Principal Criteria for Evaluating the Quality, Safety and Efficacy of hMSC-Based Products in Clinical Practice: Current Approaches and Challenges

 , , ,
, , , 
Abstract
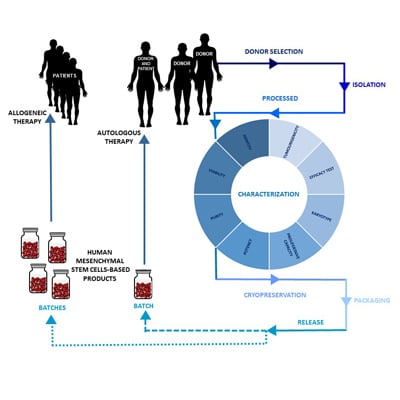
:
1. Introduction
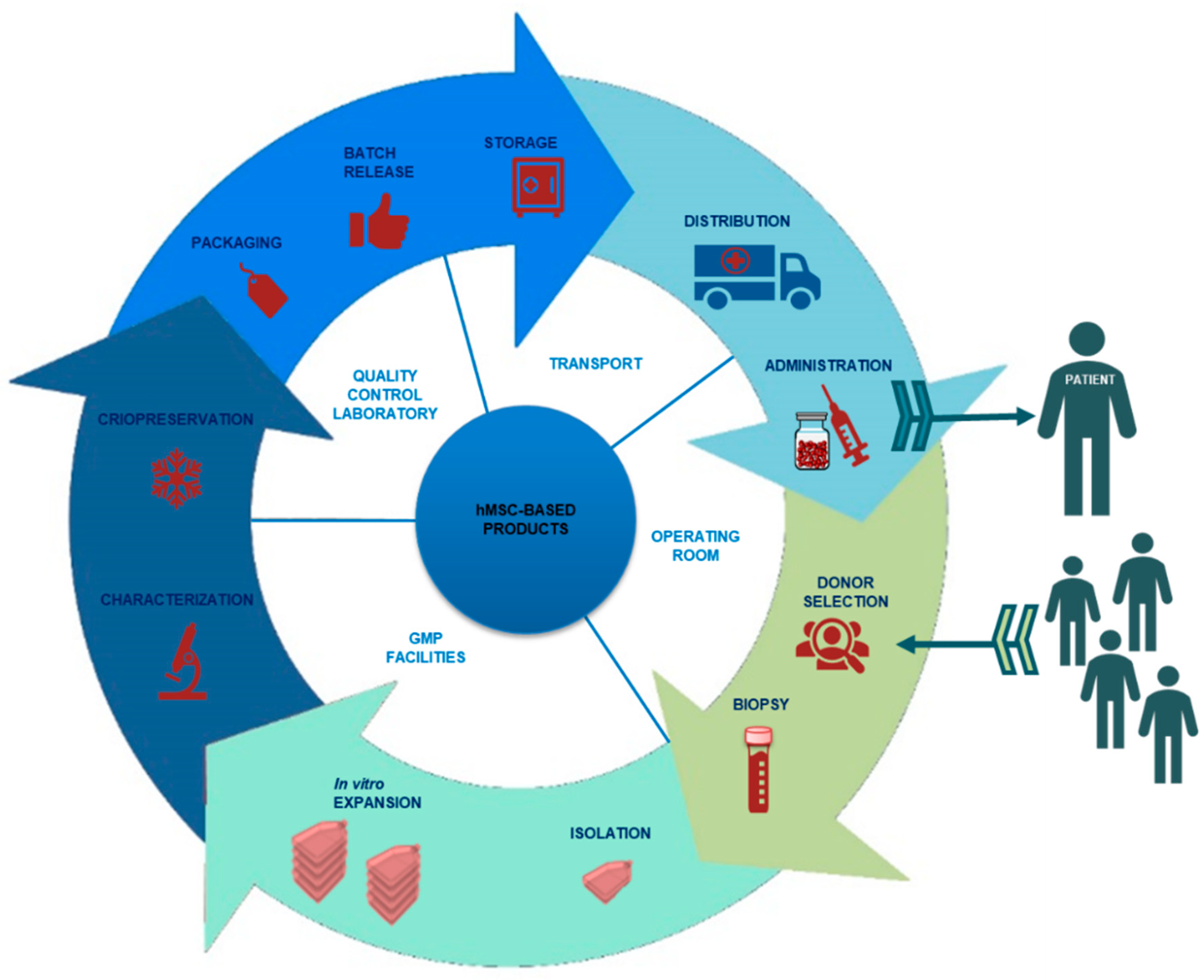
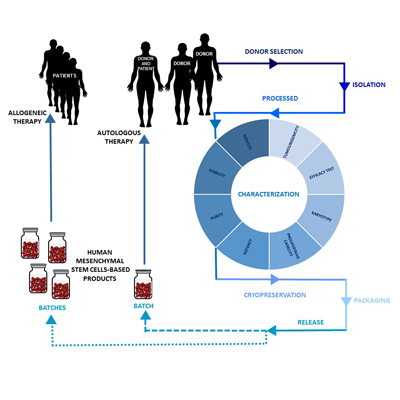
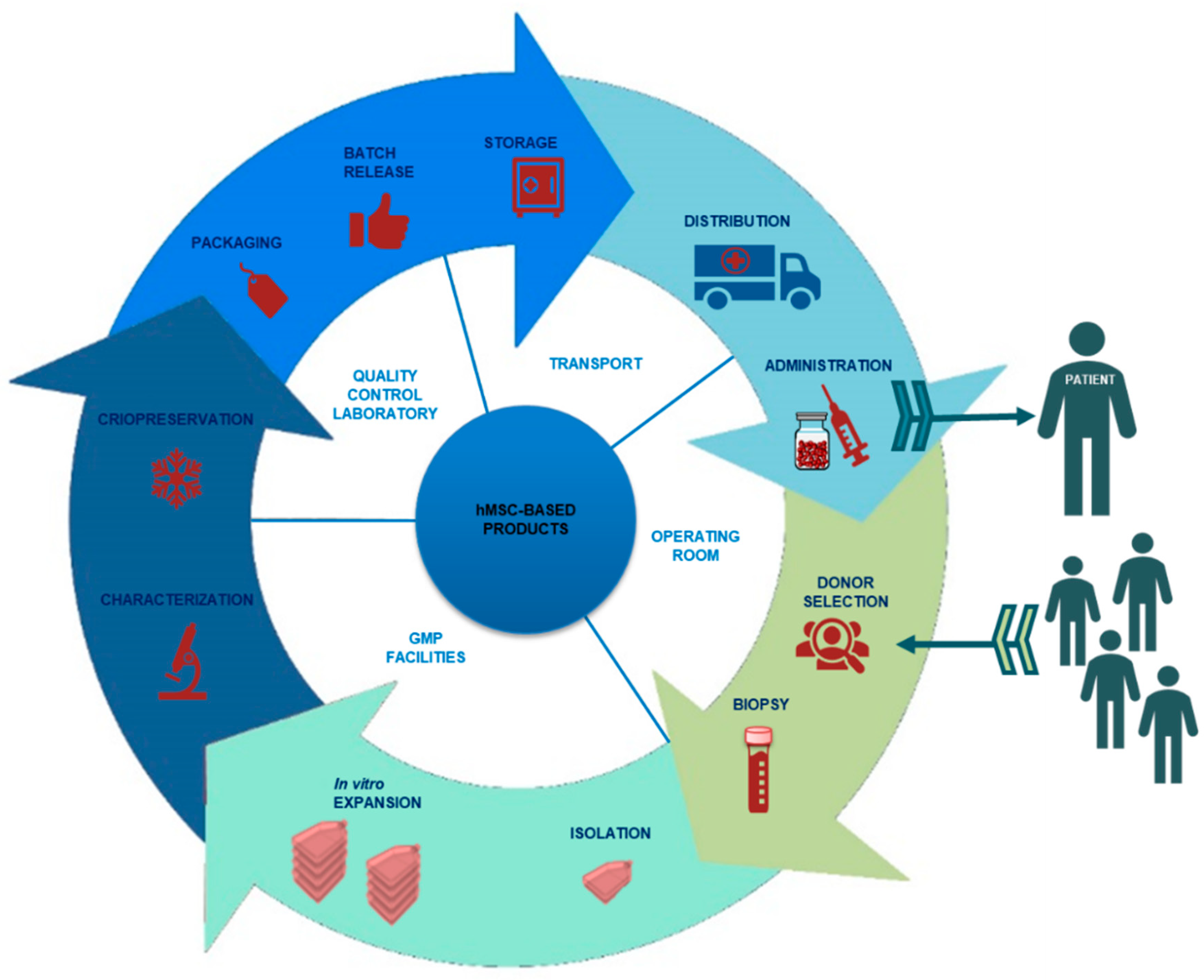
2. Design of an hMSC-Based Product Manufacturing Process
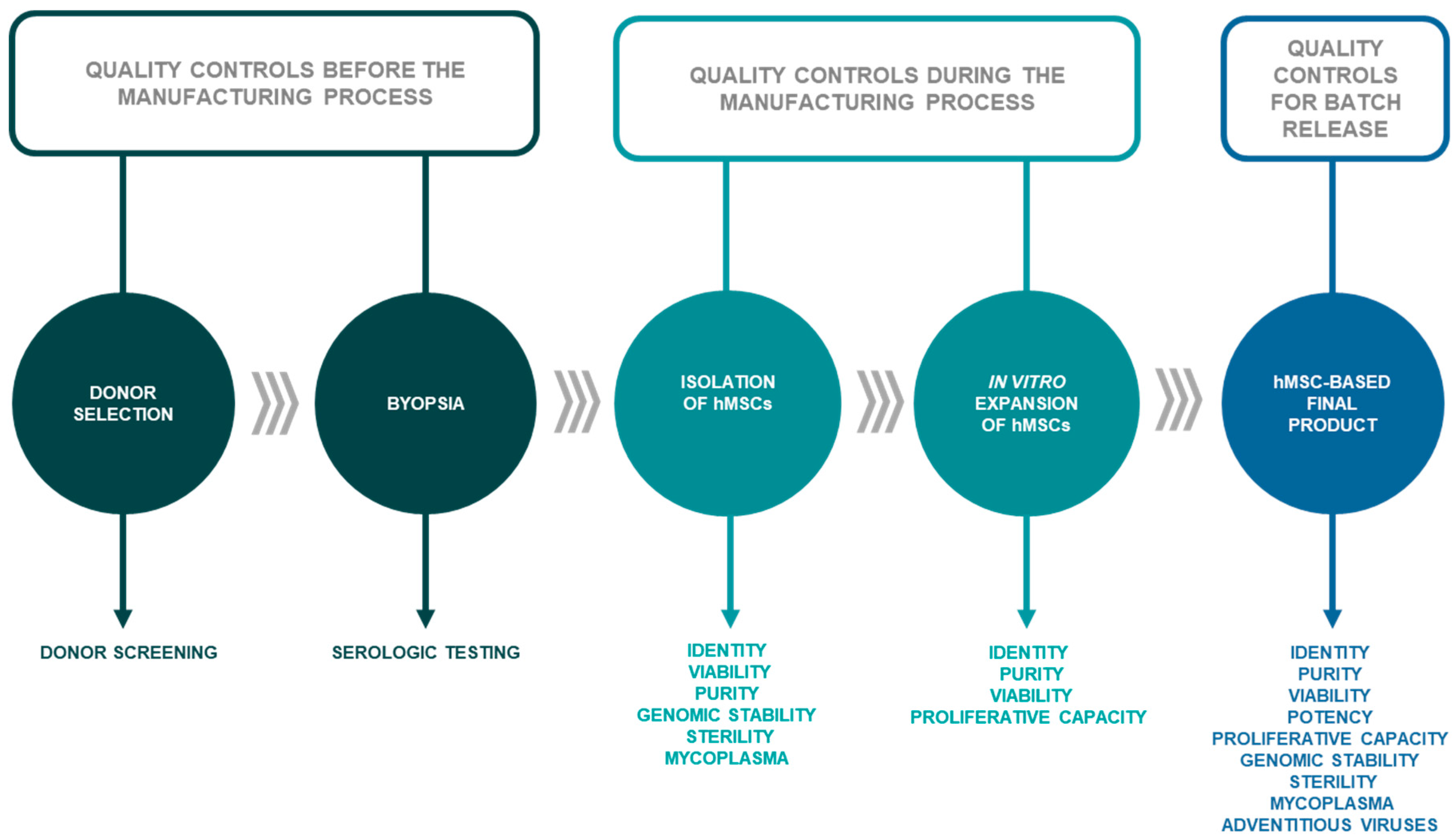
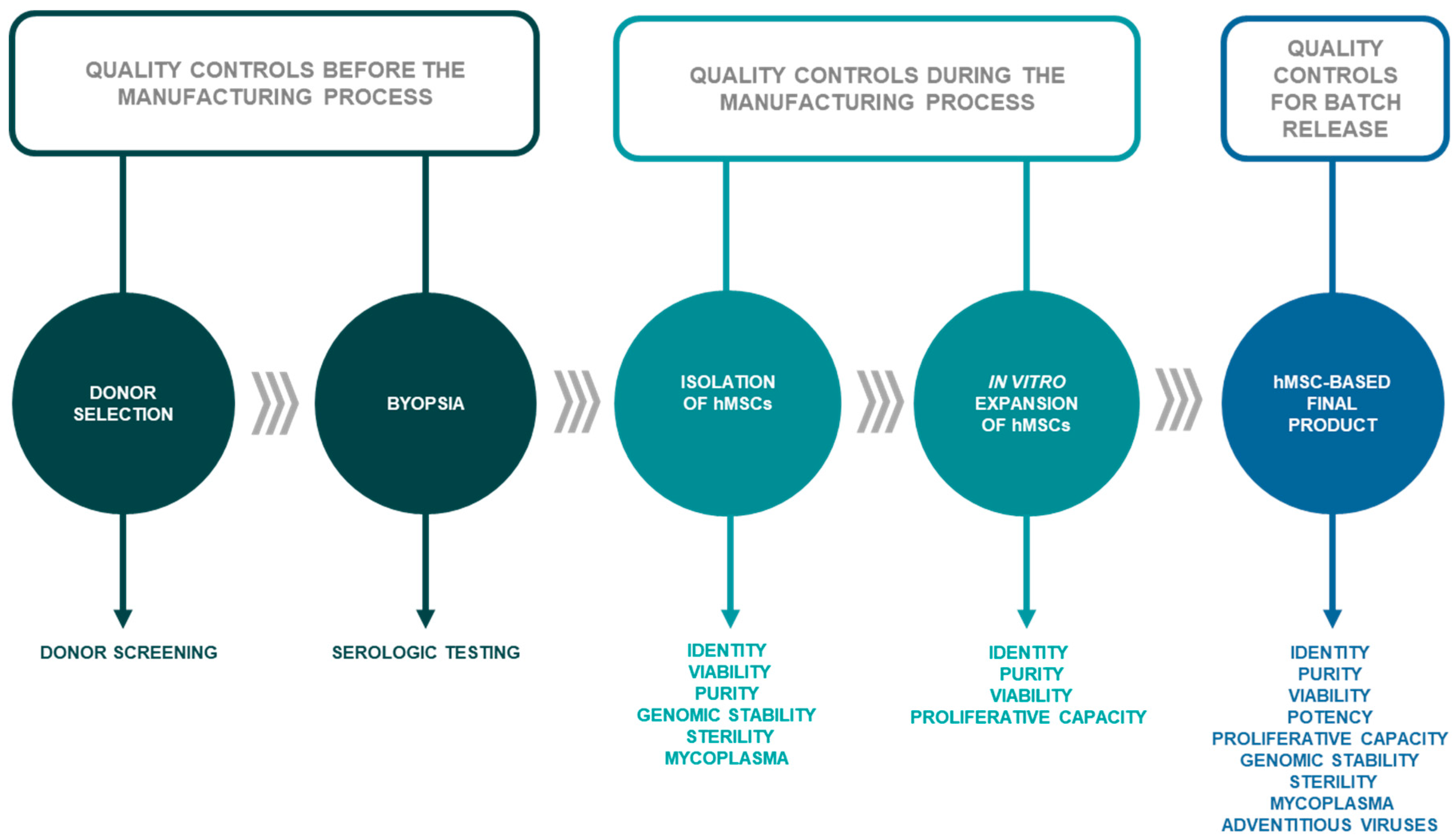
2.1. Donor Selection and Serology
2.2. Isolation and Processing of hMSCs
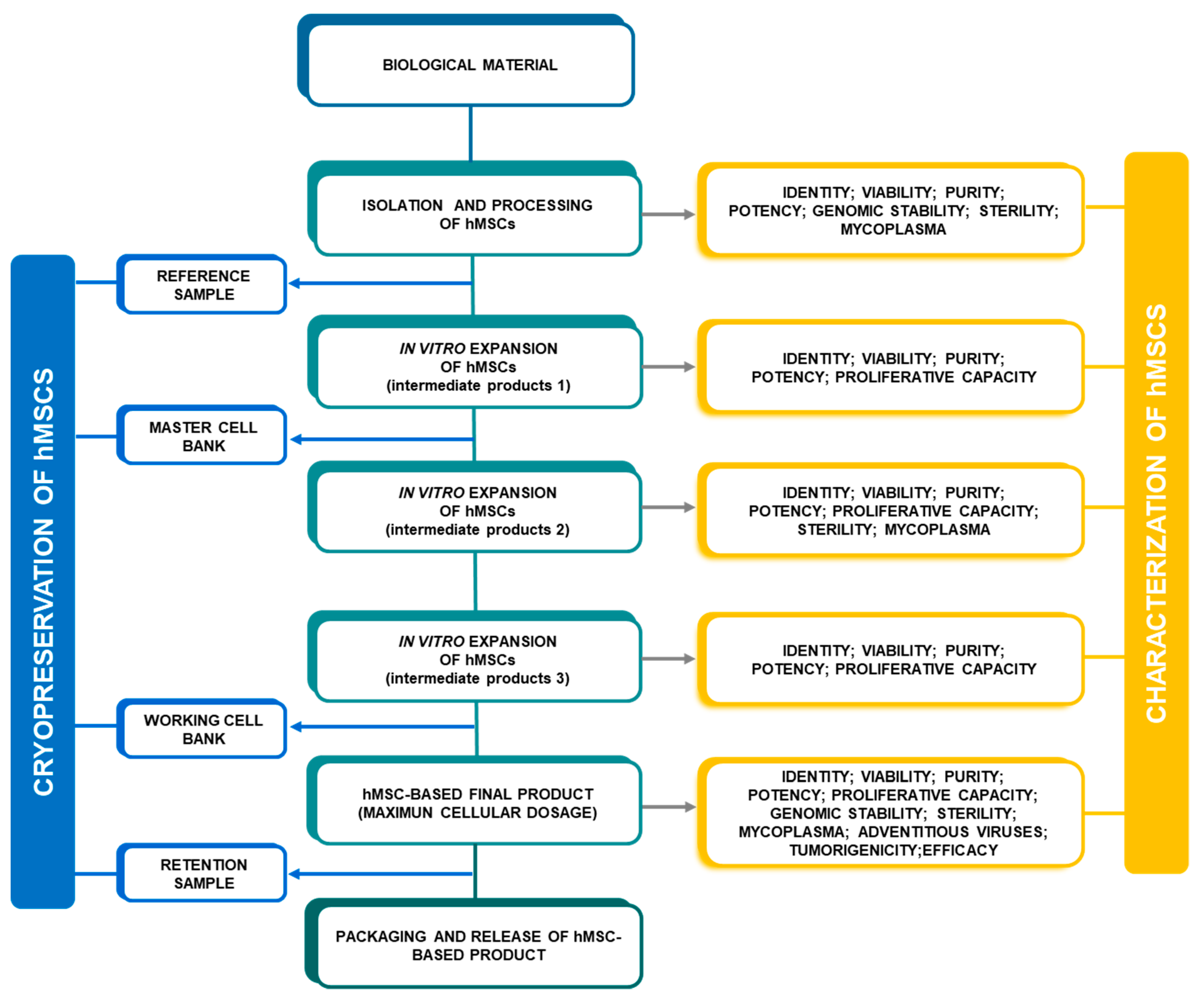
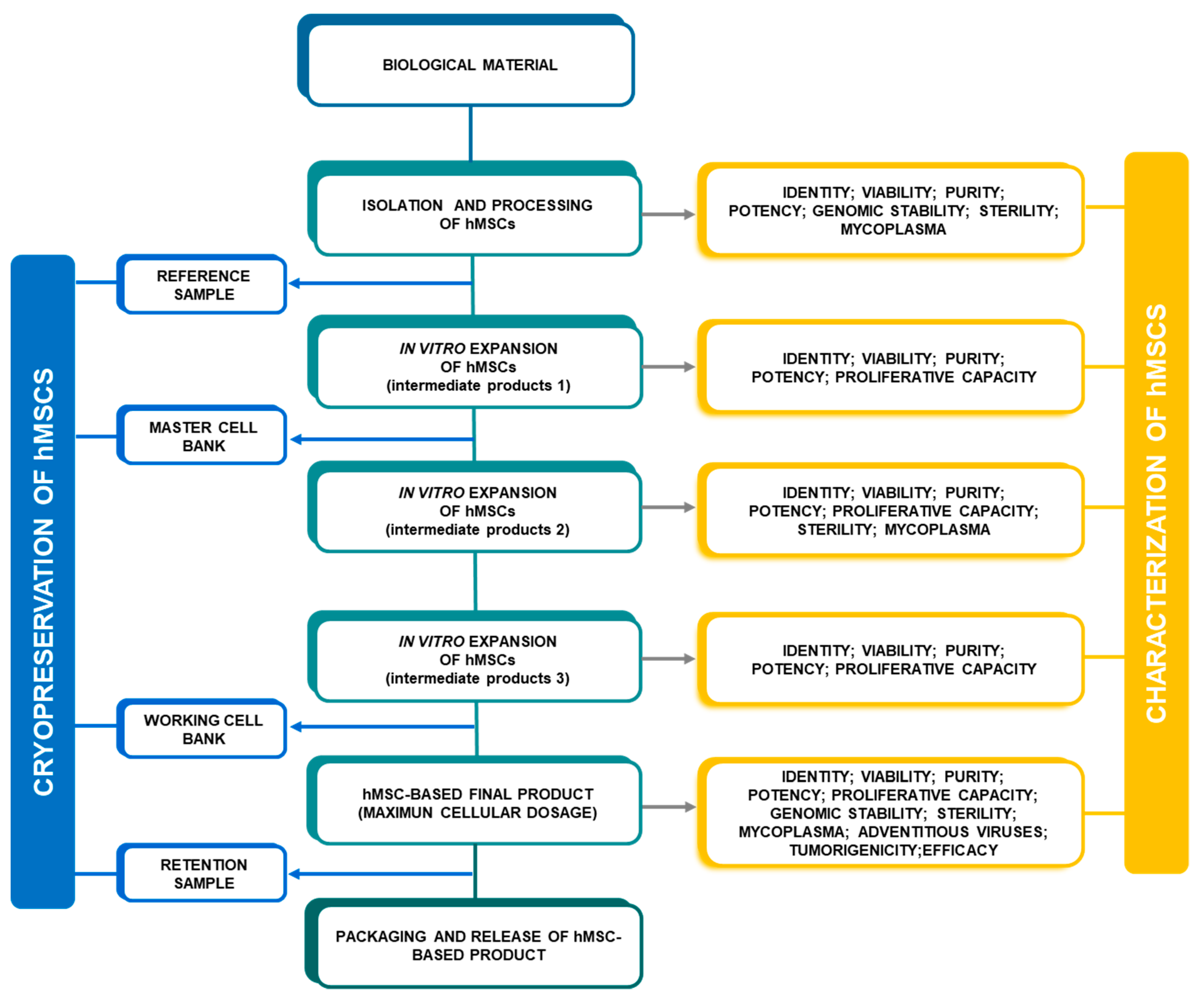
2.3. Characterization of hMSCs
2.4. Cryopreservation of hMSCs
2.5. hMSC-Based Product Packaging
2.6. Release of hMSC-Based Products
3. Minimal Criteria for hMSC Characterization
3.1. Identity
3.2. Viability
3.3. Purity
3.3.1. Cell Impurities
3.3.2. Process Impurities
3.4. Potency
3.5. Proliferative Capacity
3.6. Genomic Stability
3.7. Microbiological Quality Control
3.7.1. Sterility Test
3.7.2. Mycoplasma
3.7.3. Adventitious Viruses
3.8. Tumorigenicity
3.9. Efficacy
4. Conclusions and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Harrell, C.R.; Gazdic, M.; Fellabaum, C.; Jovicic, N.; Djonov, V.; Arsenijevic, N.; Volarevic, V. Therapeutic Potential of Amniotic Fluid Derived Mesenchymal Stem Cells Based on their Differentiation Capacity and Immunomodulatory Properties. Curr. Stem Cell Res. Ther. 2019, 14, 327–336. [Google Scholar] [CrossRef]
- Guadix, J.A.; Zugaza, J.L.; Gálvez-Martín, P. Characteristics, applications and prospects of mesenchymal stem cells in cell therapy. Med. Clin. 2017, 148, 408–414. [Google Scholar] [CrossRef]
- Von Einem, J.C.; Guenther, C.; Volk, H.-D.; Grütz, G.; Hirsch, D.; Salat, C.; Stoetzer, O.; Nelson, P.J.; Michl, M.; Modest, D.P.; et al. Treatment of advanced gastrointestinal cancer with genetically modified autologous mesenchymal stem cells: Results from the Phase 1/2 TREAT-ME-1 Trial. Int. J. Cancer 2019, 145, 1538–1546. [Google Scholar] [CrossRef]
- Fernández, O.; Izquierdo, G.; Fernández, V.; Leyva, L.; Reyes, V.; Guerrero, M.; León, A.; Arnaiz, C.; Navarro, G.; Páramo, M.D.; et al. Adipose-derived mesenchymal stem cells (AdMSC) for the treatment of secondary-progressive multiple sclerosis: A triple blinded, placebo controlled, randomized phase I/II safety and feasibility study. PLoS ONE 2018, 13, e0195891. [Google Scholar] [CrossRef]
- Lotfi, M.; Naderi-Meshkin, H.; Mahdipour, E.; Mafinezhad, A.; Bagherzadeh, R.; Sadeghnia, H.R.; Esmaily, H.; Maleki, M.; Hasssanzadeh, H.; Ghayaour-Mobarhan, M.; et al. Adipose tissue -derived mesenchymal stem cells and keratinocytes co-culture on Gelatin/Chitosan/β-Glycerol Phosphate Nanoscaffold in skin regeneration. Cell Biol. Int. 2019. [Google Scholar] [CrossRef]
- Cuende, N.; Rasko, J.E.J.; Koh, M.B.C.; Dominici, M.; Ikonomou, L. Cell, tissue and gene products with marketing authorization in 2018 worldwide. Cytotherapy 2018, 20, 1401–1413. [Google Scholar] [CrossRef]
- Mastrolia, I.; Foppiani, E.M.; Murgia, A.; Candini, O.; Samarelli, A.V.; Grisendi, G.; Veronesi, E.; Horwitz, E.M.; Dominici, M. Concise Review: Challenges in Clinical Development of Mesenchymal Stromal/Stem Cells. Stem Cells Transl. Med. 2019. [Google Scholar] [CrossRef]
- Squillaro, T.; Peluso, G.; Galderisi, U. Clinical Trials with Mesenchymal Stem Cells: An Update. Cell Transplant. 2016, 25, 829–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gálvez, P.; Clares, B.; Hmadcha, A.; Ruiz, A.; Soria, B. Development of a cell-based medicinal product: Regulatory structures in the European Union. Br. Med. Bull. 2013, 105, 85–105. [Google Scholar] [CrossRef] [PubMed]
- Nagai, S. Flexible and Expedited Regulatory Review Processes for Innovative Medicines and Regenerative Medical Products in the US, the EU, and Japan. Int. J. Mol. Sci. 2019, 20, 3801. [Google Scholar] [CrossRef] [PubMed]
- Hoogduijn, M.J.; Lombardo, E. Concise Review: Mesenchymal Stromal Cells Anno 2019: Dawn of the Therapeutic Era? Stem Cells Transl. Med. 2019. [Google Scholar] [CrossRef]
- Jost, N.; Schüssler-Lenz, M.; Ziegele, B.; Reinhardt, J. Scientific advice by the national and European approval authorities concerning advanced therapy medicinal products. Bundesgesundheitsblatt. Gesundheitsforschung. Gesundheitsschutz 2015, 58, 1207–1214. [Google Scholar] [CrossRef]
- FDA Guidance for Industry: Current Good Tissue Practice (CGTP) and Additional Requirements for Manufacturers of Human Cells, Tissues, and Cellular and Tissue-Based Products (HCT/Ps). 2011. Available online: https://www.fda.gov/downloads/biologicsbloodvaccines/guidancecomplianceregulatoryinformation/guidances/tissue/ucm285223.pdf (accessed on 2 September 2019).
- EMA. European Commission Guideline on Human Cell-Based Medicinal Products. 2008. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-human-cell-based-medicinal-products_en.pdf (accessed on 29 August 2019).
- EMA EudraLex The Rules Governing Medicinal Products in the European Union Volume 4 Good Manufacturing Practice Guidelines on Good Manufacturing Practice specific to Advanced Therapy Medicinal Products. 2017. Available online: https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-4/2017_11_22_guidelines_gmp_for_atmps.pdf (accessed on 24 August 2019).
- Trounson, A.; Thakar, R.G.; Lomax, G.; Gibbons, D. Clinical trials for stem cell therapies. BMC Med. 2011, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Martín, P.G.; Martinez, A.R.; Lara, V.G.; Naveros, B.C. Regulatory considerations in production of a cell therapy medicinal product in Europe to clinical research. Clin. Exp. Med. 2014, 14, 25–33. [Google Scholar] [CrossRef]
- Galvez-Martin, P.; Sabata, R.; Verges, J.; Zugaza, J.L.; Ruiz, A.; Clares, B. Mesenchymal Stem Cells as Therapeutics Agents: Quality and Environmental Regulatory Aspects. Stem Cells Int. 2016, 2016, 9783408. [Google Scholar] [CrossRef] [PubMed]
- Serra, M.; Roseti, L.; Bassi, A. Media fill for validation of a good manufacturing practice-compliant cell production process. Methods Mol. Biol. 2015, 1283, 161–169. [Google Scholar]
- Wuchter, P.; Bieback, K.; Schrezenmeier, H.; Bornhäuser, M.; Müller, L.P.; Bönig, H.; Wagner, W.; Meisel, R.; Pavel, P.; Tonn, T.; et al. Standardization of Good Manufacturing Practice-compliant production of bone marrow-derived human mesenchymal stromal cells for immunotherapeutic applications. Cytotherapy 2015, 17, 128–139. [Google Scholar] [CrossRef]
- FDA Guidance for Industry: Eligibility Determination for Donors of Human Cells, Tissues, and Cellular and Tissue-Based Products (HCT/Ps). 2007. Available online: https://www.fda.gov/downloads/biologicsbloodvaccines/guidancecomplianceregulatoryinformation/guidances/tissue/ucm091345.pdf (accessed on 29 August 2019).
- EMA. European Commission Note for Guidance on Minimising the Risk of Transmitting Animal Spongiform Encephalopathy Agents via Human and Veterinary Medicinal Products; EMA/410/01 rev.3. European Union: Brussels, Belgium, 2011; Volume C 73/1-18. [Google Scholar]
- Klingemann, H.; Matzilevich, D.; Marchand, J. Mesenchymal Stem Cells—Sources and Clinical Applications. Transfus. Med. Hemother. 2008, 35, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.; McKee, C.; Bakshi, S.; Walker, K.; Hakman, E.; Halassy, S.; Svinarich, D.; Dodds, R.; Govind, C.K.; Chaudhry, G.R. Mesenchymal stem cells: Cell therapy and regeneration potential. J. Tissue Eng. Regen. Med. 2019, 13, 1738–1755. [Google Scholar] [CrossRef]
- Bieback, K.; Schallmoser, K.; Klüter, H.; Strunk, D. Clinical Protocols for the Isolation and Expansion of Mesenchymal Stromal Cells. Transfus. Med. Hemother. 2008, 35, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Insausti, C.L.; Blanquer, M.B.; Olmo, L.M.; López-Martínez, M.C.; Ruiz, X.F.; Lozano, F.J.R.; Perianes, V.C.; Funes, C.; Nicolás, F.J.; Majado, M.J.; et al. Isolation and characterization of mesenchymal stem cells from the fat layer on the density gradient separated bone marrow. Stem Cells Dev. 2012, 21, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Mohamed-Ahmed, S.; Fristad, I.; Lie, S.A.; Suliman, S.; Mustafa, K.; Vindenes, H.; Idris, S.B. Adipose-derived and bone marrow mesenchymal stem cells: A donor-matched comparison. Stem Cell Res. Ther. 2018, 9, 168. [Google Scholar] [CrossRef] [PubMed]
- Bunpetch, V.; Wu, H.; Zhang, S.; Ouyang, H. From “Bench to Bedside”: Current Advancement on Large-Scale Production of Mesenchymal Stem Cells. Stem Cells Dev. 2017, 26, 1662–1673. [Google Scholar] [CrossRef] [PubMed]
- Papadaki, M. Adaptation through Collaboration: Developing Novel Platforms to Advance the Delivery of Advanced Therapies to Patients. Front. Med. 2017, 4, 56. [Google Scholar] [CrossRef]
- Fekete, N.; Gadelorge, M.; Fürst, D.; Maurer, C.; Dausend, J.; Fleury-Cappellesso, S.; Mailänder, V.; Lotfi, R.; Ignatius, A.; Sensebé, L.; et al. Platelet lysate from whole blood-derived pooled platelet concentrates and apheresis-derived platelet concentrates for the isolation and expansion of human bone marrow mesenchymal stromal cells: Production process, content and identification of active comp. Cytotherapy 2012, 14, 540–554. [Google Scholar] [CrossRef]
- Ståhle, M.U.; Brandhorst, D.; Korsgren, O.; Knutson, F. Pathogen inactivation of human serum facilitates its clinical use for islet cell culture and subsequent transplantation. Cell Transplant. 2011, 20, 775–781. [Google Scholar] [CrossRef]
- Castiglia, S.; Mareschi, K.; Labanca, L.; Lucania, G.; Leone, M.; Sanavio, F.; Castello, L.; Rustichelli, D.; Signorino, E.; Gunetti, M.; et al. Inactivated human platelet lysate with psoralen: A new perspective for mesenchymal stromal cell production in Good Manufacturing Practice conditions. Cytotherapy 2014, 16, 750–763. [Google Scholar] [CrossRef]
- Chen, M.-S.; Wang, T.-J.; Lin, H.-C.; Burnouf, T. Four types of human platelet lysate, including one virally inactivated by solvent-detergent, can be used to propagate Wharton jelly mesenchymal stromal cells. N Biotechnol. 2019, 49, 151–160. [Google Scholar] [CrossRef]
- Tonti, G.A.; Mannello, F. From bone marrow to therapeutic applications: Different behaviour and genetic/epigenetic stability during mesenchymal stem cell expansion in autologous and foetal bovine sera? Int. J. Dev. Biol. 2008, 52, 1023–1032. [Google Scholar] [CrossRef]
- Phinney, D.G. Functional heterogeneity of mesenchymal stem cells: Implications for cell therapy. J. Cell. Biochem. 2012, 113, 2806–2812. [Google Scholar] [CrossRef] [PubMed]
- Ra, J.C.; Shin, I.S.; Kim, S.H.; Kang, S.K.; Kang, B.C.; Lee, H.Y.; Kim, Y.J.; Jo, J.Y.; Yoon, E.J.; Choi, H.J.; et al. Safety of Intravenous Infusion of Human Adipose Tissue-Derived Mesenchymal Stem Cells in Animals and Humans. Stem Cells Dev. 2011, 20, 1297–1308. [Google Scholar] [CrossRef] [PubMed]
- FDA Guidance for Industry: Guidance for Human Somatic Cell Therapy and Gene Therapy. 1998. Available online: https://www.fda.gov/downloads/biologicsbloodvaccines/guidancecomplianceregulatoryinformation/guidances/cellularandgenetherapy/ucm081670.pdf (accessed on 28 August 2019).
- EMA. European Commission Guideline on Virus Safety Evaluation of Biotechnological Investigational Medicinal Products. 2006. Available online: https://www.ema.europa.eu/documents/scientific-guideline/guideline-virus-safety-evaluation-biotechnological-investigational-medicinal-products_en.pdf (accessed on 24 August 2019).
- Geraghty, R.J.; Capes-Davis, A.; Davis, J.M.; Downward, J.; Freshney, R.I.; Knezevic, I.; Lovell-Badge, R.; Masters, J.R.W.; Meredith, J.; Stacey, G.N.; et al. Guidelines for the use of cell lines in biomedical research. Br. J. Cancer 2014, 111, 1021–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Z.; Lourenco, S.D.S.; Sage, E.K.; Kolluri, K.K.; Lowdell, M.W.; Janes, S.M. Cryopreservation of human mesenchymal stromal cells expressing TRAIL for human anti-cancer therapy. Cytotherapy 2016, 18, 860–869. [Google Scholar] [CrossRef] [Green Version]
- Pegg, D.E. Principles of cryopreservation. Methods Mol. Biol. 2015, 1257, 3–19. [Google Scholar]
- Balci, D.; Can, A. The assessment of cryopreservation conditions for human umbilical cord stroma-derived mesenchymal stem cells towards a potential use for stem cell banking. Curr. Stem Cell Res. Ther. 2013, 8, 60–72. [Google Scholar] [CrossRef]
- Ginis, I.; Grinblat, B.; Shirvan, M.H. Evaluation of bone marrow-derived mesenchymal stem cells after cryopreservation and hypothermic storage in clinically safe medium. Tissue Eng. Part C. Methods 2012, 18, 453–463. [Google Scholar] [CrossRef]
- Antebi, B.; Asher, A.M.; Rodriguez, L.A.; Moore, R.K.; Mohammadipoor, A.; Cancio, L.C. Cryopreserved mesenchymal stem cells regain functional potency following a 24-h acclimation period. J. Transl. Med. 2019, 17, 297. [Google Scholar] [CrossRef]
- Gramlich, O.W.; Burand, A.J.; Brown, A.J.; Deutsch, R.J.; Kuehn, M.H.; Ankrum, J.A. Cryopreserved Mesenchymal Stromal Cells Maintain Potency in a Retinal Ischemia/Reperfusion Injury Model: Toward an off-the-shelf Therapy. Sci. Rep. 2016, 6, 26463. [Google Scholar] [CrossRef]
- Cruz, F.F.; Borg, Z.D.; Goodwin, M.; Sokocevic, D.; Wagner, D.; McKenna, D.H.; Rocco, P.R.M.; Weiss, D.J. Freshly thawed and continuously cultured human bone marrow-derived mesenchymal stromal cells comparably ameliorate allergic airways inflammation in immunocompetent mice. Stem Cells Transl. Med. 2015, 4, 615–624. [Google Scholar] [CrossRef]
- Hoogduijn, M.J.; de Witte, S.F.H.; Luk, F.; van den Hout-van Vroonhoven, M.C.G.N.; Ignatowicz, L.; Catar, R.; Strini, T.; Korevaar, S.S.; van IJcken, W.F.J.; Betjes, M.G.H.; et al. Effects of Freeze-Thawing and Intravenous Infusion on Mesenchymal Stromal Cell Gene Expression. Stem Cells Dev. 2016, 25, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Galipeau, J.; Sensébé, L. Mesenchymal Stromal Cells: Clinical Challenges and Therapeutic Opportunities. Cell Stem Cell 2018, 22, 824–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, T.R.; Ng, K.S.; Lock, L.T.; Ahsan, T.; Rowley, J.A. Peak MSC-Are We There Yet? Front. Med. 2018, 5, 178. [Google Scholar] [CrossRef] [PubMed]
- Gálvez-Martín, P.; Hmadcha, A.; Soria, B.; Calpena-Campmany, A.C.; Clares-Naveros, B. Study of the stability of packaging and storage conditions of human mesenchymal stem cell for intra-arterial clinical application in patient with critical limb ischemia. Eur. J. Pharm. Biopharm. 2014, 86, 459–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirabel, C.; Puente-Massaguer, E.; Del Mazo-Barbara, A.; Reyes, B.; Morton, P.; Gòdia, F.; Vives, J. Stability enhancement of clinical grade multipotent mesenchymal stromal cell-based products. J. Transl. Med. 2018, 16, 291. [Google Scholar] [CrossRef]
- ICH Expert Working Group. ICH Harmonised Tripartite Guideline Quality of Biotechnological Products: Stability Testing of Biotechnological/Biological Products Q5C. 1995. Available online: https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q5C/Step4/Q5C_Guideline.pdf (accessed on 2 September 2019).
- ICH Guidance on Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products. 1999. Available online: https://www.ich.org/products/guidelines/quality/quality-single/article/specifications-test-procedures-and-acceptance-criteria-for-biotechnologicalbiological-products.html (accessed on 2 September 2019).
- Panés, J.; García-Olmo, D.; Van Assche, G.; Colombel, J.F.; Reinisch, W.; Baumgart, D.C.; Dignass, A.; Nachury, M.; Ferrante, M.; Kazemi-Shirazi, L.; et al. Expanded allogeneic adipose-derived mesenchymal stem cells (Cx601) for complex perianal fistulas in Crohn’s disease: A phase 3 randomised, double-blind controlled trial. Lancet 2016, 388, 1281–1290. [Google Scholar] [CrossRef]
- EMA. European Commission Guidelines on Principles of Good Distribution Practice of Active Substances for Medicinal Products for Human Use; European Union: Brussels, Belgium, 2013; Volume C 343/1-14. [Google Scholar]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Soria-Juan, B.; Escacena, N.; Capilla-González, V.; Aguilera, Y.; Llanos, L.; Tejedo, J.R.; Bedoya, F.J.; Juan, V.; De la Cuesta, A.; Ruiz-Salmerón, R.; et al. Cost-Effective, Safe, and Personalized Cell Therapy for Critical Limb Ischemia in Type 2 Diabetes Mellitus. Front. Immunol. 2019, 10, 1151. [Google Scholar] [CrossRef]
- Grau-Vorster, M.; Laitinen, A.; Nystedt, J.; Vives, J. HLA-DR expression in clinical-grade bone marrow-derived multipotent mesenchymal stromal cells: A two-site study. Stem Cell Res. Ther. 2019, 10, 164. [Google Scholar] [CrossRef]
- Grau-Vorster, M.; Rodríguez, L.; Torrents-Zapata, S.; Vivas, D.; Codinach, M.; Blanco, M.; Oliver-Vila, I.; García-López, J.; Vives, J. Levels of IL-17F and IL-33 correlate with HLA-DR activation in clinical-grade human bone marrow-derived multipotent mesenchymal stromal cell expansion cultures. Cytotherapy 2019, 21, 32–40. [Google Scholar] [CrossRef]
- Torre, M.L.; Lucarelli, E.; Guidi, S.; Ferrari, M.; Alessandri, G.; De Girolamo, L.; Pessina, A.; Ferrero, I. Gruppo Italiano Staminali Mesenchimali (GISM) Ex vivo expanded mesenchymal stromal cell minimal quality requirements for clinical application. Stem Cells Dev. 2015, 24, 677–685. [Google Scholar] [CrossRef]
- Galocha-León, C.; Clares-Naveros, B.; Gálvez-Martín, P. Main Analytical Methods for the Viability Assessment of Mesenchymal Stem Cells for Use as Cellular Medicines. Curr. Pharm. Anal. 2018, 14, 427–436. [Google Scholar] [CrossRef]
- Council of Europe. 2.7.29. Nucleated cell count and viability. In The European Pharmacopoeia, 9.0. ed.; EDQM: Strasbourg, France, 2017. [Google Scholar]
- Seaver, S. A new United States Pharmacopeia (USP) Chapter 1046: Cell and gene therapy products. Cytotherapy 2000, 2, 45–49. [Google Scholar] [CrossRef]
- Sousa, B.R.; Parreira, R.C.; Fonseca, E.A.; Amaya, M.J.; Tonelli, F.M.P.; Lacerda, S.M.S.N.; Lalwani, P.; Santos, A.K.; Gomes, K.N.; Ulrich, H.; et al. Human adult stem cells from diverse origins: An overview from multiparametric immunophenotyping to clinical applications. Cytometry. A 2014, 85, 43–77. [Google Scholar] [CrossRef]
- EMA. European Commission Guideline on the Use of Bovine Serum in the Manufacture of Human Biological Medicinal Products. 2014. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-use-bovine-serum-manufacture-human-biological-medicinal-products_en.pdf (accessed on 24 August 2019).
- EMA. European Commission Guideline on the Use of Porcine Trypsin Used in the Manufacture of Human Biological Medicinal Products. 2014. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-use-porcine-trypsin-used-manufacture-human-biological-medicinal-products_en.pdf (accessed on 24 August 2019).
- Chen, X.; Thibeault, S. Effect of DMSO concentration, cell density and needle gauge on the viability of cryopreserved cells in three dimensional hyaluronan hydrogel. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2013, 2013, 6228–6231. [Google Scholar] [Green Version]
- Gálvez, P.; Clares, B.; Bermejo, M.; Hmadcha, A.; Soria, B. Standard requirement of a microbiological quality control program for the manufacture of human mesenchymal stem cells for clinical use. Stem Cells Dev. 2014, 23, 1074–1083. [Google Scholar] [CrossRef]
- FDA Guidance for Industry: Potency Tests for Cellular and Gene Therapy Products. 2011. Available online: http://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/CellularandGeneTherapy/UCM243392.pdf (accessed on 28 August 2019).
- Mendicino, M.; Bailey, A.M.; Wonnacott, K.; Puri, R.K.; Bauer, S.R. MSC-based product characterization for clinical trials: An FDA perspective. Cell Stem Cell 2014, 14, 141–145. [Google Scholar] [CrossRef]
- Salgado, A.J.B.O.G.; Reis, R.L.G.; Sousa, N.J.C.; Gimble, J.M. Adipose tissue derived stem cells secretome: Soluble factors and their roles in regenerative medicine. Curr. Stem Cell Res. Ther. 2010, 5, 103–110. [Google Scholar] [CrossRef]
- Bartaula-Brevik, S.; Pedersen, T.O.; Finne-Wistrand, A.; Bolstad, A.I.; Mustafa, K. Angiogenic and Immunomodulatory Properties of Endothelial and Mesenchymal Stem Cells. Tissue Eng. Part A 2016, 22, 244–252. [Google Scholar] [CrossRef] [Green Version]
- Galipeau, J.; Krampera, M.; Barrett, J.; Dazzi, F.; Deans, R.J.; DeBruijn, J.; Dominici, M.; Fibbe, W.E.; Gee, A.P.; Gimble, J.M.; et al. International Society for Cellular Therapy perspective on immune functional assays for mesenchymal stromal cells as potency release criterion for advanced phase clinical trials. Cytotherapy 2016, 18, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Lehman, N.; Cutrone, R.; Raber, A.; Perry, R.; Van’t Hof, W.; Deans, R.; Ting, A.E.; Woda, J. Development of a surrogate angiogenic potency assay for clinical-grade stem cell production. Cytotherapy 2012, 14, 994–1004. [Google Scholar] [CrossRef] [PubMed]
- Bonfield, T.L.; Nolan Koloze, M.T.; Lennon, D.P.; Caplan, A.I. Defining human mesenchymal stem cell efficacy in vivo. J. Inflamm. 2010, 7, 51. [Google Scholar] [CrossRef]
- Bloom, D.D.; Centanni, J.M.; Bhatia, N.; Emler, C.A.; Drier, D.; Leverson, G.E.; McKenna, D.H.; Gee, A.P.; Lindblad, R.; Hei, D.J.; et al. A reproducible immunopotency assay to measure mesenchymal stromal cell-mediated T-cell suppression. Cytotherapy 2015, 17, 140–151. [Google Scholar] [CrossRef]
- Tao, H.; Han, Z.; Han, Z.C.; Li, Z. Proangiogenic Features of Mesenchymal Stem Cells and Their Therapeutic Applications. Stem Cells Int. 2016, 2016, 1314709. [Google Scholar] [CrossRef]
- Jiao, J.; Milwid, J.M.; Yarmush, M.L.; Parekkadan, B. A mesenchymal stem cell potency assay. Methods Mol. Biol. 2011, 677, 221–231. [Google Scholar]
- Roddy, G.W.; Oh, J.Y.; Lee, R.H.; Bartosh, T.J.; Ylostalo, J.; Coble, K.; Rosa, R.H.; Prockop, D.J. Action at a distance: Systemically administered adult stem/progenitor cells (MSCs) reduce inflammatory damage to the cornea without engraftment and primarily by secretion of TNF-α stimulated gene/protein 6. Stem Cells 2011, 29, 1572–1579. [Google Scholar] [CrossRef]
- De Wolf, C.; van de Bovenkamp, M.; Hoefnagel, M. Regulatory perspective on in vitro potency assays for human mesenchymal stromal cells used in immunotherapy. Cytotherapy 2017, 19, 784–797. [Google Scholar] [CrossRef]
- Uccelli, A.; Moretta, L.; Pistoia, V. Mesenchymal stem cells in health and disease. Nat. Rev. Immunol. 2008, 8, 726–736. [Google Scholar] [CrossRef]
- Heldring, N.; Mäger, I.; Wood, M.J.A.; Le Blanc, K.; Andaloussi, S.E.L. Therapeutic Potential of Multipotent Mesenchymal Stromal Cells and Their Extracellular Vesicles. Hum. Gene Ther. 2015, 26, 506–517. [Google Scholar] [CrossRef]
- Soleymaninejadian, E.; Pramanik, K.; Samadian, E. Immunomodulatory properties of mesenchymal stem cells: Cytokines and factors. Am. J. Reprod. Immunol. 2012, 67, 1–8. [Google Scholar] [CrossRef]
- Trento, C.; Dazzi, F. Mesenchymal stem cells and innate tolerance: Biology and clinical applications. Swiss Med. Wkly. 2010, 140, w13121. [Google Scholar]
- Murgia, A.; Veronesi, E.; Candini, O.; Caselli, A.; D’souza, N.; Rasini, V.; Giorgini, A.; Catani, F.; Iughetti, L.; Dominici, M.; et al. Potency Biomarker Signature Genes from Multiparametric Osteogenesis Assays: Will cGMP Human Bone Marrow Mesenchymal Stromal Cells Make Bone? PLoS ONE 2016, 11, e0163629. [Google Scholar]
- Samsonraj, R.M.; Rai, B.; Sathiyanathan, P.; Puan, K.J.; Rötzschke, O.; Hui, J.H.; Raghunath, M.; Stanton, L.W.; Nurcombe, V.; Cool, S.M. Establishing criteria for human mesenchymal stem cell potency. Stem Cells 2015, 33, 1878–1891. [Google Scholar] [CrossRef]
- Deans, R. Towards the creation of a standard MSC line as a calibration tool. Cytotherapy 2015, 17, 1167–1168. [Google Scholar] [CrossRef]
- Rich, I.N. Measurement of hematopoietic stem cell proliferation, self-renewal, and expansion potential. Methods Mol. Biol. 2015, 1235, 7–17. [Google Scholar]
- Wang, Y.; Han, Z.; Song, Y.; Han, Z.C. Safety of Mesenchymal Stem Cells for Clinical Application. Stem Cells Int. 2012, 2012, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Greenwood, S.K.; Hill, R.B.; Sun, J.T.; Armstrong, M.J.; Johnson, T.E.; Gara, J.P.; Galloway, S.M. Population doubling: A simple and more accurate estimation of cell growth suppression in the in vitro assay for chromosomal aberrations that reduces irrelevant positive results. Environ. Mol. Mutagen. 2004, 43, 36–44. [Google Scholar] [CrossRef]
- Barkholt, L.; Flory, E.; Jekerle, V.; Lucas-Samuel, S.; Ahnert, P.; Bisset, L.; Büscher, D.; Fibbe, W.; Foussat, A.; Kwa, M.; et al. Risk of tumorigenicity in mesenchymal stromal cell-based therapies--bridging scientific observations and regulatory viewpoints. Cytotherapy 2013, 15, 753–759. [Google Scholar] [CrossRef]
- Stenderup, K.; Justesen, J.; Clausen, C.; Kassem, M. Aging is associated with decreased maximal life span and accelerated senescence of bone marrow stromal cells. Bone 2003, 33, 919–926. [Google Scholar] [CrossRef]
- Hayflick, L. Antecedents of cell aging research. Exp. Gerontol. 1989, 24, 355–365. [Google Scholar] [CrossRef]
- Jung, S.; Panchalingam, K.M.; Rosenberg, L.; Behie, L.A. Ex vivo expansion of human mesenchymal stem cells in defined serum-free media. Stem Cells Int. 2012, 2012, 123030. [Google Scholar] [CrossRef] [PubMed]
- Schellenberg, A.; Stiehl, T.; Horn, P.; Joussen, S.; Pallua, N.; Ho, A.D.; Wagner, W. Population dynamics of mesenchymal stromal cells during culture expansion. Cytotherapy 2012, 14, 401–411. [Google Scholar] [CrossRef]
- Dhanasekaran, M.; Indumathi, S.; Lissa, R.P.; Harikrishnan, R.; Rajkumar, J.S.; Sudarsanam, D. A comprehensive study on optimization of proliferation and differentiation potency of bone marrow derived mesenchymal stem cells under prolonged culture condition. Cytotechnology 2013, 65, 187–197. [Google Scholar] [CrossRef]
- Horwitz, E.M.; Gordon, P.L.; Koo, W.K.K.; Marx, J.C.; Neel, M.D.; McNall, R.Y.; Muul, L.; Hofmann, T. Isolated allogeneic bone marrow-derived mesenchymal cells engraft and stimulate growth in children with osteogenesis imperfecta: Implications for cell therapy of bone. Proc. Natl. Acad. Sci. USA 2002, 99, 8932–8937. [Google Scholar] [CrossRef] [Green Version]
- Zhao, K.; Lou, R.; Huang, F.; Peng, Y.; Jiang, Z.; Huang, K.; Wu, X.; Zhang, Y.; Fan, Z.; Zhou, H.; et al. Immunomodulation effects of mesenchymal stromal cells on acute graft-versus-host disease after hematopoietic stem cell transplantation. Biol. Blood Marrow Transplant. 2015, 21, 97–104. [Google Scholar] [CrossRef]
- Sareen, N.; Sequiera, G.L.; Chaudhary, R.; Abu-El-Rub, E.; Chowdhury, S.R.; Sharma, V.; Surendran, A.; Moudgil, M.; Fernyhough, P.; Ravandi, A.; et al. Early passaging of mesenchymal stem cells does not instigate significant modifications in their immunological behavior. Stem Cell Res. Ther. 2018, 9, 121. [Google Scholar] [CrossRef] [Green Version]
- Bochkov, N.P.; Nikitina, V.A.; Buyanovskaya, O.A.; Voronina, E.S.; Goldstein, D.V.; Kuleshov, N.P.; Rzhaninova, A.A.; Chaushev, I.N. Aneuploidy of stem cells isolated from human adipose tissue. Bull. Exp. Biol. Med. 2008, 146, 344–347. [Google Scholar] [CrossRef]
- Bochkov, N.P.; Voronina, E.S.; Kosyakova, N.V.; Liehr, T.; Rzhaninova, A.A.; Katosova, L.D.; Platonova, V.I.; Gol’dshtein, D. V Chromosome variability of human multipotent mesenchymal stromal cells. Bull. Exp. Biol. Med. 2007, 143, 122–126. [Google Scholar] [CrossRef]
- Bernardo, M.E.; Zaffaroni, N.; Novara, F.; Cometa, A.M.; Avanzini, M.A.; Moretta, A.; Montagna, D.; Maccario, R.; Villa, R.; Daidone, M.G.; et al. Human bone marrow derived mesenchymal stem cells do not undergo transformation after long-term in vitro culture and do not exhibit telomere maintenance mechanisms. Cancer Res. 2007, 67, 9142–9149. [Google Scholar] [CrossRef]
- Boozer, S.; Lehman, N.; Lakshmipathy, U.; Love, B.; Raber, A.; Maitra, A.; Deans, R.; Rao, M.S.; Ting, A.E. Global Characterization and Genomic Stability of Human MultiStem, A Multipotent Adult Progenitor Cell. J. Stem Cells 2009, 4, 17–28. [Google Scholar]
- Røsland, G.V.; Svendsen, A.; Torsvik, A.; Sobala, E.; McCormack, E.; Immervoll, H.; Mysliwietz, J.; Tonn, J.-C.; Goldbrunner, R.; Lønning, P.E.; et al. Long-term cultures of bone marrow-derived human mesenchymal stem cells frequently undergo spontaneous malignant transformation. Cancer Res. 2009, 69, 5331–5339. [Google Scholar] [CrossRef]
- Duarte, D.M.; Cornélio, D.A.; Corado, C.; Medeiros, V.K.; de Araújo, L.A.; Cavalvanti, G.B., Jr.; de Medeiros, S.R. Chromosomal characterization of cryopreserved mesenchymal stem cells from the human subendothelium umbilical cord vein. Regen. Med. 2012, 7, 147–157. [Google Scholar] [CrossRef]
- Buyanovskaya, O.A.; Kuleshov, N.P.; Nikitina, V.A.; Voronina, E.S.; Katosova, L.D.; Bochkov, N.P. Spontaneous aneuploidy and clone formation in adipose tissue stem cells during different periods of culturing. Bull. Exp. Biol. Med. 2009, 148, 109–112. [Google Scholar] [CrossRef]
- Grigorian, A.S.; Kruglyakov, P.V.; Taminkina, U.A.; Efimova, O.A.; Pendina, A.A.; Voskresenskaya, A.V.; Kuznetsova, T.V.; Polyntsev, D.G. Alterations of cytological and karyological profile of human mesenchymal stem cells during in vitro culturing. Bull. Exp. Biol. Med. 2010, 150, 125–130. [Google Scholar] [CrossRef]
- Ferreira, R.J.; Irioda, A.C.; Cunha, R.C.; Francisco, J.C.; Guarita-Souza, L.C.; Srikanth, G.V.N.; Nityanand, S.; Rosati, R.; Chachques, J.C.; de Carvalho, K.A.T. Controversies about the chromosomal stability of cultivated mesenchymal stem cells: Their clinical use is it safe? Curr. Stem Cell Res. Ther. 2012, 7, 356–363. [Google Scholar] [CrossRef]
- Ross, A.L.; Leder, D.E.; Weiss, J.; Izakovic, J.; Grichnik, J.M. Genomic instability in cultured stem cells: Associated risks and underlying mechanisms. Regen. Med. 2011, 6, 653–662. [Google Scholar] [CrossRef]
- Neri, P.; Bahlis, N.J. Genomic instability in multiple myeloma: Mechanisms and therapeutic implications. Expert Opin. Biol. Ther. 2013, 13 (Suppl. S1), S69–S82. [Google Scholar] [CrossRef]
- Bernardo, M.E.; Avanzini, M.A.; Perotti, C.; Cometa, A.M.; Moretta, A.; Lenta, E.; Del Fante, C.; Novara, F.; de Silvestri, A.; Amendola, G.; et al. Optimization of in vitro expansion of human multipotent mesenchymal stromal cells for cell-therapy approaches: Further insights in the search for a fetal calf serum substitute. J. Cell. Physiol. 2007, 211, 121–130. [Google Scholar] [CrossRef]
- Zhang, Z.-X.; Guan, L.-X.; Zhang, K.; Wang, S.; Cao, P.-C.; Wang, Y.-H.; Wang, Z.; Dai, L.-J. Cytogenetic analysis of human bone marrow-derived mesenchymal stem cells passaged in vitro. Cell Biol. Int. 2007, 31, 645–648. [Google Scholar] [CrossRef]
- Tarte, K.; Gaillard, J.; Lataillade, J.-J.; Fouillard, L.; Becker, M.; Mossafa, H.; Tchirkov, A.; Rouard, H.; Henry, C.; Splingard, M.; et al. Clinical-grade production of human mesenchymal stromal cells: Occurrence of aneuploidy without transformation. Blood 2010, 115, 1549–1553. [Google Scholar] [CrossRef]
- Ben-David, U.; Mayshar, Y.; Benvenisty, N. Large-Scale Analysis Reveals Acquisition of Lineage-Specific Chromosomal Aberrations in Human Adult Stem Cells. Cell Stem Cell 2011, 9, 97–102. [Google Scholar] [CrossRef] [Green Version]
- Capelli, C.; Pedrini, O.; Cassina, G.; Spinelli, O.; Salmoiraghi, S.; Golay, J.; Rambaldi, A.; Giussani, U.; Introna, M. Frequent occurrence of non-malignant genetic alterations in clinical grade mesenchymal stromal cells expanded for cell therapy protocols. Haematologica 2014, 99, e94–e97. [Google Scholar] [CrossRef] [Green Version]
- Hastings, R.J.; Cavani, S.; Bricarelli, F.D.; Patsalis, P.C.; Kristoffersson, U. ECA PWG Co-ordinators Cytogenetic Guidelines and Quality Assurance: A common European framework for quality assessment for constitutional and acquired cytogenetic investigations. Eur. J. Hum. Genet. 2007, 15, 525–527. [Google Scholar] [CrossRef]
- Borghesi, A.; Avanzini, M.A.; Novara, F.; Mantelli, M.; Lenta, E.; Achille, V.; Cerbo, R.M.; Tzialla, C.; Longo, S.; De Silvestri, A.; et al. Genomic alterations in human umbilical cord–derived mesenchymal stromal cells call for stringent quality control before any possible therapeutic approach. Cytotherapy 2013, 15, 1362–1373. [Google Scholar] [CrossRef]
- Lysák, D.; Holubová, M.; Bergerová, T.; Vávrová, M.; Cangemi, G.C.; Ciccocioppo, R.; Kruzliak, P.; Jindra, P. Validation of shortened 2-day sterility testing of mesenchymal stem cell-based therapeutic preparation on an automated culture system. Cell Tissue Bank. 2016, 17, 1–9. [Google Scholar] [CrossRef]
- PIC/S Recommendations on Validation Master Plan, Installation and Operation Qualification, Non-Sterile Process Validation, Cleaning Validation. PI 006-3, 27 Sept 2007. Available online: file:///C:/Users/usuari/Downloads/pi_006_3_recommendation_on_validation_master_plan%20(1).pdf (accessed on 30 August 2019).
- Council of Europe. Chapter 2.6.1. Sterility. In The European Pharmacopoeia, 9.0. ed.; EDQM: Strasbourg, France, 2017. [Google Scholar]
- Council of Europe. Chapter 5.1.6. Alternative methods for control of microbiological quality. In The European Pharmacopoeia, 9.0. ed.; EDQM: Strasbourg, France, 2017. [Google Scholar]
- United States Pharmacopeial (USP). General Chapter <1223> Validation of alternative microbiological methods; United States Pharmacopeial Convention: Rockville, MD, USA, 2015; USP 38/NF33:1439. [Google Scholar]
- Sparrow, R.L. Microbial screening of UC blood units by an automated culture system: Effect of delayed testing on bacterial detection. Cytotherapy 2004, 6, 23–29. [Google Scholar] [CrossRef]
- Council of Europe. Chapter 2.6.7. Mycoplasmas. In The European Pharmacopoeia, 9.0. ed.; EDQM: Strasbourg, France, 2017. [Google Scholar]
- United States Pharmacopeial (USP). General Chapter <63> Mycoplasma Tests; United States Pharmacopeial Convention: Rockville, MD, USA, 2010; USP 38/NF28:88. [Google Scholar]
- Leland, D.S.; Ginocchio, C.C. Role of cell culture for virus detection in the age of technology. Clin. Microbiol. Rev. 2007, 20, 49–78. [Google Scholar] [CrossRef]
- Gombold, J.; Karakasidis, S.; Niksa, P.; Podczasy, J.; Neumann, K.; Richardson, J.; Sane, N.; Johnson-Leva, R.; Randolph, V.; Sadoff, J.; et al. Systematic evaluation of in vitro and in vivo adventitious virus assays for the detection of viral contamination of cell banks and biological products. Vaccine 2014, 32, 2916–2926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- EMA ICH Topic Q 5 A (R1). Quality of Biotechnological Products: Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin. Note for guidance on quality of biotechnological products: viral safety evaluation of biotechnology products derived from cell lines of human or animal origin (CPMP/ICH/295/95). 1997. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-5-r1-viral-safety-evaluation-biotechnology-products-derived-cell-lines-human-animal-origin_en.pdf. (accessed on 24 August 2019).
- Viganò, M.; Budelli, S.; Lavazza, C.; Montemurro, T.; Montelatici, E.; de Cesare, S.; Lazzari, L.; Orlandi, A.R.; Lunghi, G.; Giordano, R. Tips and Tricks for Validation of Quality Control Analytical Methods in Good Manufacturing Practice Mesenchymal Stromal Cell Production. Stem Cells Int. 2018, 2018, 3038565. [Google Scholar] [CrossRef]
- Ng, S.H.; Braxton, C.; Eloit, M.; Feng, S.F.; Fragnoud, R.; Mallet, L.; Mee, E.T.; Sathiamoorthy, S.; Vandeputte, O.; Khan, A.S. Current Perspectives on High-Throughput Sequencing (HTS) for Adventitious Virus Detection: Upstream Sample Processing and Library Preparation. Viruses 2018, 10, 566. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.S.; Benetti, L.; Blumel, J.; Deforce, D.; Egan, W.M.; Knezevic, I.; Krause, P.R.; Mallet, L.; Mayer, D.; Minor, P.D.; et al. Report of the international conference on next generation sequencing for adventitious virus detection in biologicals. Biologicals 2018, 55, 1–16. [Google Scholar] [CrossRef]
- EMA. European Commission Stem Cell-Based Products for Veterinary Use: Specific Questions on Tumorigenicity to Be Addressed by Ad Hoc Expert Group on Veterinary Novel Therapies (ADVENT). 2016. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/draft-stem-cell-based-products-veterinary-use-specific-questions-tumorigenicity-be-addressed-ad-hoc_en.pdf (accessed on 24 August 2019).
- Torsvik, A.; Røsland, G.V.; Svendsen, A.; Molven, A.; Immervoll, H.; McCormack, E.; Lønning, P.E.; Primon, M.; Sobala, E.; Tonn, J.-C.; et al. Spontaneous malignant transformation of human mesenchymal stem cells reflects cross-contamination: Putting the research field on track—Letter. Cancer Res. 2010, 70, 6393–6396. [Google Scholar] [CrossRef] [PubMed]
- Garcia, S.; Bernad, A.; Martín, M.C.; Cigudosa, J.C.; Garcia-Castro, J.; de la Fuente, R. Pitfalls in spontaneous in vitro transformation of human mesenchymal stem cells. Exp. Cell Res. 2010, 316, 1648–1650. [Google Scholar] [CrossRef] [PubMed]
- Lazennec, G.; Lam, P.Y. Recent discoveries concerning the tumor—Mesenchymal stem cell interactions. Biochim. Biophys. Acta 2016, 1866, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Medeiros Tavares Marques, J.C.; Cornélio, D.A.; Nogueira Silbiger, V.; Ducati Luchessi, A.; de Souza, S.; Batistuzzo de Medeiros, S.R. Identification of new genes associated to senescent and tumorigenic phenotypes in mesenchymal stem cells. Sci. Rep. 2017, 7, 17837. [Google Scholar] [CrossRef]
- Li, Y.; Xu, X.; Wang, L.; Liu, G.; Li, Y.; Wu, X.; Jing, Y.; Li, H.; Wang, G. Senescent mesenchymal stem cells promote colorectal cancer cells growth via galectin-3 expression. Cell Biosci. 2015, 5, 21. [Google Scholar] [CrossRef]
- Pan, Q.; Fouraschen, S.M.G.; de Ruiter, P.E.; Dinjens, W.N.M.; Kwekkeboom, J.; Tilanus, H.W.; van der Laan, L.J.W. Detection of spontaneous tumorigenic transformation during culture expansion of human mesenchymal stromal cells. Exp. Biol. Med. 2014, 239, 105–115. [Google Scholar] [CrossRef]
- Heslop, J.A.; Hammond, T.G.; Santeramo, I.; Tort Piella, A.; Hopp, I.; Zhou, J.; Baty, R.; Graziano, E.I.; Proto Marco, B.; Caron, A.; et al. Concise review: Workshop review: Understanding and assessing the risks of stem cell-based therapies. Stem Cells Transl. Med. 2015, 4, 389–400. [Google Scholar] [CrossRef]
- Caplan, A.I. Adult Mesenchymal Stem Cells: When, Where, and How. Stem Cells Int. 2015, 2015, 628767. [Google Scholar] [CrossRef]
- Krueger, T.E.G.; Thorek, D.L.J.; Denmeade, S.R.; Isaacs, J.T.; Brennen, W.N. Concise Review: Mesenchymal Stem Cell-Based Drug Delivery: The Good, the Bad, the Ugly, and the Promise. Stem Cells Transl. Med. 2018, 7, 651–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leyendecker, A.; Pinheiro, C.C.G.; Amano, M.T.; Bueno, D.F. The Use of Human Mesenchymal Stem Cells as Therapeutic Agents for the in vivo Treatment of Immune-Related Diseases: A Systematic Review. Front. Immunol. 2018, 9, 2056. [Google Scholar] [CrossRef] [PubMed]
- Yong, K.W.; Choi, J.R.; Mohammadi, M.; Mitha, A.P.; Sanati-Nezhad, A.; Sen, A. Mesenchymal Stem Cell Therapy for Ischemic Tissues. Stem Cells Int. 2018, 2018, 8179075. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-T.; Ting, C.-H.; Yen, M.-L.; Liu, K.-J.; Sytwu, H.-K.; Wu, K.K.; Yen, B.L. Human mesenchymal stem cells (MSCs) for treatment towards immune- and inflammation-mediated diseases: Review of current clinical trials. J. Biomed. Sci. 2016, 23, 76. [Google Scholar] [CrossRef] [PubMed]
- Waterman, R.S.; Tomchuck, S.L.; Henkle, S.L.; Betancourt, A.M. A new mesenchymal stem cell (MSC) paradigm: Polarization into a pro-inflammatory MSC1 or an Immunosuppressive MSC2 phenotype. PLoS ONE 2010, 5, e10088. [Google Scholar] [CrossRef] [PubMed]
- Boregowda, S.V.; Phinney, D.G. Quantifiable Metrics for Predicting MSC Therapeutic Efficacy. J. Stem Cell Res. Ther. 2016, 6, 365. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Characterization Criteria | Assays | Methods | Release Criteria | According to Different Frameworks | |
|---|---|---|---|---|---|
| FDA | EMA | ||||
| Identity | Morphology | Microscopy |
| N/A | N/A |
| Phenotypic markers | Flow cytometry, FACS analysis |
| N/A | N/A | |
| Ability to differentiate | Stain | Differentiate into osteoblasts, adipocytes or chondroblasts | N/A | N/A | |
| Purity | Endotoxin detection | LAL test | ≤0.5 EU/ml | ICH Guideline Q4B Annex 14 | |
|
| ||||
| Potency | Paracrine secretion (cytokines) |
|
| ICH Q6B | |
|
| ||||
| Viability | Living/dead cell count |
|
|
|
|
| Proliferative capacity | Total cumulative population doubling | Total number of cellular divisions | No senescence | N/A | N/A |
| Genomic stability | Karyotype |
(at least 20metaphases) |
| N/A | N/A |
| Microbiological quality control | Sterility test | Direct inoculation | Negative (no haze in the media) | ICH guideline Q4B Annex 8 | |
|
| ||||
| Mycoplasma test | Real-time PCR | Negative |
|
| |
| Adventitious viruses (for allogeneic products) | In vitro adventitious viral agent test | Negative | ICH Topic Q 5 A (R1) | ||
|
| ||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guadix, J.A.; López-Beas, J.; Clares, B.; Soriano-Ruiz, J.L.; Zugaza, J.L.; Gálvez-Martín, P. Principal Criteria for Evaluating the Quality, Safety and Efficacy of hMSC-Based Products in Clinical Practice: Current Approaches and Challenges. Pharmaceutics 2019, 11, 552. https://doi.org/10.3390/pharmaceutics11110552
Guadix JA, López-Beas J, Clares B, Soriano-Ruiz JL, Zugaza JL, Gálvez-Martín P. Principal Criteria for Evaluating the Quality, Safety and Efficacy of hMSC-Based Products in Clinical Practice: Current Approaches and Challenges. Pharmaceutics. 2019; 11(11):552. https://doi.org/10.3390/pharmaceutics11110552
Chicago/Turabian StyleGuadix, Juan Antonio, Javier López-Beas, Beatriz Clares, José Luis Soriano-Ruiz, José Luis Zugaza, and Patricia Gálvez-Martín. 2019. "Principal Criteria for Evaluating the Quality, Safety and Efficacy of hMSC-Based Products in Clinical Practice: Current Approaches and Challenges" Pharmaceutics 11, no. 11: 552. https://doi.org/10.3390/pharmaceutics11110552
APA StyleGuadix, J. A., López-Beas, J., Clares, B., Soriano-Ruiz, J. L., Zugaza, J. L., & Gálvez-Martín, P. (2019). Principal Criteria for Evaluating the Quality, Safety and Efficacy of hMSC-Based Products in Clinical Practice: Current Approaches and Challenges. Pharmaceutics, 11(11), 552. https://doi.org/10.3390/pharmaceutics11110552