Macrophage Targeting pH Responsive Polymersomes for Glucocorticoid Therapy

 ,
,  ,
, 

Abstract
{kind=link}
{kind=link}
{kind=link}
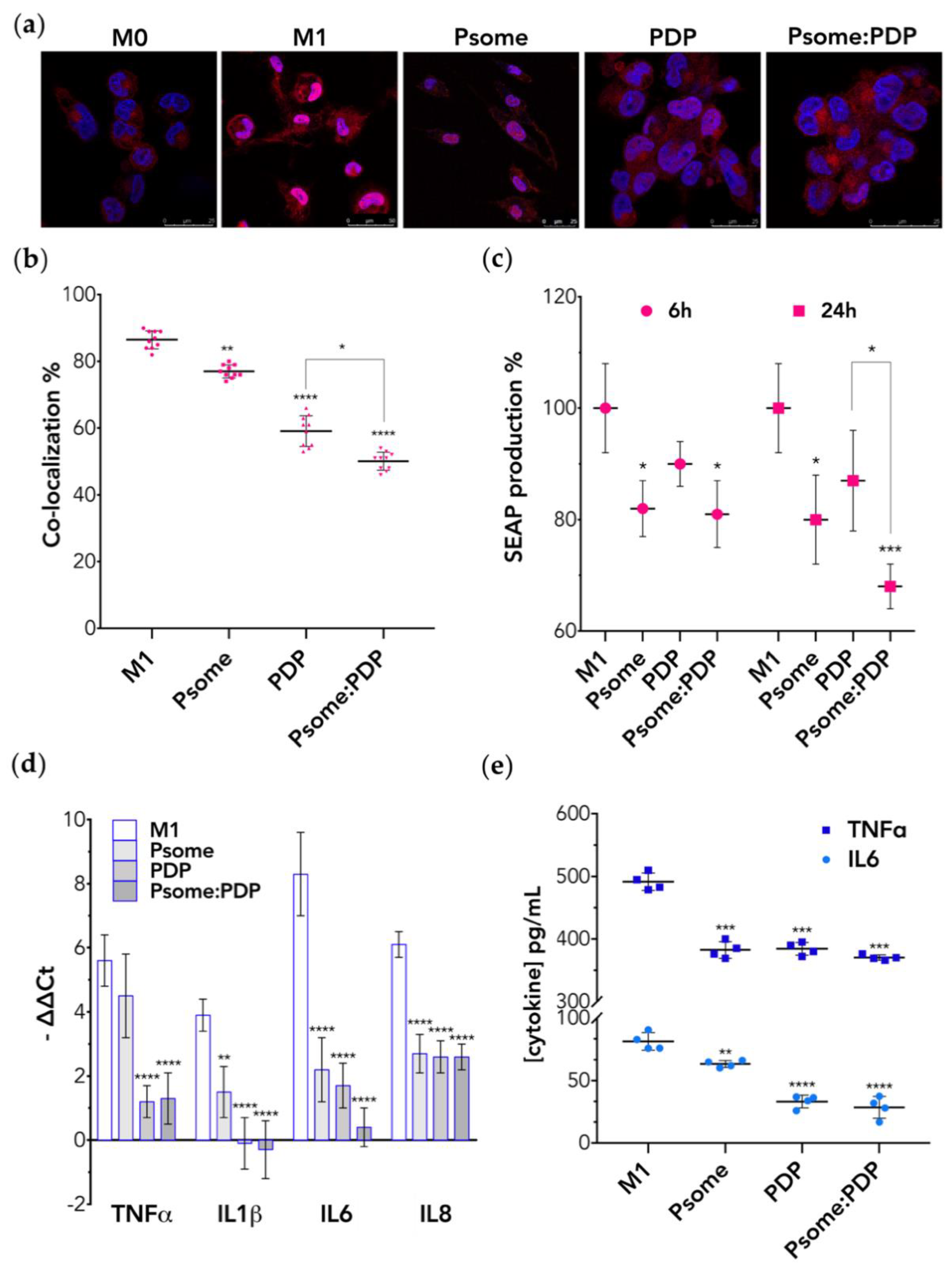{kind=link}
1. Introduction
2. Materials and Methods
2.1. Preparation of PMPC–PDPA Polymersomes
2.2. Characterisation Studies
2.1.1. Size and Size Distribution Study
2.1.2. Morphology Study
2.1.3. Polymer and Drug Quantification Study
2.1.4. Drug Release Study
2.3. In Vitro Cellular Studies
2.3.1. Cell Culture and Differentiation
2.3.2. Cell Viability Assay
2.3.3. Cell Uptake Imaging
2.3.4. NF-κB Signalling Imaging and Quantification Assay
2.3.5. RNA Extraction, Reverse Transcription, and Real-Time Quantitative Polymerase Chain Reaction
2.3.6. Enzyme Linked Immunosorbent Assay
2.4. Statistical Analysis
3. Results and Discussion
3.1. PMPC–PDPA Polymersomes Are Suitable Nanocarriers for PDP
3.2. PMPC–PDPA Polymersomes Enable a pH-Controlled Drug Release
3.3. PMPC–PDPA Polymersomes Enable a Rapid Intracellular Drug Delivery
3.4. PDP Loaded PMPC–PDPA Polymersomes Promote Inflammation Resolution In Vitro
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dunster, J. The macrophage and its role in inflammation and tissue repair: Mathematical and systems biology approaches. Wiley Interdiscip. Rev.-Syst. Biol. Med. 2016, 8, 87–99. [Google Scholar] [CrossRef]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, N.; Kobayashi, K. Macrophages in inflammation. Curr. Drug Targets Inflamm. Allergy 2005, 4, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Oishi, Y.; Manabe, I. Macrophages in inflammation, repair and regeneration. Int. Immunol. 2018, 30, 511–528. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Jung, S. Monocytes and macrophages: Developmental pathways and tissue homeostasis. Nat. Rev. Immunol. 2014, 14, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Lund, M.E.; To, J.; O’Brien, B.A.; Donnelly, S. The choice of phorbol 12-myristate 13-acetate differentiation protocol influences the response of THP-1 macrophages to a pro-inflammatory stimulus. J. Immunol. Methods 2016, 430, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2. [Google Scholar] [CrossRef]
- Barnes, P.J. Mechanisms and resistance in glucocorticoid control of inflammation. J. Steroid Biochem. Mol. Biol. 2010, 120, 76–85. [Google Scholar] [CrossRef]
- Lühder, F.; Reichardt, H.M. Novel Drug Delivery Systems Tailored for Improved Administration of Glucocorticoids. Int. J. Mol. Sci. 2017, 18, 1836. [Google Scholar] [CrossRef]
- Brasier, A.R. The nuclear factor-kappaB-interleukin-6 signalling pathway mediating vascular inflammation. Cardiovasc. Res. 2010, 86, 211–218. [Google Scholar] [CrossRef]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta 2011, 1813, 878–888. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, R.; Wada, H.; Ito, K.; Adcock, I.M. Effects of glucocorticoids on gene transcription. Eur. J. Pharmacol. 2004, 500, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Colley, H.E.; Hearnden, V.; Avila-Olias, M.; Cecchin, D.; Canton, I.; Madsen, J.; MacNeil, S.; Warren, N.; Hu, K.; McKeating, J.A.; et al. Polymersome-mediated delivery of combination anticancer therapy to head and neck cancer cells: 2D and 3D in vitro evaluation. Mol. Pharm. 2014, 11, 1176–1188. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.; Reeves, K.J.; Hearnden, V.; Colley, H.; Massignani, M.; Canton, I.; Madsen, J.; Blanazs, A.; Armes, S.P.; Lewis, A.L.; et al. Internalization and biodistribution of polymersomes into oral squamous cell carcinoma cells in vitro and in vivo. Nanomedicine (Lond.) 2010, 5, 1025–1036. [Google Scholar] [CrossRef]
- Massignani, M.; Canton, I.; Sun, T.; Hearnden, V.; Macneil, S.; Blanazs, A.; Armes, S.P.; Lewis, A.; Battaglia, G. Enhanced fluorescence imaging of live cells by effective cytosolic delivery of probes. PLoS ONE 2010, 5, e10459. [Google Scholar] [CrossRef] [PubMed]
- Rizzello, L.; Robertson, J.D.; Elks, P.M.; Poma, A.; Daneshpour, N.; Prajsnar, T.K.; Evangelopoulos, D.; Canseco, J.O.; Yona, S.; Marriott, H.M.; et al. Targeting mononuclear phagocytes for eradicating intracellular parasites. bioRxiv 2017. [Google Scholar] [CrossRef]
- Contini, C.; Pearson, R.; Wang, L.; Messager, L.; Gaitzsch, J.; Rizzello, L.; Ruiz-Perez, L.; Battaglia, G. Bottom-Up Evolution of Vesicles from Disks to High-Genus Polymersomes. iScience 2018, 7, 132–144. [Google Scholar] [CrossRef]
- Wang, L.; Chierico, L.; Little, D.; Patikarnmonthon, N.; Yang, Z.; Azzouz, M.; Madsen, J.; Armes, S.P.; Battaglia, G. Encapsulation of biomacromolecules within polymersomes by electroporation. Angew. Chem. Int. Ed. Engl. 2012, 51, 11122–11125. [Google Scholar] [CrossRef]
- Lomas, H.; Du, J.; Canton, I.; Madsen, J.; Warren, N.; Armes, S.P.; Lewis, A.L.; Battaglia, G. Efficient encapsulation of plasmid DNA in pH-sensitive PMPC-PDPA polymersomes: Study of the effect of PDPA block length on copolymer-DNA binding affinity. Macromol. Biosci. 2010, 10, 513–530. [Google Scholar] [CrossRef]
- Massignani, M.; LoPresti, C.; Blanazs, A.; Madsen, J.; Armes, S.P.; Lewis, A.L.; Battaglia, G. Controlling cellular uptake by surface chemistry, size, and surface topology at the nanoscale. Small 2009, 5, 2424–2432. [Google Scholar] [CrossRef]
- Pegoraro, C.; Cecchin, D.; Gracia, L.S.; Warren, N.; Madsen, J.; Armes, S.P.; Lewis, A.; Macneil, S.; Battaglia, G. Enhanced drug delivery to melanoma cells using PMPC-PDPA polymersomes. Cancer Lett. 2013, 334, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Guan, L.; Rizzello, L.; Battaglia, G. Polymersomes and their applications in cancer delivery and therapy. Nanomedicine (Lond.) 2015, 10, 2757–2780. [Google Scholar] [CrossRef] [PubMed]
- Lardner, A. The effects of extracellular pH on immune function. J. Leukoc. Biol. 2001, 69, 522–530. [Google Scholar] [PubMed]
- Du, J.; Tang, Y.; Lewis, A.L.; Armes, S.P. pH-sensitive vesicles based on a biocompatible zwitterionic diblock copolymer. J. Am. Chem. Soc. 2005, 127, 17982–17983. [Google Scholar] [CrossRef] [PubMed]
- Gaitzsch, J.; Delahaye, M.; Poma, A.; Du Prez, F.; Battaglia, G. Comparison of metal free polymer-dye conjugation strategies in protic solvents. Polym. Chem. 2016, 7, 3046–3055. [Google Scholar] [CrossRef]
- Robertson, J.D.; Yealland, G.; Avila-Olias, M.; Chierico, L.; Bandmann, O.; Renshaw, S.A.; Battaglia, G. pH-sensitive tubular polymersomes: Formation and applications in cellular delivery. ACS Nano 2014, 8, 4650–4661. [Google Scholar] [CrossRef]
- Battaglia, G.; Ryan, A.J. The evolution of vesicles from bulk lamellar gels. Nat. Mater. 2005, 4, 869–876. [Google Scholar] [CrossRef]
- Fetsch, C.; Gaitzsch, J.; Messager, L.; Battaglia, G.; Luxenhofer, R. Self-Assembly of Amphiphilic Block Copolypeptoids - Micelles, Worms and Polymersomes. Sci. Rep. 2016, 6, 33491. [Google Scholar] [CrossRef]
- Robertson, J.D.; Rizzello, L.; Avila-Olias, M.; Gaitzsch, J.; Contini, C.; Magoń, M.S.; Renshaw, S.A.; Battaglia, G. Purification of Nanoparticles by Size and Shape. Sci. Rep. 2016, 6, 27494. [Google Scholar] [CrossRef]
- Ruiz-Pérez, L.; Messager, L.; Gaitzsch, J.; Joseph, A.; Sutto, L.; Gervasio, F.L.; Battaglia, G. Molecular engineering of polymersome surface topology. Sci. Adv. 2016, 2, e1500948. [Google Scholar] [CrossRef]
- Gouveia, V.M.; Lopes-de-Araujo, J.; Costa Lima, S.A.; Nunes, C.; Reis, S. Hyaluronic acid-conjugated pH-sensitive liposomes for targeted delivery of prednisolone on rheumatoid arthritis therapy. Nanomedicine (Lond.) 2018, 13, 1037–1049. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Poma, A.; Rizzello, L.; Gouveia, V.M.; Ruiz-Perez, L.; Battaglia, G.; Williams, C.K. Metabolically Active, Fully Hydrolysable Polymersomes. Angew. Chem. Int. Ed. Engl. 2019, 58, 4581–4586. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J.D.; Ward, J.R.; Avila-Olias, M.; Battaglia, G.; Renshaw, S.A. Targeting Neutrophilic Inflammation Using Polymersome-Mediated Cellular Delivery. J. Immunol. 2017, 198, 3596–3604. [Google Scholar] [CrossRef] [PubMed]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef]
- Canton, I.; Battaglia, G. Endocytosis at the nanoscale. Chem. Soc. Rev. 2012, 41, 2718–2739. [Google Scholar] [CrossRef]
- Mircioiu, C.; Voicu, V.; Anuta, V.; Tudose, A.; Celia, C.; Paolino, D.; Fresta, M.; Sandulovici, R.; Mircioiu, I. Mathematical Modeling of Release Kinetics from Supramolecular Drug Delivery Systems. Pharmaceutics 2019, 11, 140. [Google Scholar] [CrossRef]
- Mhlanga, N.; Ray, S.S. Kinetic models for the release of the anticancer drug doxorubicin from biodegradable polylactide/metal oxide-based hybrids. Int. J. Biol. Macromol. 2015, 72, 1301–1307. [Google Scholar] [CrossRef]
- Chanput, W.; Mes, J.J.; Wichers, H.J. THP-1 cell line: An in vitro cell model for immune modulation approach. Int. Immunopharmacol. 2014, 23, 37–45. [Google Scholar] [CrossRef]
- Skov, L.; Beurskens, F.J.; Zachariae, C.O.; Reitamo, S.; Teeling, J.; Satijn, D.; Knudsen, K.M.; Boot, E.P.; Hudson, D.; Baadsgaard, O.; et al. IL-8 as antibody therapeutic target in inflammatory diseases: Reduction of clinical activity in palmoplantar pustulosis. J. Immunol. 2008, 181, 669–679. [Google Scholar] [CrossRef]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef]
- Treede, I.; Braun, A.; Sparla, R.; Kuhnel, M.; Giese, T.; Turner, J.; Anes, E.; Kulaksiz, H.; Fullekrug, J.; Stremmel, W.; et al. Anti-inflammatory effects of phosphatidylcholine. J. Biol. Chem. 2007, 282, 27155–27164. [Google Scholar] [CrossRef] [PubMed]
- Linton, M.F.; Tao, H.; Linton, E.F.; Yancey, P.G. SR-BI: A Multifunctional Receptor in Cholesterol Homeostasis and Atherosclerosis. Trends Endocrinol. Metab. 2017, 28, 461–472. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gouveia, V.M.; Rizzello, L.; Nunes, C.; Poma, A.; Ruiz-Perez, L.; Oliveira, A.; Reis, S.; Battaglia, G. Macrophage Targeting pH Responsive Polymersomes for Glucocorticoid Therapy. Pharmaceutics 2019, 11, 614. https://doi.org/10.3390/pharmaceutics11110614
Gouveia VM, Rizzello L, Nunes C, Poma A, Ruiz-Perez L, Oliveira A, Reis S, Battaglia G. Macrophage Targeting pH Responsive Polymersomes for Glucocorticoid Therapy. Pharmaceutics. 2019; 11(11):614. https://doi.org/10.3390/pharmaceutics11110614
Chicago/Turabian StyleGouveia, Virgínia M., Loris Rizzello, Claudia Nunes, Alessandro Poma, Lorena Ruiz-Perez, António Oliveira, Salette Reis, and Giuseppe Battaglia. 2019. "Macrophage Targeting pH Responsive Polymersomes for Glucocorticoid Therapy" Pharmaceutics 11, no. 11: 614. https://doi.org/10.3390/pharmaceutics11110614
APA StyleGouveia, V. M., Rizzello, L., Nunes, C., Poma, A., Ruiz-Perez, L., Oliveira, A., Reis, S., & Battaglia, G. (2019). Macrophage Targeting pH Responsive Polymersomes for Glucocorticoid Therapy. Pharmaceutics, 11(11), 614. https://doi.org/10.3390/pharmaceutics11110614