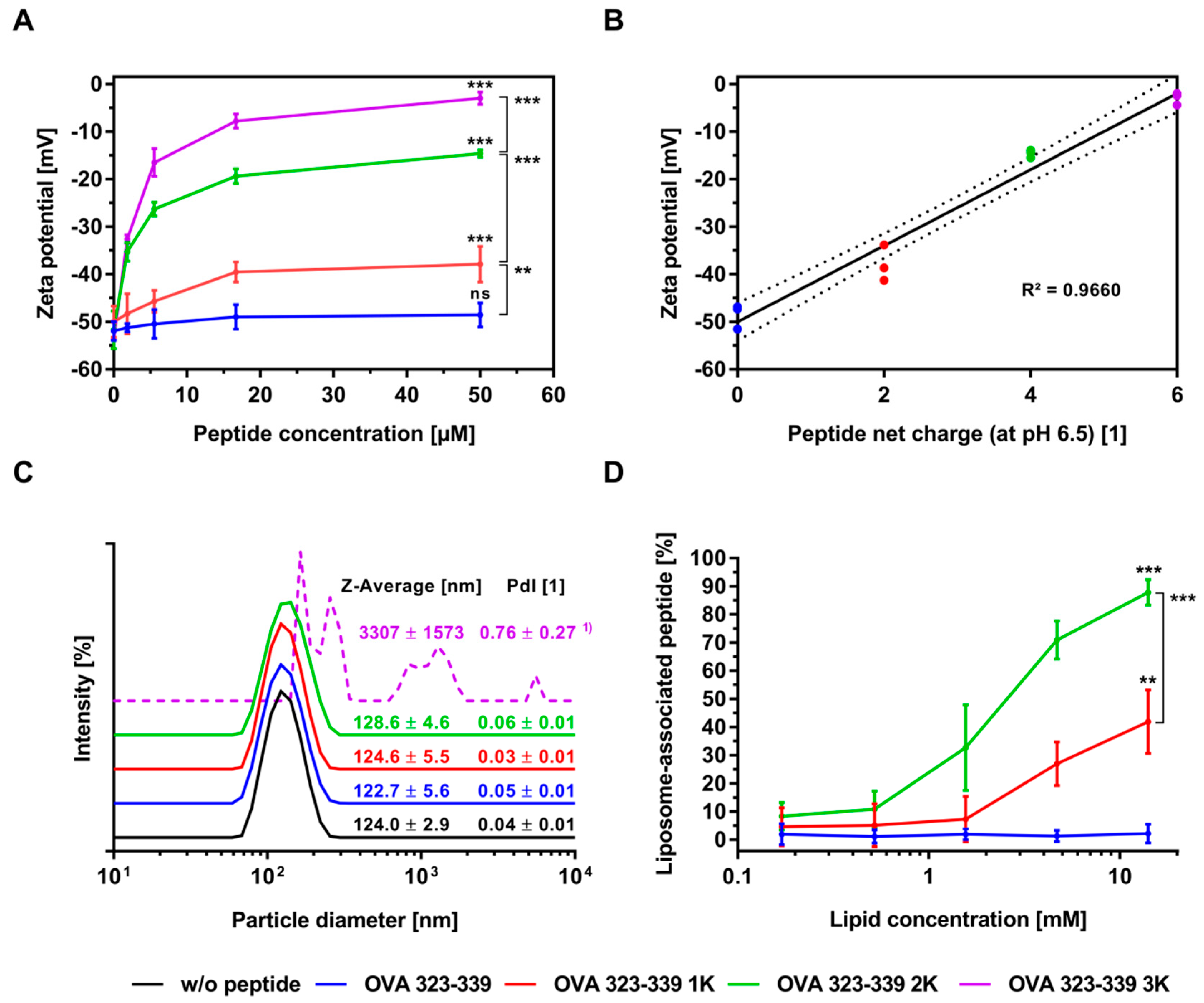
2.4. Particle Size and Zeta Potential
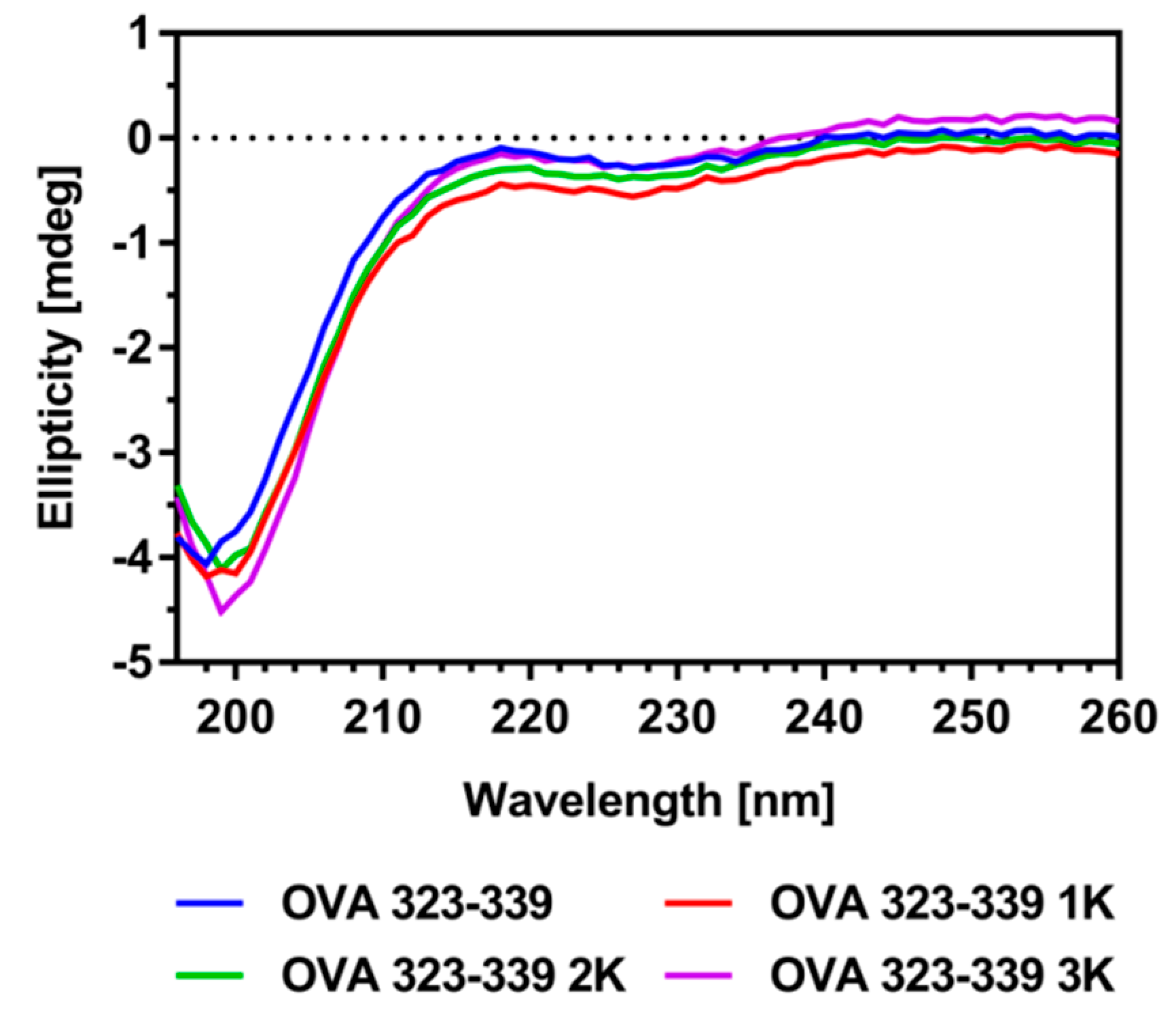
2.5. CD Spectroscopy
2.6. HPLC
2.6.1. General
2.6.2. Peptide Quantification
2.6.3. Peptide Extraction
2.6.4. Lipid Quantification
2.7. Peptide Binding
2.8. Encapsulation Efficiency
2.9. In Vitro Stability Study
2.10. Mice
2.11. Cell Isolation and Purification
2.12. CD4+ T-Cell Proliferation Assay
3. Results and Discussion
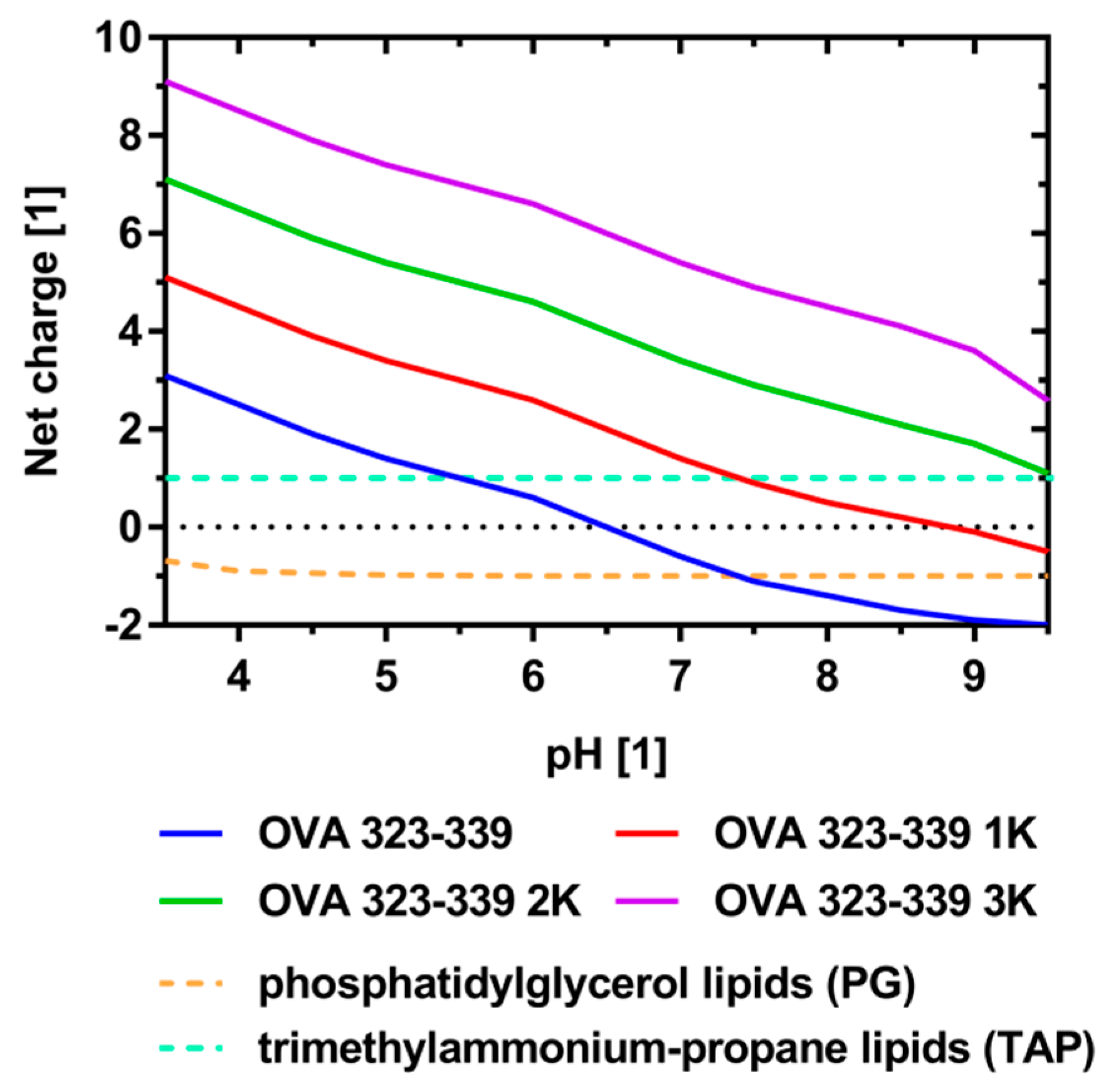
3.1. Peptide Design and Characterization
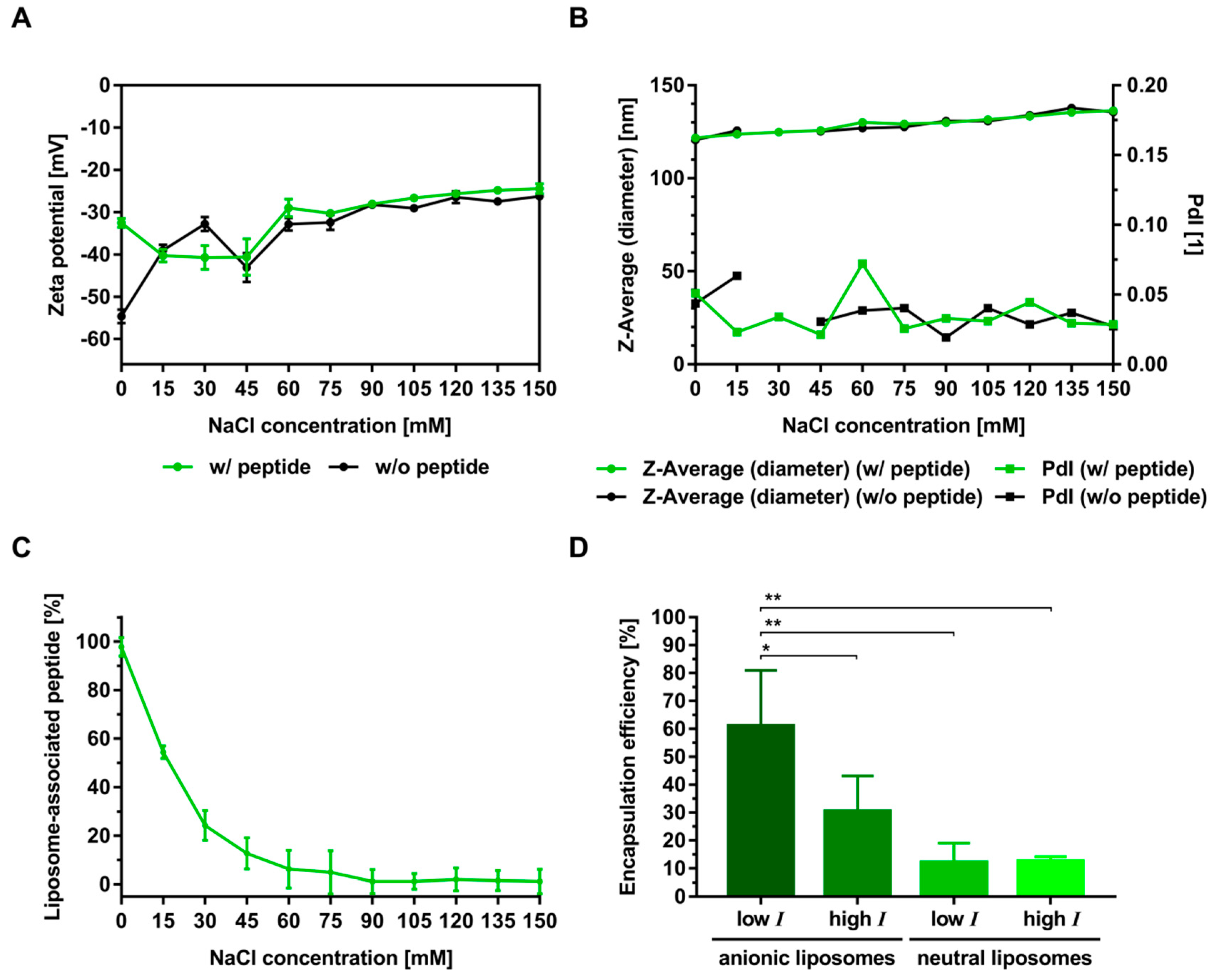
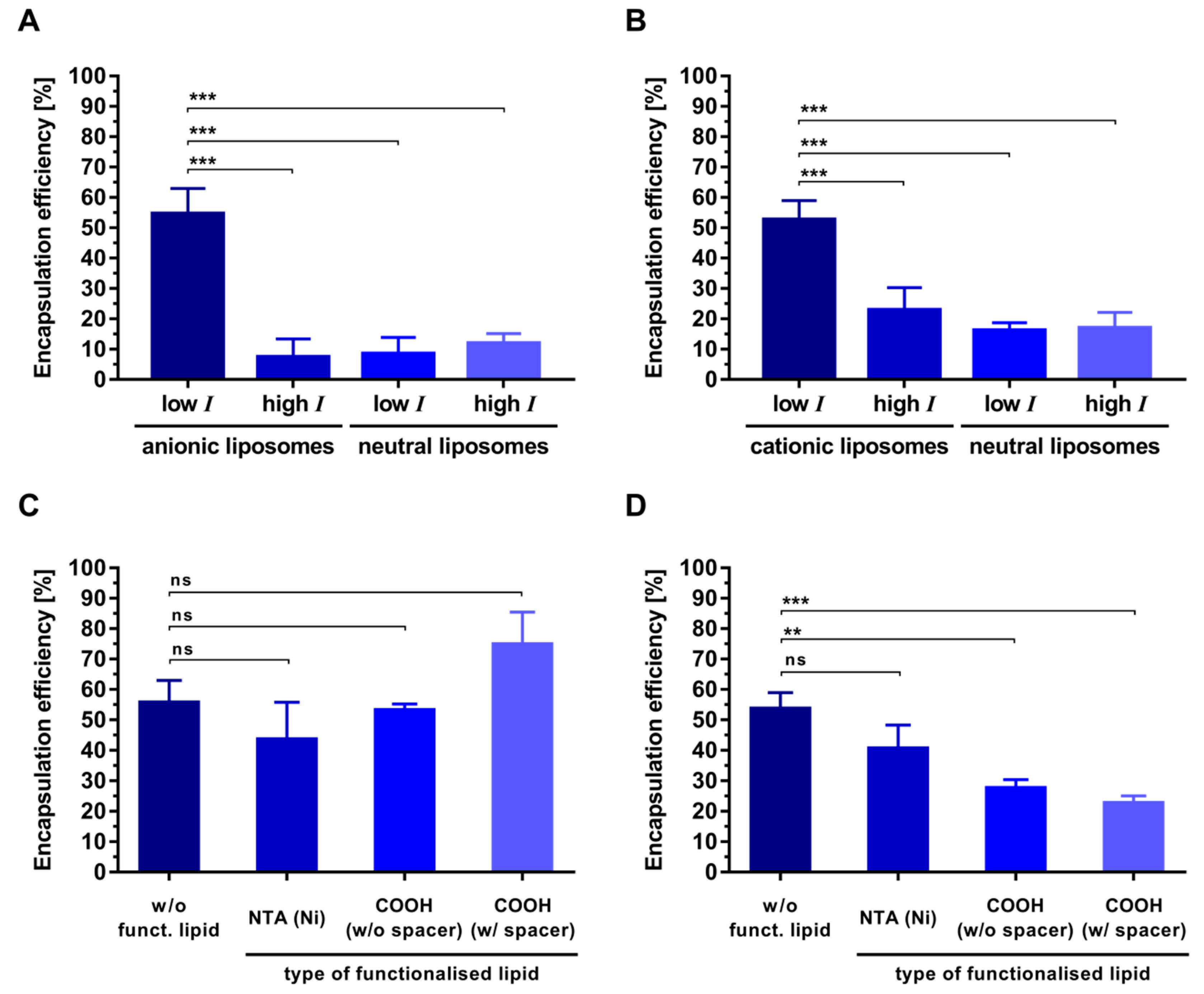
3.2. Electrostatic Interactions of Peptides with Liposomes
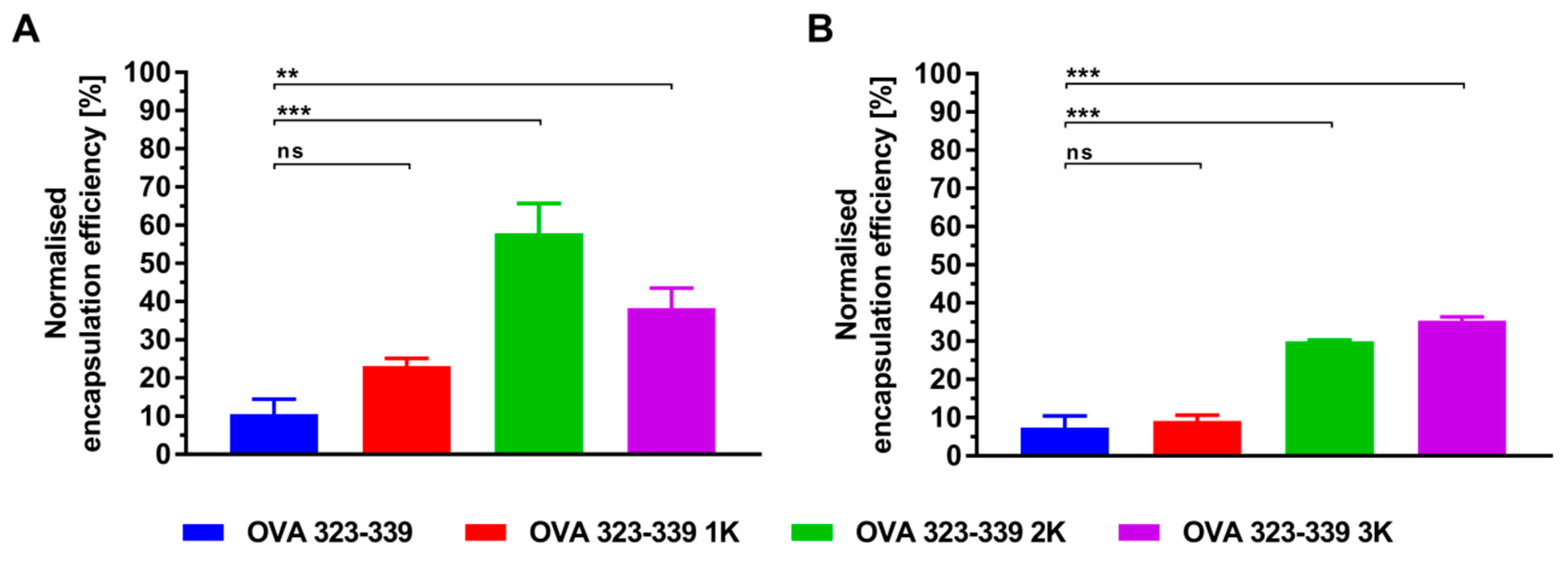
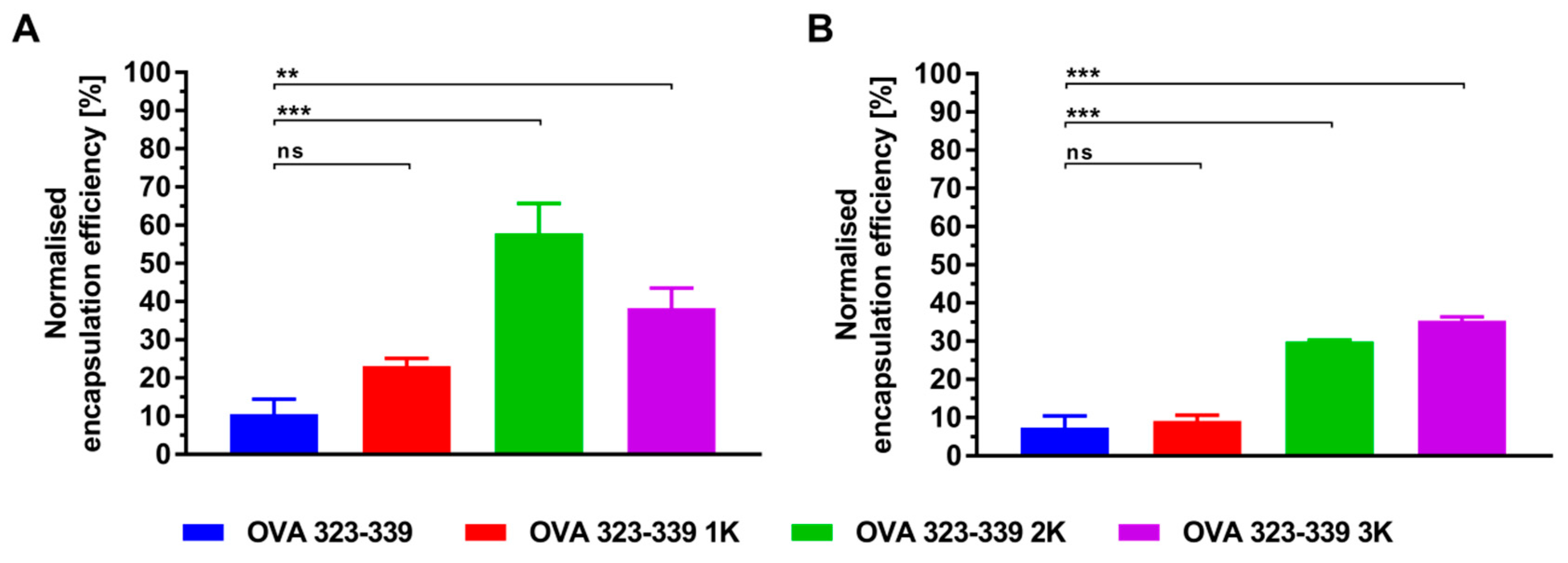
3.3. Electrostatic-Driven Encapsulation of Peptides
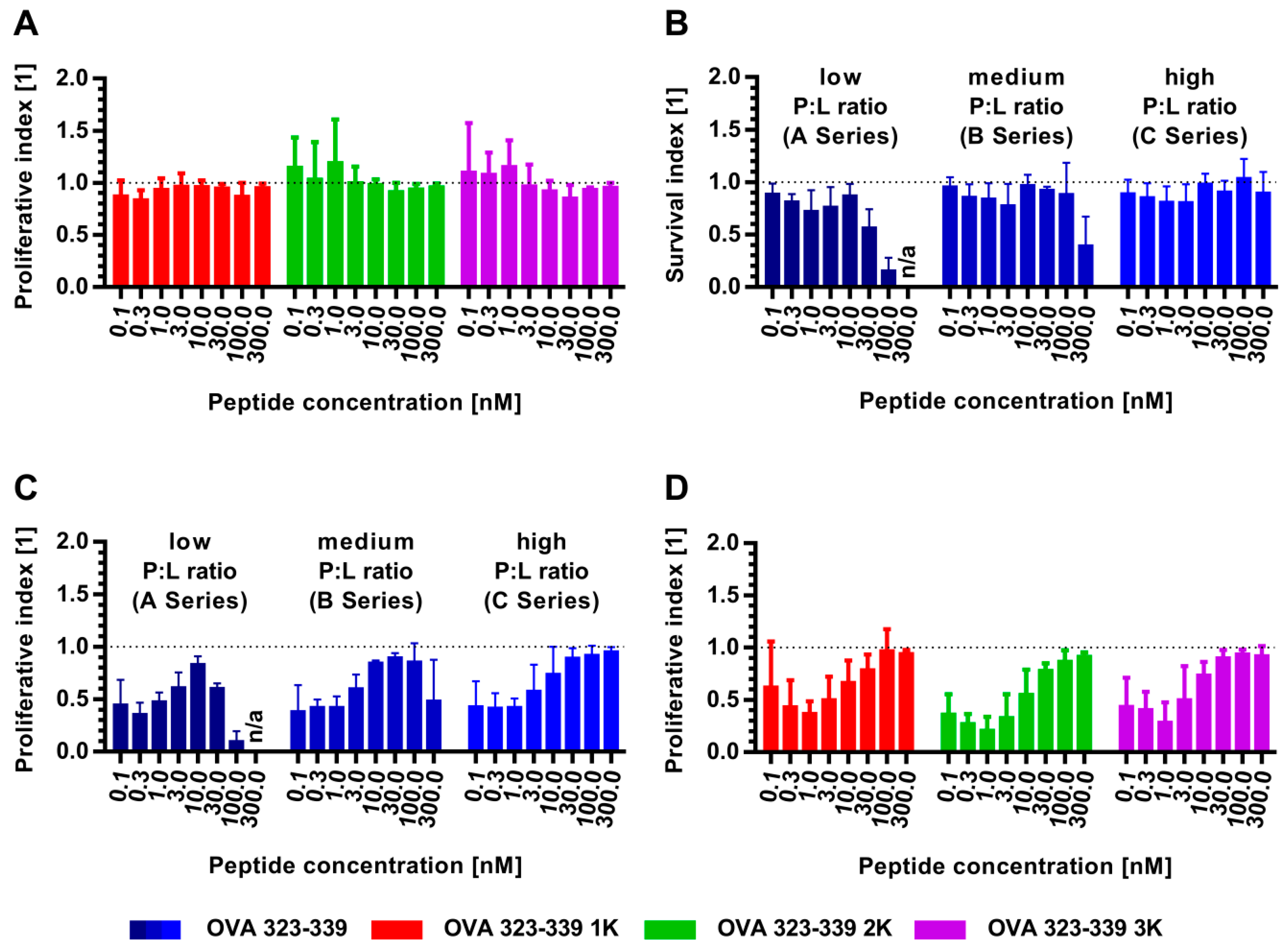
3.4. In Vitro Co-Cultivation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Li, W.; Joshi, M.; Singhania, S.; Ramsey, K.; Murthy, A. Peptide Vaccine: Progress and Challenges. Vaccines 2014, 2, 515–536. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorganic Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef] [PubMed]
- Skwarczynski, M.; Toth, I. Peptide-based synthetic vaccines. Chem. Sci. 2016, 7, 842–854. [Google Scholar] [CrossRef] [PubMed]
- Du, A.W.; Stenzel, M.H. Drug Carriers for the Delivery of Therapeutic Peptides. Biomacromolecules 2014, 15, 1097–1114. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Chen, M.; Wang, T. Liposomes used as a vaccine adjuvant-delivery system: From basics to clinical immunization. J. Control. Release 2019, 303, 130–150. [Google Scholar] [CrossRef]
- De Serrano, L.O.; Burkhart, D.J. Liposomal vaccine formulations as prophylactic agents: Design considerations for modern vaccines. J. Nanobiotechnol. 2017, 15, 1–23. [Google Scholar] [CrossRef]
- Perrie, Y.; Crofts, F.; Devitt, A.; Griffiths, H.R.; Kastner, E.; Nadella, V. Designing liposomal adjuvants for the next generation of vaccines. Adv. Drug Deliv. Rev. 2016, 99, 85–96. [Google Scholar] [CrossRef]
- Carugo, D.; Bottaro, E.; Owen, J.; Stride, E.; Nastruzzi, C. Liposome production by microfluidics: Potential and limiting factors. Sci. Rep. 2016, 6, 1–15. [Google Scholar] [CrossRef]
- Wagner, A.; Vorauer-Uhl, K. Liposome Technology for Industrial Purposes. J. Drug Deliv. 2011, 2011, 591325. [Google Scholar] [CrossRef]
- Heuts, J.; Varypataki, E.M.; van der Maaden, K.; Romeijn, S.; Drijfhout, J.W.; van Scheltinga, A.T.; Ossendorp, F.; Jiskoot, W. Cationic Liposomes: A Flexible Vaccine Delivery System for Physicochemically Diverse Antigenic Peptides. Pharm. Res. 2018, 35. [Google Scholar] [CrossRef]
- Hwang, S.Y.; Kim, H.K.; Choo, J.; Seong, G.H.; Hien, T.B.D.; Lee, E.K. Effects of operating parameters on the efficiency of liposomal encapsulation of enzymes. Colloids Surf. B Biointerfaces 2012, 94, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Colletier, J.P.; Chaize, B.; Winterhalter, M.; Fournier, D. Protein encapsulation in liposomes: Efficiency depends on interactions between protein and phospholipid bilayer. BMC Biotechnol. 2002, 2, 9. [Google Scholar] [CrossRef] [PubMed]
- Brgles, M.; Jurašin, D.; Sikirić, M.D.; Frkanec, R.; Tomašić, J. Entrapment of Ovalbumin into Liposomes—Factors Affecting Entrapment Efficiency, Liposome Size, and Zeta Potential. J. Liposome Res. 2008, 18, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Mori, T.; Tomita, M.; Tsumoto, K. pH Switching That Crosses over the Isoelectric Point (pI) Can Improve the Entrapment of Proteins within Giant Liposomes by Enhancing Protein−Membrane Interaction. Langmuir 2014, 30, 554–563. [Google Scholar] [CrossRef]
- Seelig, J. Thermodynamics of lipid – peptide interactions. Biochim. Biophys. Acta Biomembr. 2004, 1666, 40–50. [Google Scholar] [CrossRef]
- Kim, J.; Mosior, M.; Chung, L.A.; Wu, H.; McLaughlin, S. Binding of peptides with basic residues to membranes containing acidic phospholipids. Biophys. J. 1991, 60, 135–148. [Google Scholar] [CrossRef]
- Murray, D.; Arbuzova, A.; Hangyás-Mihályné, G.; Gambhir, A.; Ben-Tal, N.; Honig, B.; McLaughlin, S. Electrostatic Properties of Membranes Containing Acidic Lipids and Adsorbed Basic Peptides: Theory and Experiment. Biophys. J. 1999, 77, 3176–3188. [Google Scholar] [CrossRef]
- Van Hoof, B.; Markvoort, A.J.; Van Santen, R.A.; Hilbers, P.A.J. Molecular Simulation of Protein Encapsulation in Vesicle Formation. J. Phys. Chem. B 2014, 118, 3346–3354. [Google Scholar] [CrossRef]
- Xu, X.; Khan, M.A.; Burgess, D.J. Predicting hydrophilic drug encapsulation inside unilamellar liposomes. Int. J. Pharm. 2012, 423, 410–418. [Google Scholar] [CrossRef]
- Forbes, N.; Hussain, M.T.; Briuglia, M.L.; Edwards, D.P.; Ter Horst, J.H.; Szita, N.; Perrie, Y. Rapid and scale-independent microfluidic manufacture of liposomes entrapping protein incorporating in-line purification and at-line size monitoring. Int. J. Pharm. 2019, 556, 68–81. [Google Scholar] [CrossRef]
- Temchura, V.; Überla, K. Intrastructural help: improving the HIV-1 envelope antibody response induced by virus-like particle vaccines. Curr. Opin. HIV AIDS 2017, 12, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Elsayed, H.; Nabi, G.; McKinstry, W.J.; Khoo, K.K.; Mak, J.; Salazar, A.M.; Tenbusch, M.; Temchura, V.; Überla, K. Intrastructural Help: Harnessing T Helper Cells Induced by Licensed Vaccines for Improvement of HIV Env Antibody Responses to Virus-Like Particle Vaccines. J. Virol. 2018, 92, 1–15. [Google Scholar] [CrossRef] [PubMed]
- genannt Bonsmann, M.S.; Niezold, T.; Temchura, V.; Pissani, F.; Ehrhardt, K.; Brown, E.P.; Osei-Owusu, N.Y.; Hannaman, D.; Hengel, H.; Ackerman, M.E.; et al. Enhancing the Quality of Antibodies to HIV-1 Envelope by GagPol-Specific Th Cells. J. Immunol. 2015, 195, 4861–4872. [Google Scholar] [CrossRef] [PubMed]
- Hills, T.; Jakeman, P.G.; Carlisle, R.C.; Klenerman, P.; Seymour, L.W.; Cawood, R. A Rapid-Response Humoral Vaccine Platform Exploiting Pre-Existing Non-Cognate Populations of Anti-Vaccine or Anti-Viral CD4+ T Helper Cells to Confirm B Cell Activation. PLoS ONE 2016, 11, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Zaman, M.; Ozberk, V.; Langshaw, E.L.; McPhun, V.; Powell, J.L.; Phillips, Z.N.; Ho, M.F.; Calcutt, A.; Batzloff, M.R.; Toth, I.; et al. Novel platform technology for modular mucosal vaccine that protects against streptococcus. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef]
- Lu, H.; Wang, J.; Bai, Y.; Lang, J.W.; Liu, S.; Lin, Y.; Cheng, J. Ionic polypeptides with unusual helical stability. Nat. Commun. 2011, 2, 206–209. [Google Scholar] [CrossRef]
- Robison, A.D.; Sun, S.; Poyton, M.F.; Johnson, G.A.; Pellois, J.P.; Jungwirth, P.; Vazdar, M.; Cremer, P.S. Polyarginine Interacts More Strongly and Cooperatively than Polylysine with Phospholipid Bilayers. J. Phys. Chem. B 2016, 120, 9287–9296. [Google Scholar] [CrossRef]
- Yin, L.M.; Edwards, M.A.; Li, J.; Yip, C.M.; Deber, C.M. Roles of Hydrophobicity and Charge Distribution of Cationic Antimicrobial Peptides in Peptide-Membrane Interactions. J. Biol. Chem. 2012, 287, 7738–7745. [Google Scholar] [CrossRef]
- Wilms, D.; Andrä, J. Comparison of patient-derived high and low phosphatidylserine-exposing colorectal carcinoma cells in their interaction with anti-cancer peptides. J. Pept. Sci. 2017, 23, 56–67. [Google Scholar] [CrossRef]
- Manzini, M.C.; Perez, K.R.; Riske, K.A.; Bozelli, J.C.; Santos, T.L.; Da Silva, M.A.; Saraiva, G.K.V.; Politi, M.J.; Valente, A.P.; Almeida, F.C.L.; et al. Peptide:lipid ratio and membrane surface charge determine the mechanism of action of the antimicrobial peptide BP100. Conformational and functional studies. Biochim. Biophys. Acta Biomembr. 2014, 1838, 1985–1999. [Google Scholar] [CrossRef]
- Johnson, J.E.; Xie, M.; Singh, L.M.R.; Edge, R.; Cornell, R.B. Both Acidic and Basic Amino Acids in an Amphitropic Enzyme, CTP:Phosphocholine Cytidylyltransferase, Dictate Its Selectivity for Anionic Membranes. J. Biol. Chem. 2003, 278, 514–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penwell, A.; Sharp, K.; Mansour, M.; Sammatur, L. Development and validation of an HPLC/UV assay for separation and quantification of peptide antigens from a liposomal vaccine delivery platform. J. Pharm. Biomed. Anal. 2012, 66, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.T.; Forbes, N.; Perrie, Y. Comparative analysis of protein quantification methods for the rapid determination of protein loading in liposomal formulations. Pharmaceutics 2019, 11, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Temchura, V.; Tenbusch, M.; Nchinda, G.; Nabi, G.; Tippler, B.; Zelenyuk, M.; Wildner, O.; Überla, K.; Kuate, S. Enhancement of immunostimulatory properties of exosomal vaccines by incorporation of fusion-competent G protein of vesicular stomatitis virus. Vaccine 2008, 26, 3662–3672. [Google Scholar] [CrossRef] [PubMed]
- Temchura, V.; Kalinin, S.; Nabi, G.; Tippler, B.; Niezold, T.; Überla, K. Divergence of Primary Cognate B- and T-Cell Proliferative Responses to Subcutaneous and Intravenous Immunization with Virus-Like Particles. Viruses 2014, 6, 3334–3347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2009; pp. 571–607. ISBN 978-1-58829-343-5. [Google Scholar]
- Protein Calculator v3. Available online: http://protcalc.sourceforge.net/ (accessed on 28 May 2019).
- Marsh, D. Handbook of Lipid Bilayers, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2013; ISBN 978-1-4200-8833-5. [Google Scholar]
- Simberg, D.; Weisman, S.; Talmon, Y.; Barenholz, Y. DOTAP (and other cationic lipids): chemistry, biophysics, and transfection. Crit. Rev. Ther. Drug Carrier Syst. 2004, 21, 257–317. [Google Scholar] [CrossRef]
- Volodkin, D.; Ball, V.; Schaaf, P.; Voegel, J.; Mohwald, H. Complexation of phosphocholine liposomes with polylysine. Stabilization by surface coverage versus aggregation. Biochem. Biophys. Acta 2007, 1768, 280–290. [Google Scholar] [CrossRef] [Green Version]
- Volodkin, D.; Mohwald, H.; Voegel, J.; Ball, V. Coating of negatively charged liposomes by polylysine: Drug release study. J. Control. Release 2007, 117, 111–120. [Google Scholar] [CrossRef]
- Lamazière, A.; Burlina, F.; Wolf, C.; Chassaing, G.; Trugnan, G.; Ayala-Sanmartin, J. Non-Metabolic Membrane Tubulation and Permeability Induced by Bioactive Peptides. PLoS ONE 2007, 2. [Google Scholar] [CrossRef]
- Suleiman, E. Development of a workflow for formulation and manufacturing process development of liposomal drug delivery systems. Master’s Thesis, University of Veterinary Medicine, Vienna, Wien, Austria, 2016. [Google Scholar]
- Leng, J.; Egelhaaf, S.U.; Cates, M.E. Kinetics of the Micelle-to-Vesicle Transition: Aqueous Lecithin-Bile Salt Mixtures. Biophys. J. 2003, 85, 1624–1646. [Google Scholar] [CrossRef] [Green Version]
- Garidel, P.; Blume, A. 1,2-Dimyristoyl-sn-glycero-3-phosphoglycerol (DMPG) monolayers: influence of temperature, pH, ionic strength and binding of alkaline earth cations. Chem. Phys. Lipids 2005, 138, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; Shao, Q.; Williams, H.; Hilty, C.; Gao, Y.Q. Methanol Strengthens Hydrogen Bonds and Weakens Hydrophobic Interactions in Proteins - A Combined Molecular Dynamics and NMR study. J. Phys. Chem. B 2011, 115, 6653–6660. [Google Scholar] [CrossRef] [PubMed]
- Guida, V. Thermodynamics and kinetics of vesicles formation processes. Adv. Colloid Interface Sci. 2010, 161, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Quinn, D.; Sadovsky, Y.; Suresh, S.; Hsia, K.J. Formation and size distribution of self-assembled vesicles. Proc. Natl. Acad. Sci. USA 2017, 114, 2910–2915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varypataki, E.M.; Silva, A.L.; Barnier-Quer, C.; Collin, N.; Ossendorp, F.; Jiskoot, W. Synthetic long peptide-based vaccine formulations for induction of cell mediated immunity: A comparative study of cationic liposomes and PLGA nanoparticles. J. Control. Release 2016, 226, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Grit, M.; Underberg, W.J.M.; Crommelin, D.J.A. Hydrolysis of Saturated Soybean Phosphatidylcholine in Aqueous Liposome Dispersions. J. Pharm. Sci. 1993, 82, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.; Maki, K.; Ebina, T.; Kuwajima, K.; Soda, K.; Kuroda, Y. Mutational Analysis of Protein Solubility Enhancement Using Short Peptide Tags. Biopolymers 2006, 85, 12–18. [Google Scholar] [CrossRef]
- Barnden, M.J.; Allison, J.; Heath, W.R.; Carbone, F.R. Defective TCR expression in transgenic mice constructed using cDNA- based α- and β-chain genes under the control of heterologous regulatory elements. Immunol. Cell Biol. 1998, 76, 34–40. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Variant | Sequence | GRAVY [1] 1 | Isoelectric Point [1] 2 |
|---|---|---|---|
| OVA 323-339 | ISQAVHAAHAEINEAGR | −0.299 | 6.5 |
| OVA 323-339 1K | KISQAVHAAHAEINEAGRK | −0.616 | 8.9 |
| OVA 323-339 2K | KKISQAVHAAHAEINEAGRKK | −0.929 | 10.0 |
| OVA 323-339 3K | KKKISQAVHAAHAEINEAGRKKK | −1.187 | 10.3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suleiman, E.; Damm, D.; Batzoni, M.; Temchura, V.; Wagner, A.; Überla, K.; Vorauer-Uhl, K. Electrostatically Driven Encapsulation of Hydrophilic, Non-Conformational Peptide Epitopes into Liposomes. Pharmaceutics 2019, 11, 619. https://doi.org/10.3390/pharmaceutics11110619
Suleiman E, Damm D, Batzoni M, Temchura V, Wagner A, Überla K, Vorauer-Uhl K. Electrostatically Driven Encapsulation of Hydrophilic, Non-Conformational Peptide Epitopes into Liposomes. Pharmaceutics. 2019; 11(11):619. https://doi.org/10.3390/pharmaceutics11110619
Chicago/Turabian StyleSuleiman, Ehsan, Dominik Damm, Mirjam Batzoni, Vladimir Temchura, Andreas Wagner, Klaus Überla, and Karola Vorauer-Uhl. 2019. "Electrostatically Driven Encapsulation of Hydrophilic, Non-Conformational Peptide Epitopes into Liposomes" Pharmaceutics 11, no. 11: 619. https://doi.org/10.3390/pharmaceutics11110619