Development of Injectable PEGylated Liposome Encapsulating Disulfiram for Colorectal Cancer Treatment

 ,
, 
Abstract
:
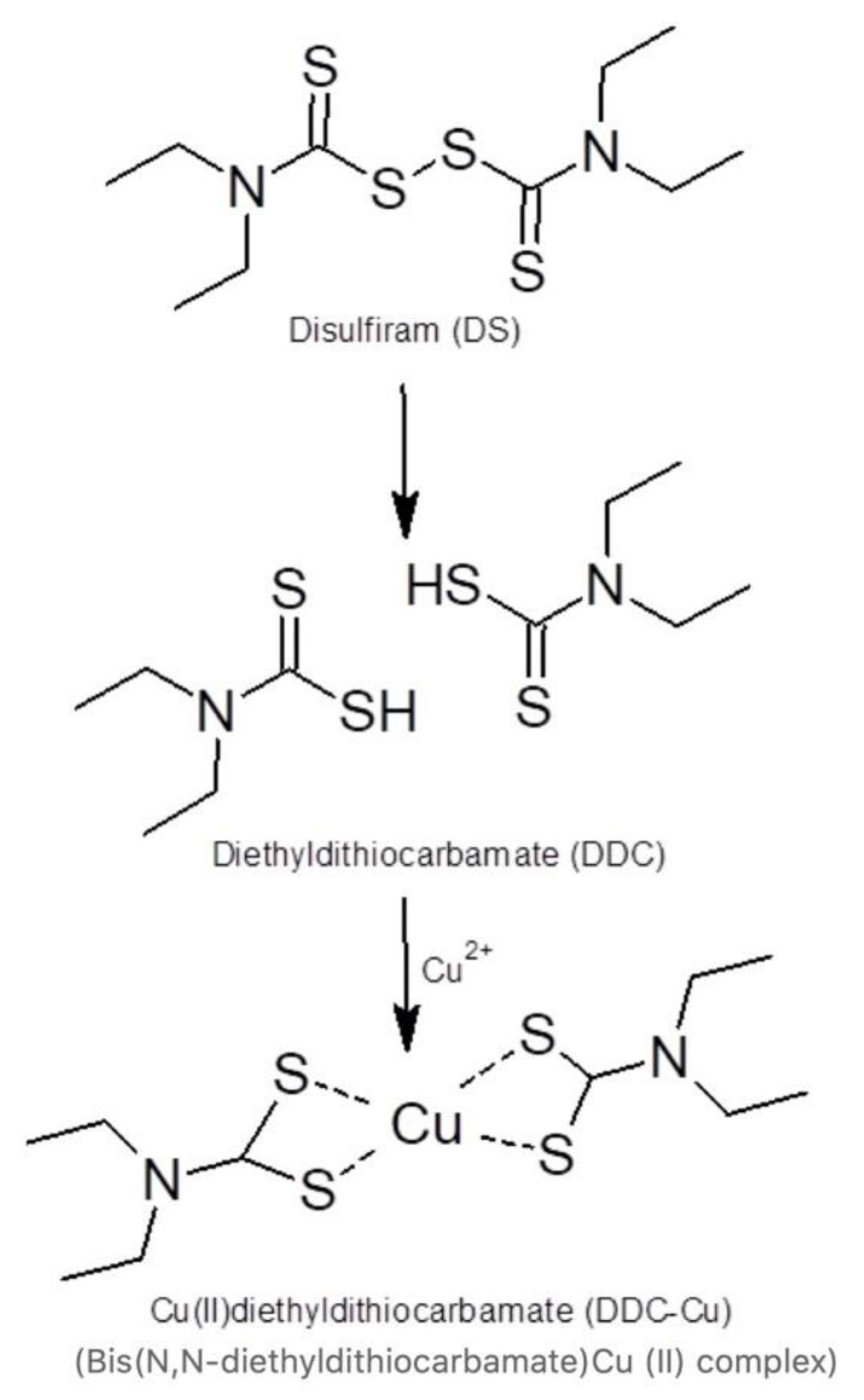
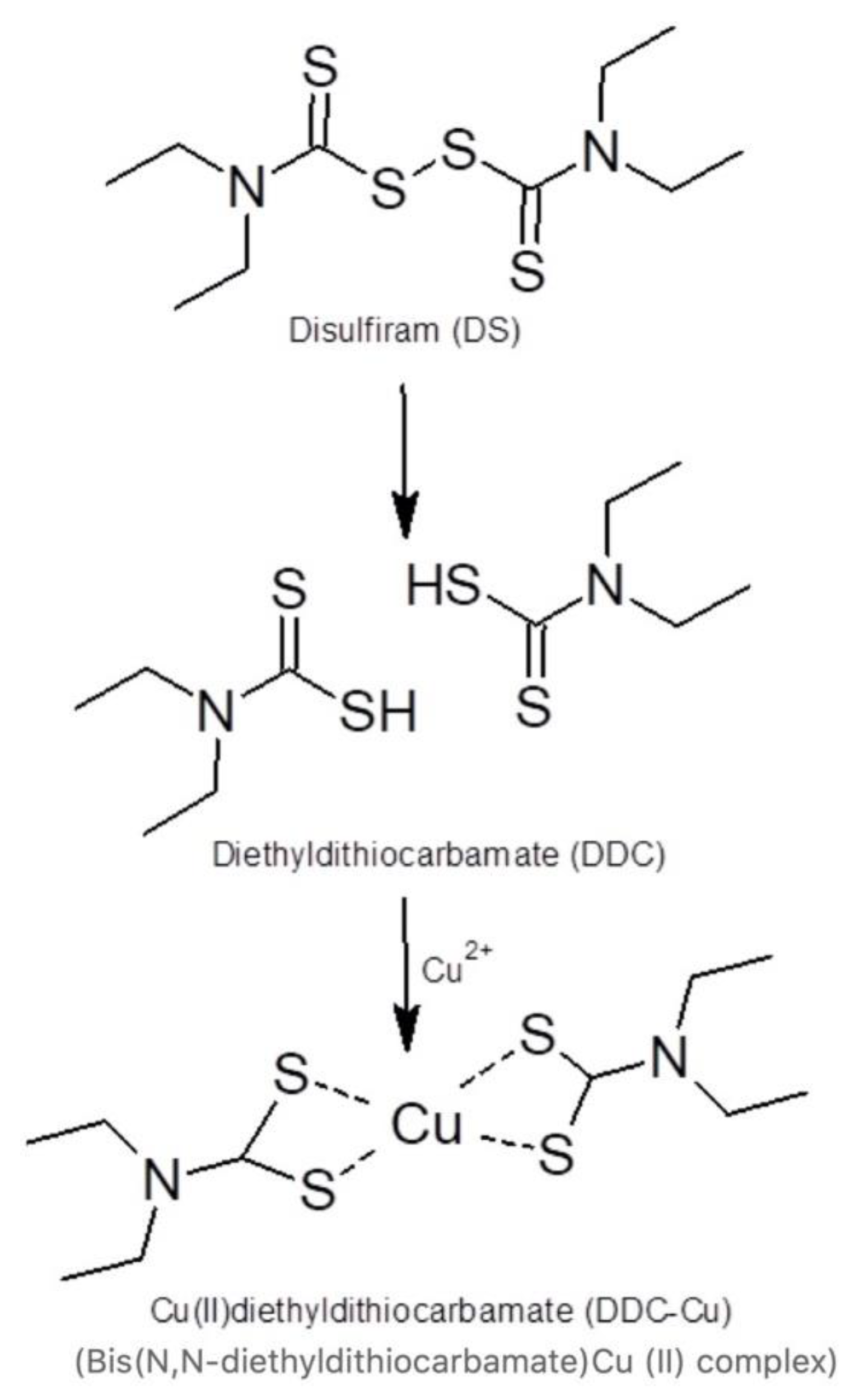
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
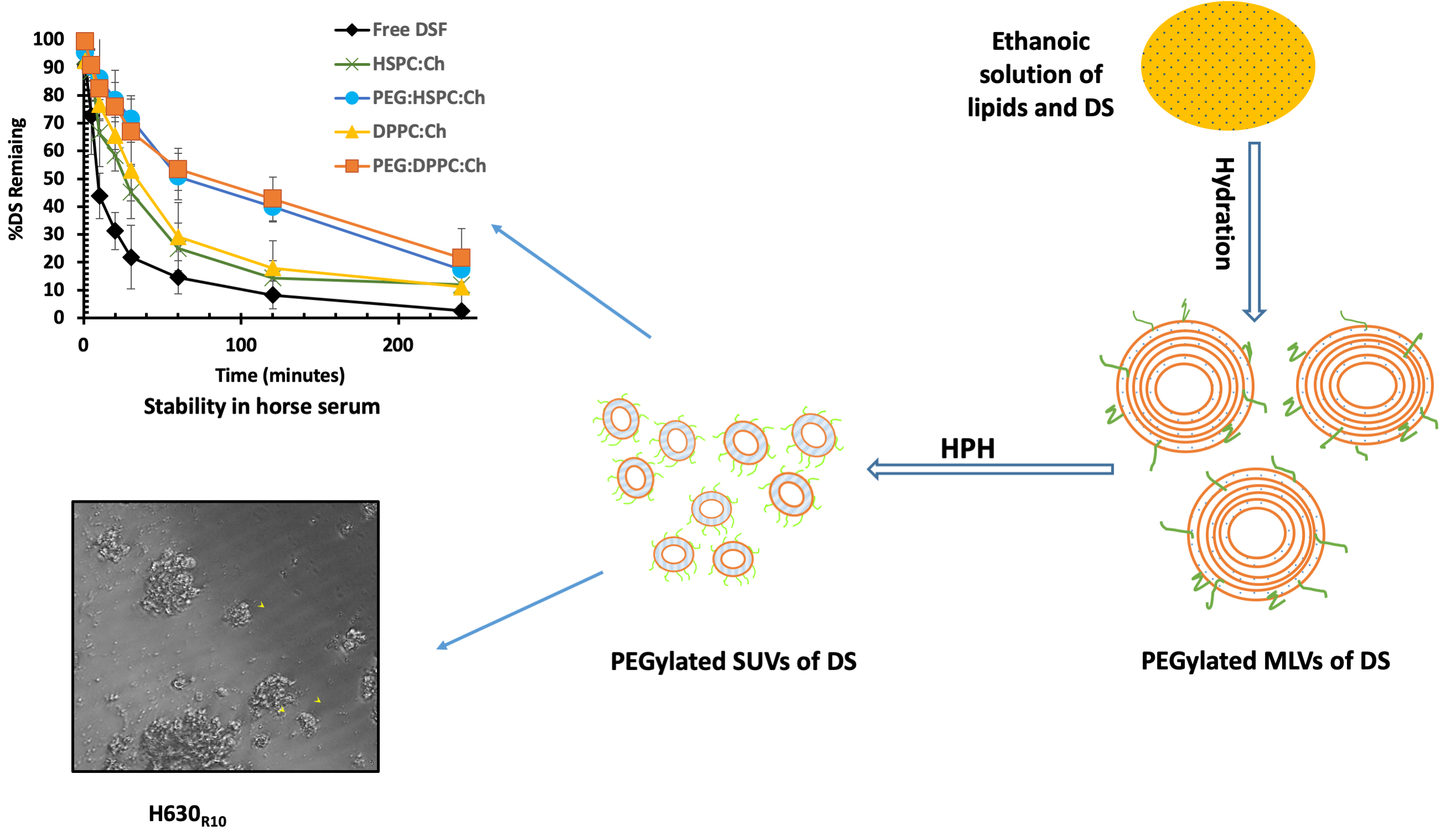
2.2.1. Preparation of Liposomes
2.2.2. Size Reduction of Liposomes
2.2.3. Photon Correlation Spectroscopy (PCS) Analysis after Size Reduction
2.2.4. Determination of Drug Entrapment Efficiency
2.2.5. Stability of Encapsulated Disulfiram in Horse Serum
2.2.6. Cytotoxicity Study (MTT Assay)
2.2.7. Statistical Analysis
3. Results and Discussion
3.1. Size Analysis of Liposomes
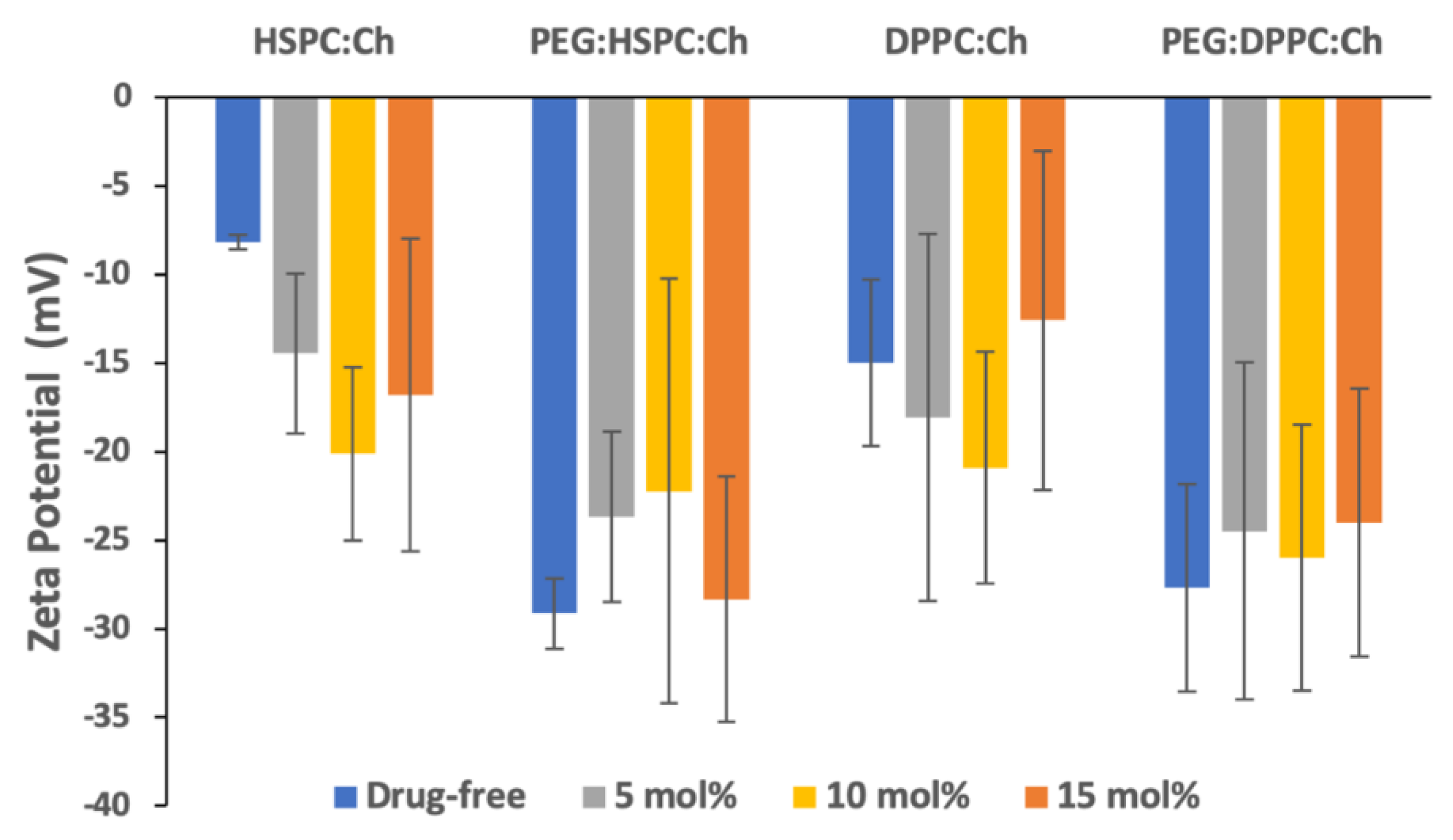
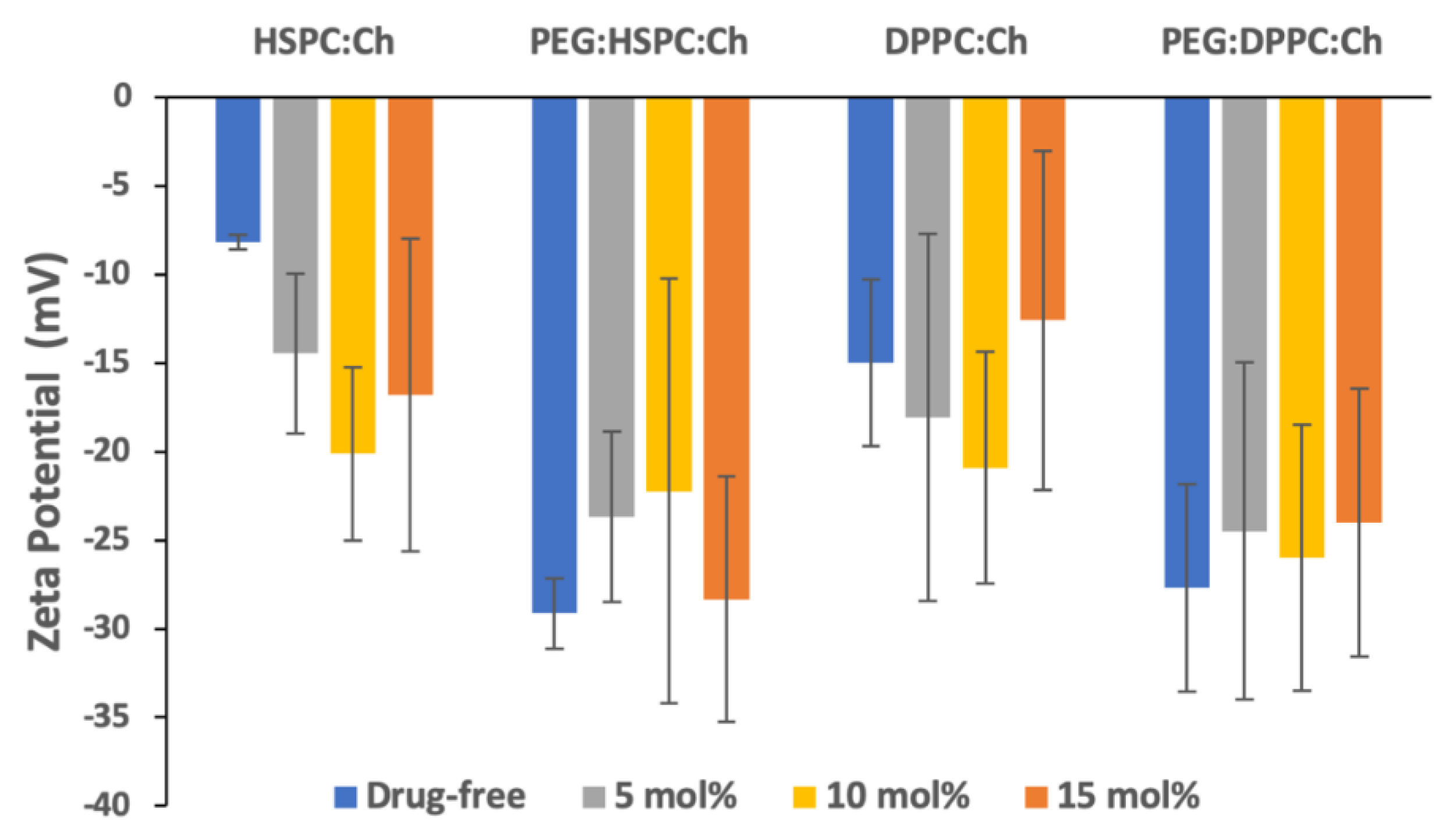
3.2. Zeta Potential Analysis
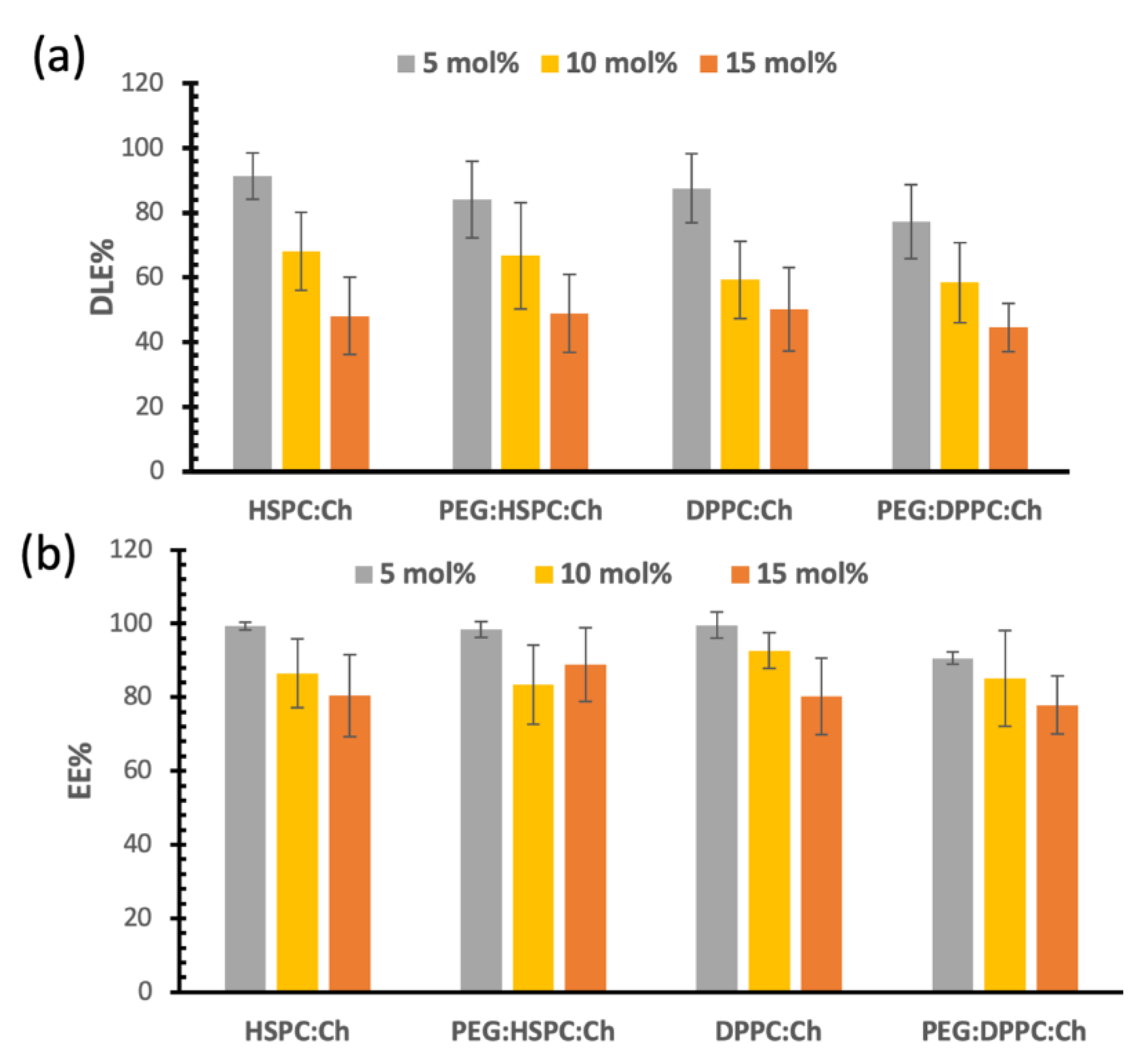
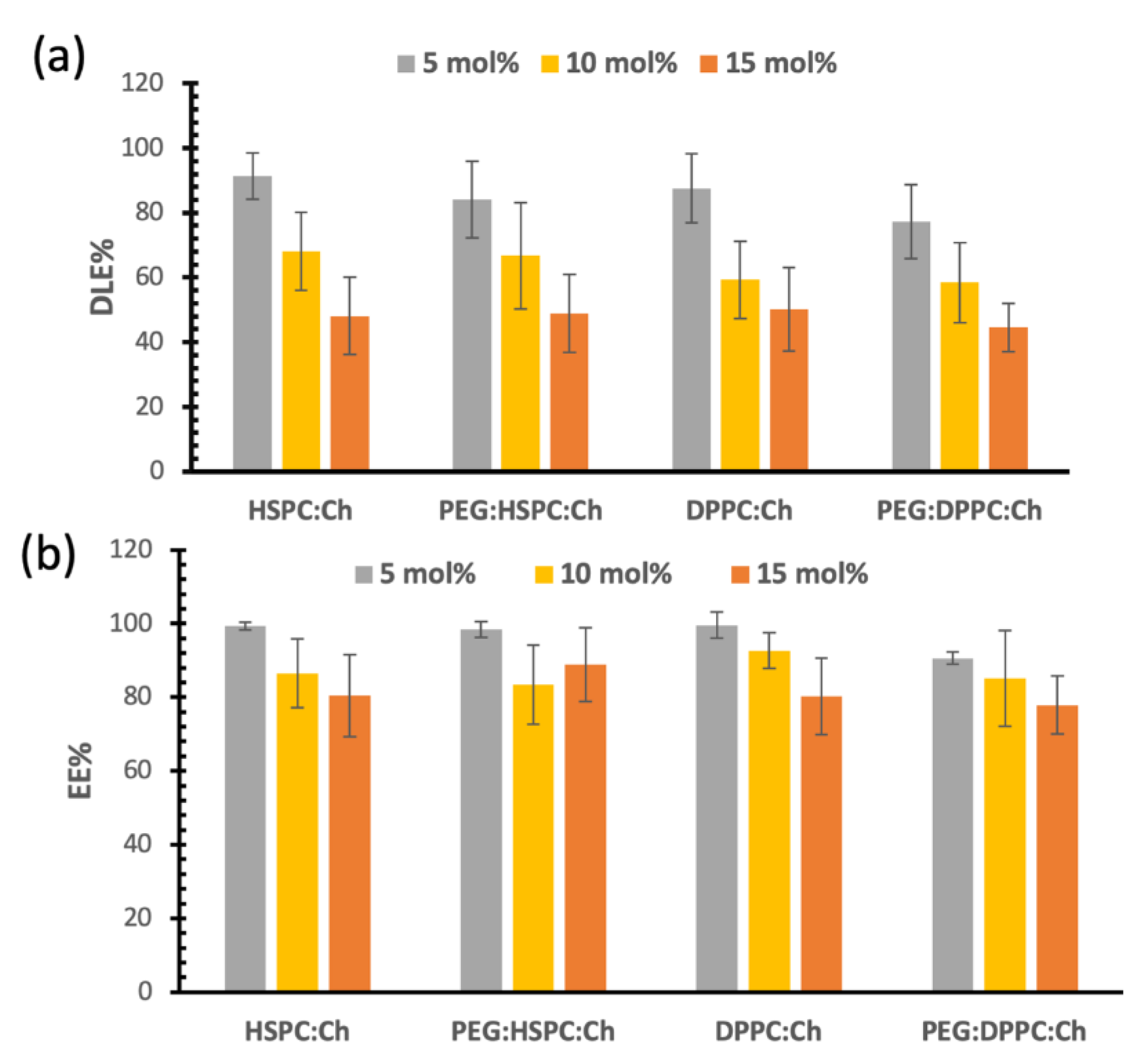
3.3. Drug Loading and Entrapment Efficiencies of DS in Liposomes
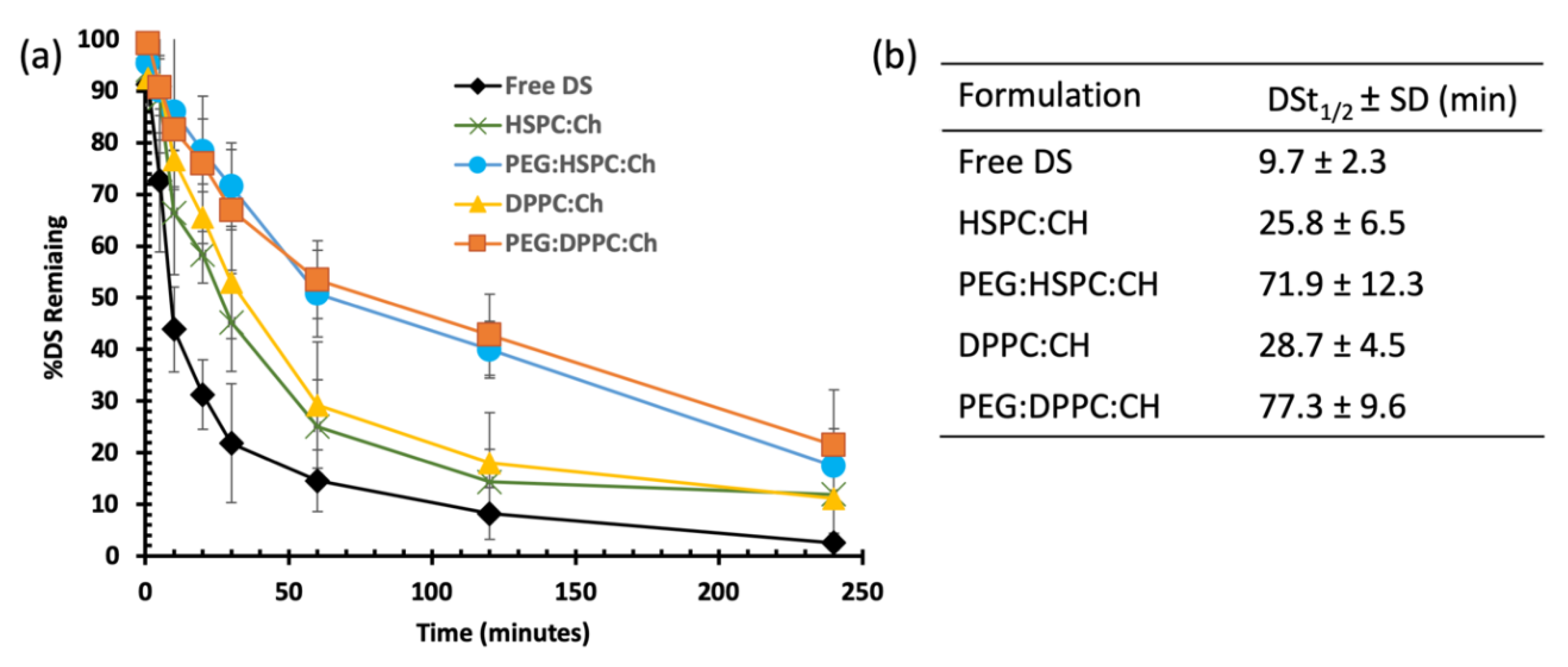
3.4. Stability Studies in Horse Serum
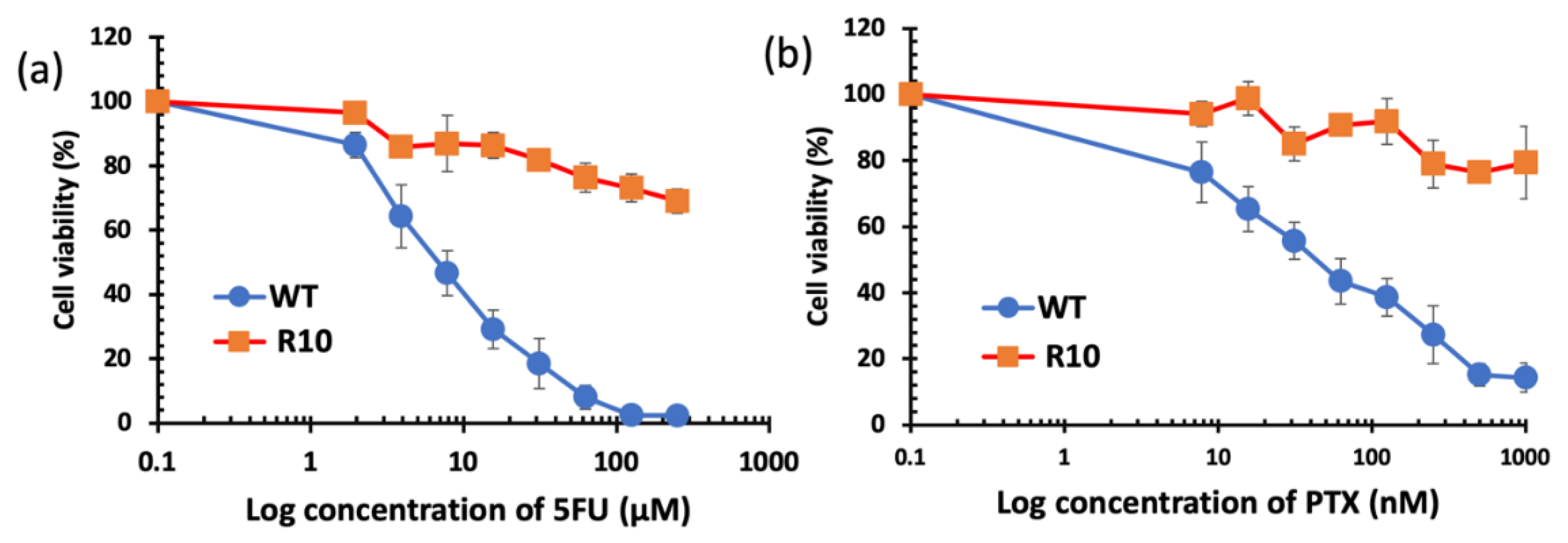
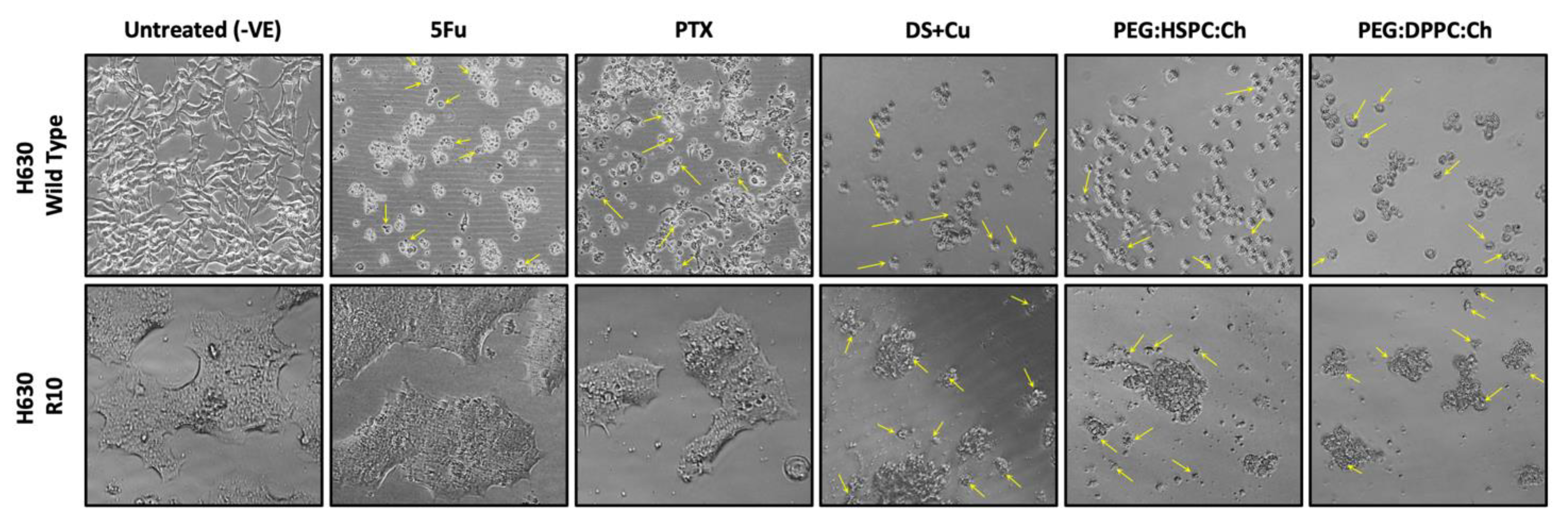
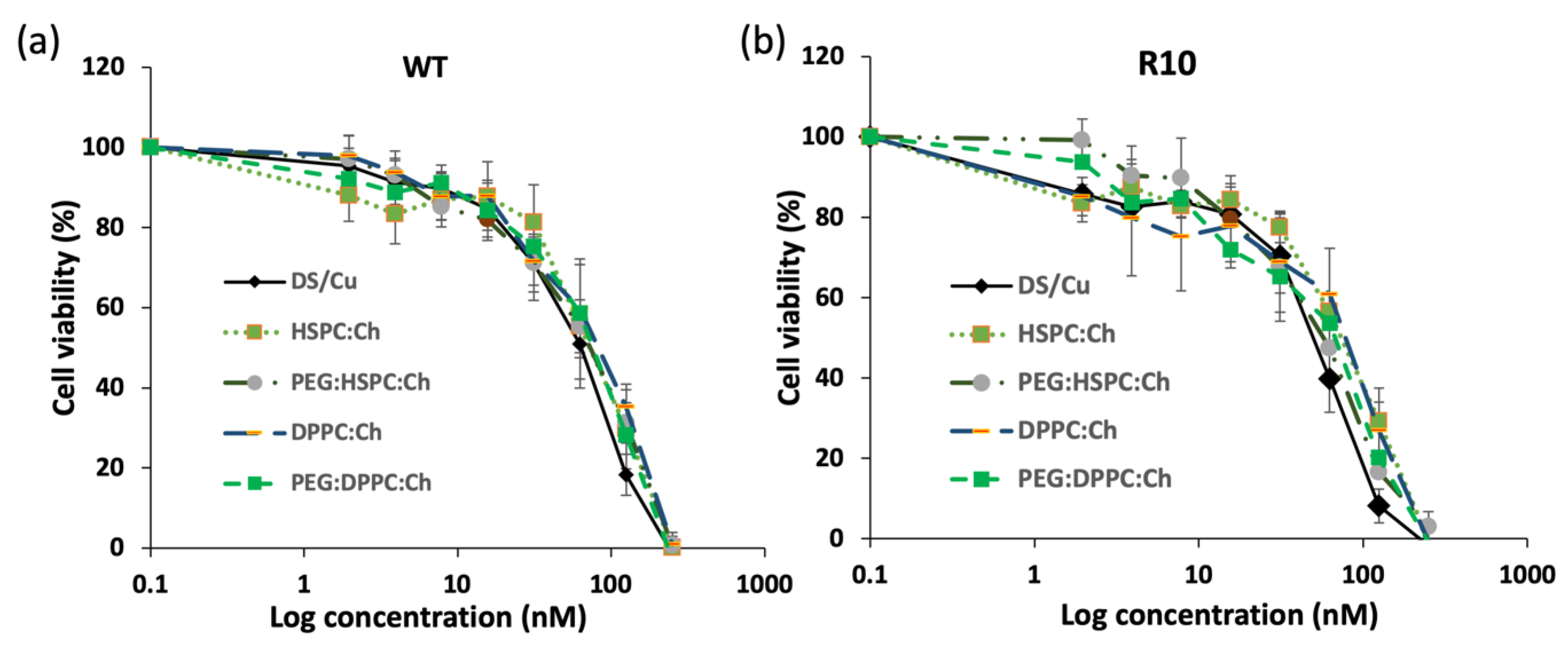
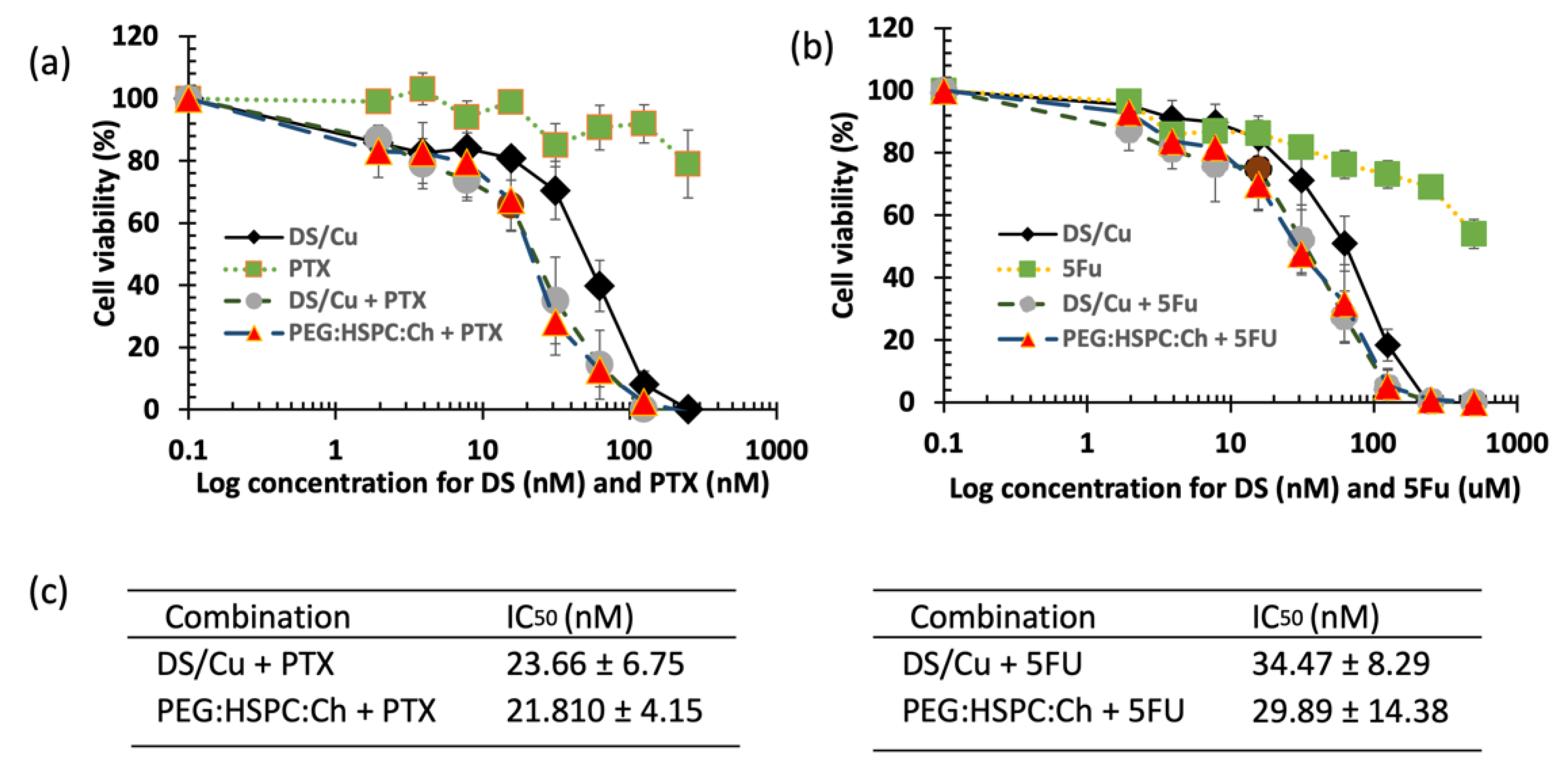
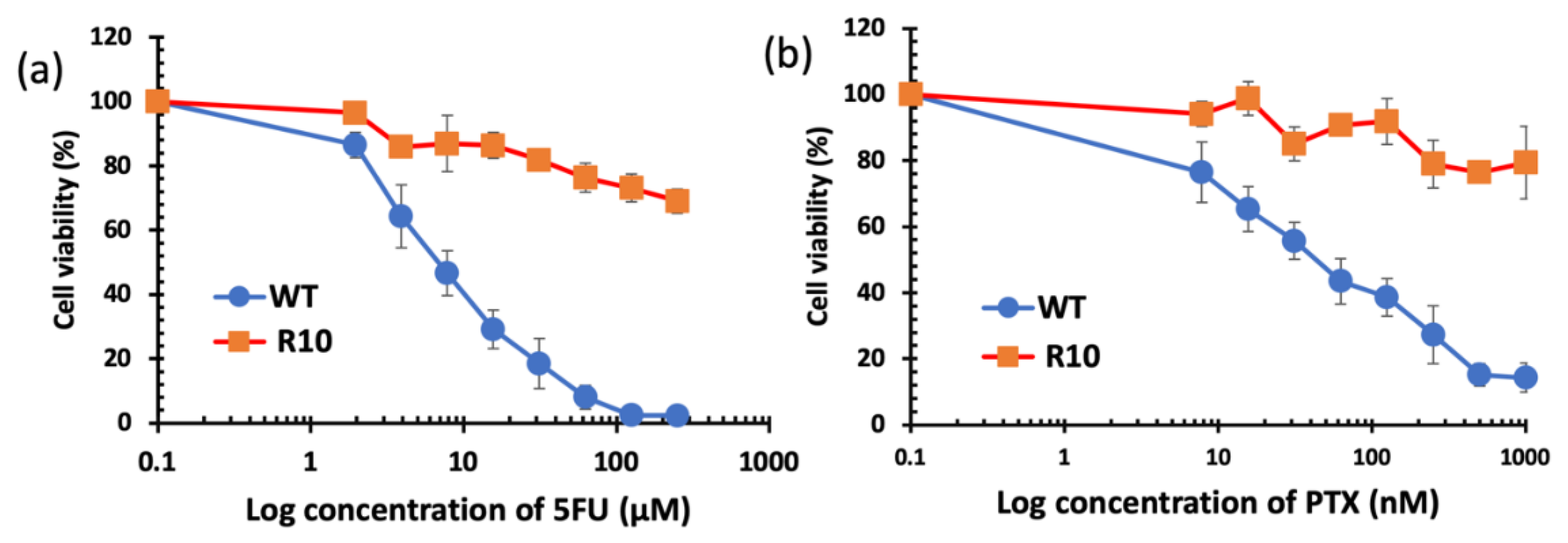
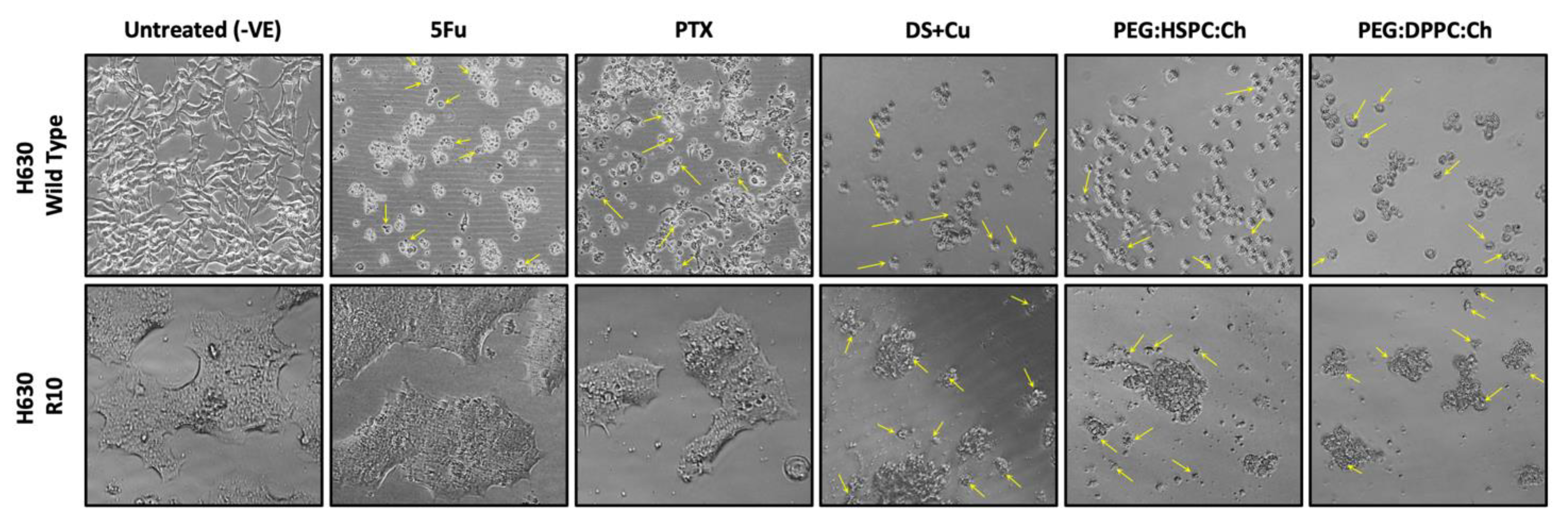
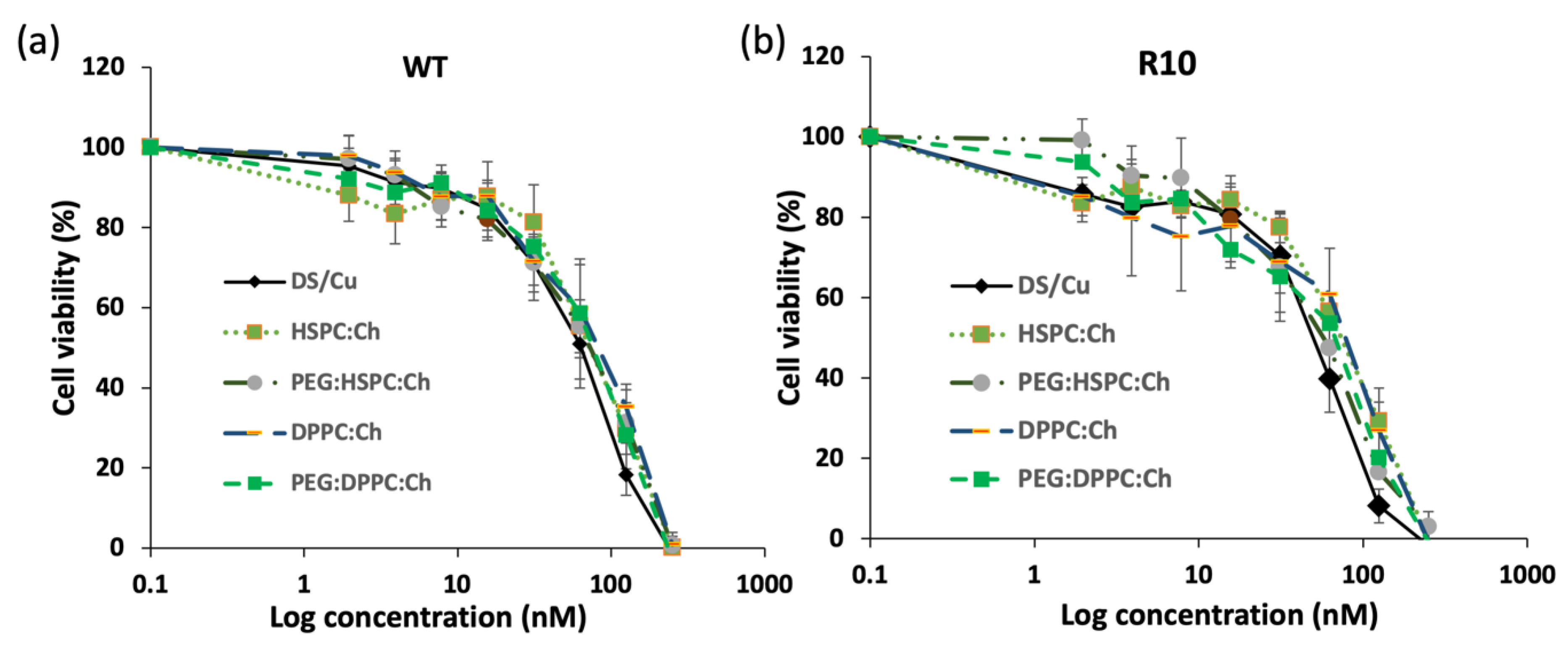
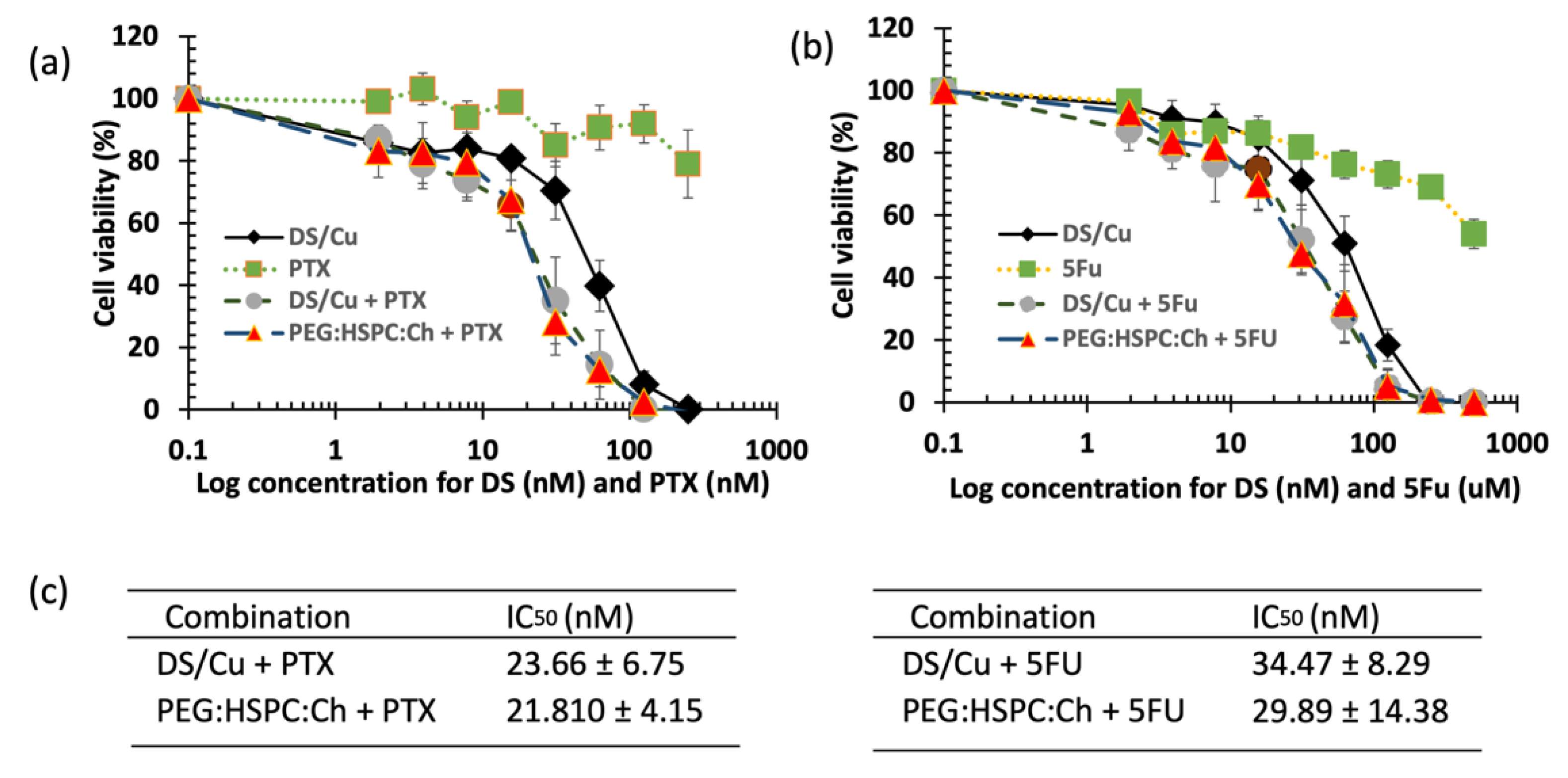
3.5. Cytotoxicity Studies
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Walker, I.; Newell, H. Do molecularly targeted agents in oncology have reduced attrition rates? Nat. Rev. Drug Discov. 2009, 8, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Reed, M.D. The Rescue and Repurposing of Pharmaceuticals: Augmenting the Drug Development Paradigm. J. Pediatr. Pharmacol. Ther. 2016, 21, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Wang, Z.; Brown, S.; Kannappan, V.; Tawari, P.E.; Jiang, W.; Irache, J.M.; Tang, J.Z.; Britland, S.; Armesilla, A.L.; et al. Liposome encapsulated Disulfiram inhibits NFκB pathway and targets breast cancer stem cells in vitro and in vivo. Oncotarget 2014, 5, 7471–7485. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Kumar, I.S.; Brown, S.; Kannappan, V.; Tawari, P.E.; Tang, J.Z.; Jiang, W.; Armesilla, A.L.; Darling, J.L.; Wang, W. Disulfiram targets cancer stem-like cells and reverses resistance and cross-resistance in acquired paclitaxel-resistant triple-negative breast cancer cells. Br. J. Cancer 2013, 109, 1876–1885. [Google Scholar] [CrossRef]
- Guo, X.; Xu, B.; Pandey, S.; Goessl, E.; Brown, J.; Armesilla, A.L.; Darling, J.L.; Wang, W. Disulfiram/copper complex inhibiting NFkappaB activity and potentiating cytotoxic effect of gemcitabine on colon and breast cancer cell lines. Cancer Lett. 2010, 290, 104–113. [Google Scholar] [CrossRef]
- Liu, P.; Brown, S.; Goktug, T.; Channathodiyil, P.; Kannappan, V.; Hugnot, J.-P.; Guichet, P.-O.; Bian, X.; Armesilla, A.L.; Darling, J.L.; et al. Cytotoxic effect of disulfiram/copper on human glioblastoma cell lines and ALDH-positive cancer-stem-like cells. Br. J. Cancer 2012, 107, 1488–1497. [Google Scholar] [CrossRef]
- Viola-Rhenals, M.; Patel, K.R.; Jaimes-Santamaria, L.; Wu, G.; Liu, J.; Dou, Q.P. Recent Advances in Antabuse (Disulfiram): The Importance of its Metal-binding Ability to its Anticancer Activity. Curr. Med. Chem. 2018, 25, 506–524. [Google Scholar] [CrossRef]
- Tawari, P.E.; Wang, Z.; Najlah, M.; Tsang, C.W.; Kannappan, V.; Liu, P.; McConville, C.; He, B.; Armesilla, A.L.; Wang, W. The cytotoxic mechanisms of disulfiram and copper(ii) in cancer cells. Toxicol. Res. 2015, 4, 1439–1442. [Google Scholar] [CrossRef]
- Wang, F.; Jiao, P.; Qi, M.; Frezza, M.; Dou, Q.P.; Yan, B. Turning Tumor-Promoting Copper into an Anti-Cancer Weapon via High-Throughput Chemistry. Curr. Med. Chem. 2010, 17, 2685–2698. [Google Scholar] [CrossRef]
- Safi, R.; Nelson, E.R.; Chitneni, S.K.; Franz, K.J.; George, D.J.; Zalutsky, M.R.; McDonnell, D.P. Copper signaling axis as a target for prostate cancer therapeutics. Cancer Res. 2014, 74, 5819–5831. [Google Scholar] [CrossRef]
- Johansson, B. A review of the pharmacokinetics and pharmacodynamics of disulfiram and its metabolites. Acta Psychiatr. Scand. 1992, 86, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tan, J.; McConville, C.; Kannappan, V.; Tawari, P.E.; Brown, J.; Ding, J.; Armesilla, A.L.; Irache, J.M.; Mei, Q.-B.; et al. Poly lactic-co-glycolic acid controlled delivery of disulfiram to target liver cancer stem-like cells. Nanomedicine 2017, 13, 641–657. [Google Scholar] [CrossRef] [PubMed]
- Butcher, K.; Kannappan, V.; Kilari, R.S.; Morris, M.R.; McConville, C.; Armesilla, A.L.; Wang, W. Investigation of the key chemical structures involved in the anticancer activity of disulfiram in A549 non-small cell lung cancer cell line. BMC Cancer 2018, 18, 753. [Google Scholar] [CrossRef] [PubMed]
- Najlah, M.; Ahmed, Z.; Iqbal, M.; Wang, Z.; Tawari, P.; Wang, W.; McConville, C. Development and characterisation of disulfiram-loaded PLGA nanoparticles for the treatment of non-small cell lung cancer. Eur. J. Pharm. Biopharm. 2017, 112, 224–233. [Google Scholar] [CrossRef]
- Koudelka, S.; Turánek, J. Liposomal paclitaxel formulations. J. Control. Release 2012, 163, 322–334. [Google Scholar] [CrossRef]
- Brown, S.; Khan, D.R. The treatment of breast cancer using liposome technology. J. Drug Deliv. 2012, 2012, 212965. [Google Scholar] [CrossRef]
- Pauli, G.; Tang, W.-L.; Li, S.-D. Development and Characterization of the Solvent-Assisted Active Loading Technology (SALT) for Liposomal Loading of Poorly Water-Soluble Compounds. Pharmaceutics 2019, 11, 465. [Google Scholar] [CrossRef]
- Palchetti, S.; Caputo, D.; Digiacomo, L.; Capriotti, A.L.; Coppola, R.; Pozzi, D.; Caracciolo, G. Protein Corona Fingerprints of Liposomes: New Opportunities for Targeted Drug Delivery and Early Detection in Pancreatic Cancer. Pharmaceutics 2019, 11, 31. [Google Scholar] [CrossRef]
- Wang, W. Disulfiram Formulation and Uses Thereof. WO2012076897A1, 14 June 2012. [Google Scholar]
- Najlah, M.; Jain, M.; Wan, K.-W.; Ahmed, W.; Alhnan, M.A.; Phoenix, D.A.; Taylor, K.M.G.; Elhissi, A. Ethanol-based proliposome delivery systems of paclitaxel for in vitro application against brain cancer cells. J. Liposome Res. 2018, 28, 74–85. [Google Scholar] [CrossRef]
- Perrett, S.; Golding, M.; Williams, W.P. A Simple Method for the Preparation of Liposomes for Pharmaceutical Applications: Characterization of the Liposomes. J. Pharm. Pharmacol. 1991, 43, 154–161. [Google Scholar] [CrossRef]
- Subramanian, S.; Khan, I.; Korale, O.; Alhnan, M.A.; Ahmed, W.; Najlah, M.; Taylor, K.M.; Elhissi, A. A simple approach to predict the stability of phospholipid vesicles to nebulization without performing aerosolization studies. Int. J. Pharm. 2016, 502, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Najlah, M.; Hidayat, K.; Omer, H.K.; Mwesigwa, E.; Ahmed, W.; AlObaidy, K.G.; Phoenix, D.A.; Elhissi, A. A facile approach to manufacturing non-ionic surfactant nanodipsersions using proniosome technology and high-pressure homogenization. J. Liposome Res. 2015, 25, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Nii, T.; Ishii, F. Encapsulation efficiency of water-soluble and insoluble drugs in liposomes prepared by the microencapsulation vesicle method. Int. J. Pharm. 2005, 298, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Ishii, F.; Nagasaka, Y. Simple and Convenient Method for Estimation of Marker Entrapped in Liposomes. J. Dispers. Sci. Technol. 2001, 22, 97–101. [Google Scholar] [CrossRef]
- Ramadhani, N.; Shabir, M.; McConville, C. Preparation and characterisation of Kolliphor® P 188 and P 237 solid dispersion oral tablets containing the poorly water soluble drug disulfiram. Int. J. Pharm. 2014, 475, 514–522. [Google Scholar] [CrossRef]
- Shergill, M.; Patel, M.; Khan, S.; Bashir, A.; McConville, C. Development and characterisation of sustained release solid dispersion oral tablets containing the poorly water soluble drug disulfiram. Int. J. Pharm. 2016, 497, 3–11. [Google Scholar] [CrossRef]
- Song, W.; Tang, Z.; Lei, T.; Wen, X.; Wang, G.; Zhang, D.; Deng, M.; Tang, X.; Chen, X. Stable loading and delivery of disulfiram with mPEG-PLGA/PCL mixed nanoparticles for tumor therapy. Nanomed.: Nanotechnol. Biol. Med. 2016, 12, 377–386. [Google Scholar] [CrossRef]
- Plumb, J.A.; Milroy, R.; Kaye, S.B. Effects of the pH dependence of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide-formazan absorption on chemosensitivity determined by a novel tetrazolium-based assay. Cancer Res. 1989, 49, 4435–4440. [Google Scholar]
- Urakami, K.; Kobayashi, C.; Miyazaki, Y.; Nishijima, K.; Yoshimura, Y.; Hashimoto, K. Degradation products generated by sonication of benzyl alcohol, a sample preparation solvent for the determination of residual solvents in pharmaceutical bulks, on capillary gas chromatography. Chem. Pharm. Bull. 2000, 48, 1299–1303. [Google Scholar] [CrossRef]
- Mui, B.; Chow, L.; Hope, M.J. Extrusion technique to generate liposomes of defined size. Meth. Enzymol. 2003, 367, 3–14. [Google Scholar]
- Barnadas Rodríguez, R.; Sabés Xamaní, M. Liposomes prepared by high-pressure homogenizers. Meth. Enzymol. 2003, 367, 28–46. [Google Scholar] [PubMed]
- Sriwongsitanont, S.; Ueno, M. Effect of a PEG lipid (DSPE-PEG2000) and freeze-thawing process on phospholipid vesicle size and lamellarity. Colloid Polym. Sci. 2004, 282, 753–760. [Google Scholar] [CrossRef]
- Kontogiannopoulos, K.N.; Tsermentseli, S.K.; Assimopoulou, A.N.; Papageorgiou, V.P. Sterically stabilized liposomes as a potent carrier for shikonin. J. Liposome Res. 2014, 24, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Tenzel, R.A.; Aitcheson, D.F. Preparation of Uniform-Size Liposomes and Other Lipid Structures. WO1989011335A1, 30 November 1989. [Google Scholar]
- Dadashzadeh, S.; Mirahmadi, N.; Babaei, M.H.; Vali, A.M. Peritoneal retention of liposomes: Effects of lipid composition, PEG coating and liposome charge. J. Control. Release 2010, 148, 177–186. [Google Scholar] [CrossRef]
- Tsermentseli, S.K.; Kontogiannopoulos, K.N.; Papageorgiou, V.P.; Assimopoulou, A.N. Comparative Study of PEGylated and Conventional Liposomes as Carriers for Shikonin. Fluids 2018, 3, 36. [Google Scholar] [CrossRef] [Green Version]
- Howard, F.B.; Levin, I.W. Lipid vesicle aggregation induced by cooling. Int. J. Mol. Sci. 2010, 11, 754–761. [Google Scholar] [CrossRef]
- Wang, C.-H.; Huang, Y.-Y. Encapsulating protein into preformed liposomes by ethanol-destabilized method. Artif. Cell Blood Substit. Immobil. Biotechnol 2003, 31, 303–312. [Google Scholar] [CrossRef]
- Khan, I.; Yousaf, S.; Subramanian, S.; Korale, O.; Alhnan, M.A.; Ahmed, W.; Taylor, K.M.G.; Elhissi, A. Proliposome powders prepared using a slurry method for the generation of beclometasone dipropionate liposomes. Int. J. Pharm. 2015, 496, 342–350. [Google Scholar] [CrossRef]
- Chen, J.; He, C.-Q.; Lin, A.-H.; Xu, F.; Wang, F.; Zhao, B.; Liu, X.; Chen, Z.-P.; Cai, B. Brucine-loaded liposomes composed of HSPC and DPPC at different ratios: In vitro and in vivo evaluation. Drug Dev. Ind. Pharm. 2014, 40, 244–251. [Google Scholar] [CrossRef]
- Torchilin, V.P.; Trubetskoy, V.S. Which polymers can make nanoparticulate drug carriers long-circulating? Adv. Drug Deliv. Rev. 1995, 16, 141–155. [Google Scholar] [CrossRef]
- Wang, X.; Song, Y.; Su, Y.; Tian, Q.; Li, B.; Quan, J.; Deng, Y. Are PEGylated liposomes better than conventional liposomes? A special case for vincristine. Drug Deliv. 2016, 23, 1092–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Immordino, M.L.; Dosio, F.; Cattel, L. Stealth liposomes: Review of the basic science, rationale, and clinical applications, existing and potential. Int. J. Nanomed. 2006, 1, 297–315. [Google Scholar]
- Barenholz, Y. Doxil®—The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Yang, L.; Yang, C.; Liu, Y.; Chen, Q.; Pan, W.; Cai, Q.; Luo, L.; Liu, L.; Jiang, S.; et al. Membrane Loaded Copper Oleate PEGylated Liposome Combined with Disulfiram for Improving Synergistic Antitumor Effect In Vivo. Pharm. Res. 2018, 35, 147. [Google Scholar] [CrossRef]
- Al-Suwayeh, S.A.; Tebbett, I.R.; Wielbo, D.; Brazeau, G.A. In Vitro-In Vivo Myotoxicity of Intramuscular Liposomal Formulations. Pharm. Res. 1996, 13, 1384–1388. [Google Scholar] [CrossRef]
- Liu, Y.; Ji, M.; Wong, M.K.; Joo, K.-I.; Wang, P. Enhanced therapeutic efficacy of iRGD-conjugated crosslinked multilayer liposomes for drug delivery. Biomed Res. Int. 2013, 2013, 378380. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ingredients | DSPE-PEG2000 | HSPC | DPPC | Ch | DS | |
|---|---|---|---|---|---|---|
| Formulation | ||||||
| HSPC:Ch | 0.0 (mol% *) | - | 1 ** | - | 1 | - |
| 5.0 | - | 1 | - | 1 | 0.11 | |
| 10.0 | - | 1 | - | 1 | 0.22 | |
| 15.0 | - | 1 | - | 1 | 0.36 | |
| PEG:HSPC:Ch | 0.0 | 0.1 | 0.9 | - | 1 | - |
| 5.0 | 0.1 | 0.9 | - | 1 | 0.11 | |
| 10.0 | 0.1 | 0.9 | - | 1 | 0.22 | |
| 15.0 | 0.1 | 0.9 | - | 1 | 0.36 | |
| DPPC:Ch | 0.0 | - | - | 1 | 1 | - |
| 5.0 | - | - | 1 | 1 | 0.11 | |
| 10.0 | - | - | 1 | 1 | 0.22 | |
| 15.0 | - | - | 1 | 1 | 0.36 | |
| PEG:DPPC:Ch | 0.0 | 0.1 | - | 0.9 | 1 | - |
| 5.0 | 0.1 | - | 0.9 | 1 | 0.11 | |
| 10.0 | 0.1 | - | 0.9 | 1 | 0.22 | |
| 15.0 | 0.1 | - | 0.9 | 1 | 0.36 |
| IC50 | H630 WT (nM) | H630 R10 (nM) |
|---|---|---|
| 5FU | 3420 ± 630.0 | >250,000 |
| PTX | 43.63 ± 15.21 | >1000 |
| DS/Cu | 57.736 ± 11.08 | 49.092 ± 8.20 |
| HSPC:Ch | 75.544 ± 22.68 | 73.094 ± 12.28 |
| PEG:HSPC:Ch | 69.076 ± 3.95 | 56.800 ± 3.21 |
| DPPC:Ch | 76.273 ± 10.97 | 64.69 ± 10.88 |
| PEG:DPPC:Ch | 71.289 ± 10.81 | 56.165 ± 21.16 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Najlah, M.; Said Suliman, A.; Tolaymat, I.; Kurusamy, S.; Kannappan, V.; Elhissi, A.M.A.; Wang, W. Development of Injectable PEGylated Liposome Encapsulating Disulfiram for Colorectal Cancer Treatment. Pharmaceutics 2019, 11, 610. https://doi.org/10.3390/pharmaceutics11110610
Najlah M, Said Suliman A, Tolaymat I, Kurusamy S, Kannappan V, Elhissi AMA, Wang W. Development of Injectable PEGylated Liposome Encapsulating Disulfiram for Colorectal Cancer Treatment. Pharmaceutics. 2019; 11(11):610. https://doi.org/10.3390/pharmaceutics11110610
Chicago/Turabian StyleNajlah, Mohammad, Ammar Said Suliman, Ibrahim Tolaymat, Sathishkumar Kurusamy, Vinodh Kannappan, Abdelbary M. A. Elhissi, and Weiguang Wang. 2019. "Development of Injectable PEGylated Liposome Encapsulating Disulfiram for Colorectal Cancer Treatment" Pharmaceutics 11, no. 11: 610. https://doi.org/10.3390/pharmaceutics11110610
APA StyleNajlah, M., Said Suliman, A., Tolaymat, I., Kurusamy, S., Kannappan, V., Elhissi, A. M. A., & Wang, W. (2019). Development of Injectable PEGylated Liposome Encapsulating Disulfiram for Colorectal Cancer Treatment. Pharmaceutics, 11(11), 610. https://doi.org/10.3390/pharmaceutics11110610