The Mechanistic Differences in HLA-Associated Carbamazepine Hypersensitivity


 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Maintenance of Cell Lines
2.2. Production of Soluble HLA Molecules
2.3. Mass Spectrometric Sequencing of the Presented Peptides and Measurement of the Drugs
2.4. Mass Spectrometric Analysis of Drug-Induced Modifications of the Proteome
3. Results
3.1. HLA-A*31:01 and B*15:02 Differ Functionally by Their Peptide Loading
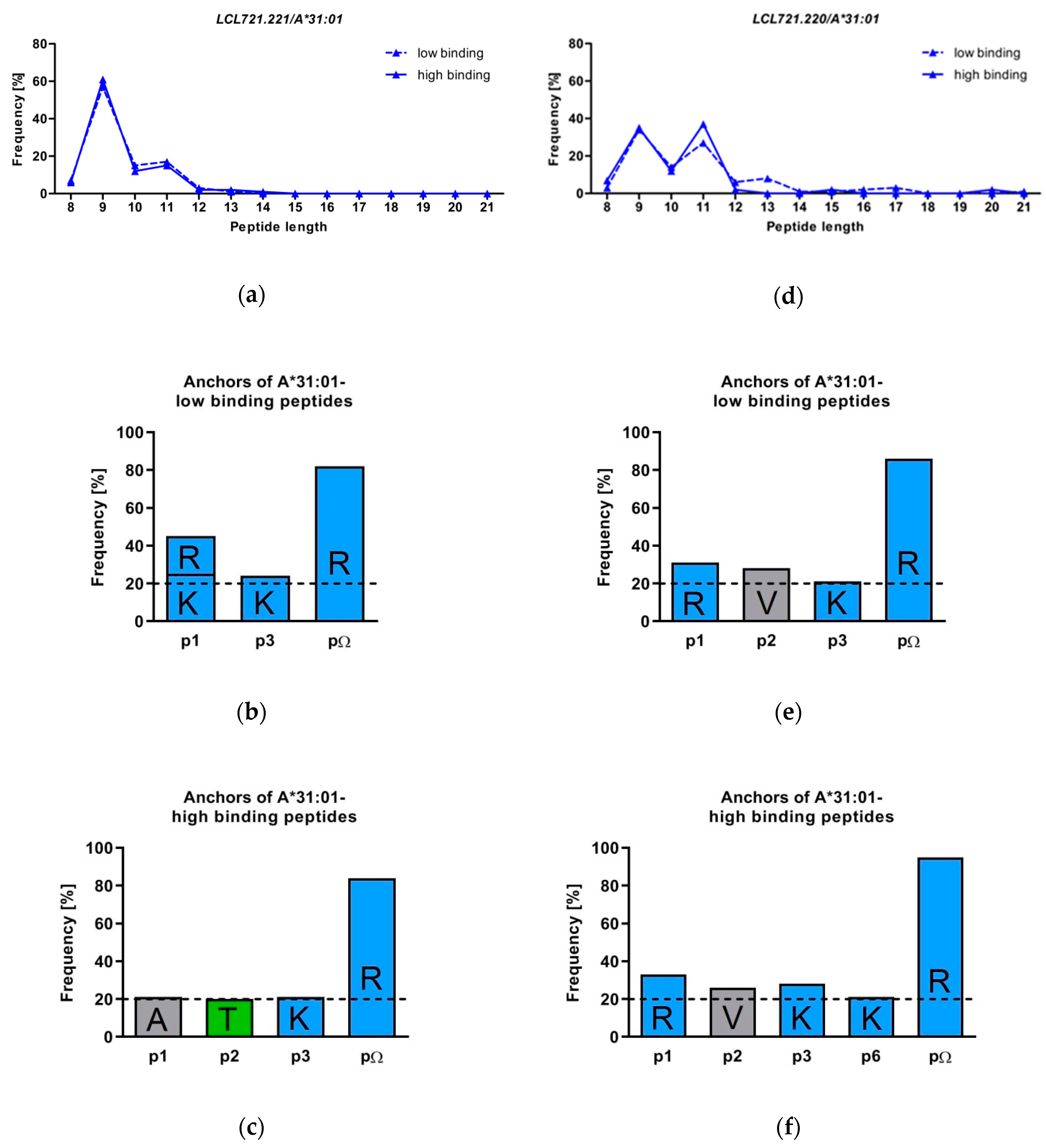
3.2. HLA-A*31:01 Shows a Preference for Presentation of 9 and 11 Mers
3.3. The Peptide Binding Motif of HLA-A*31:01 Exhibits a Strong Arginine Anchor at pΩ
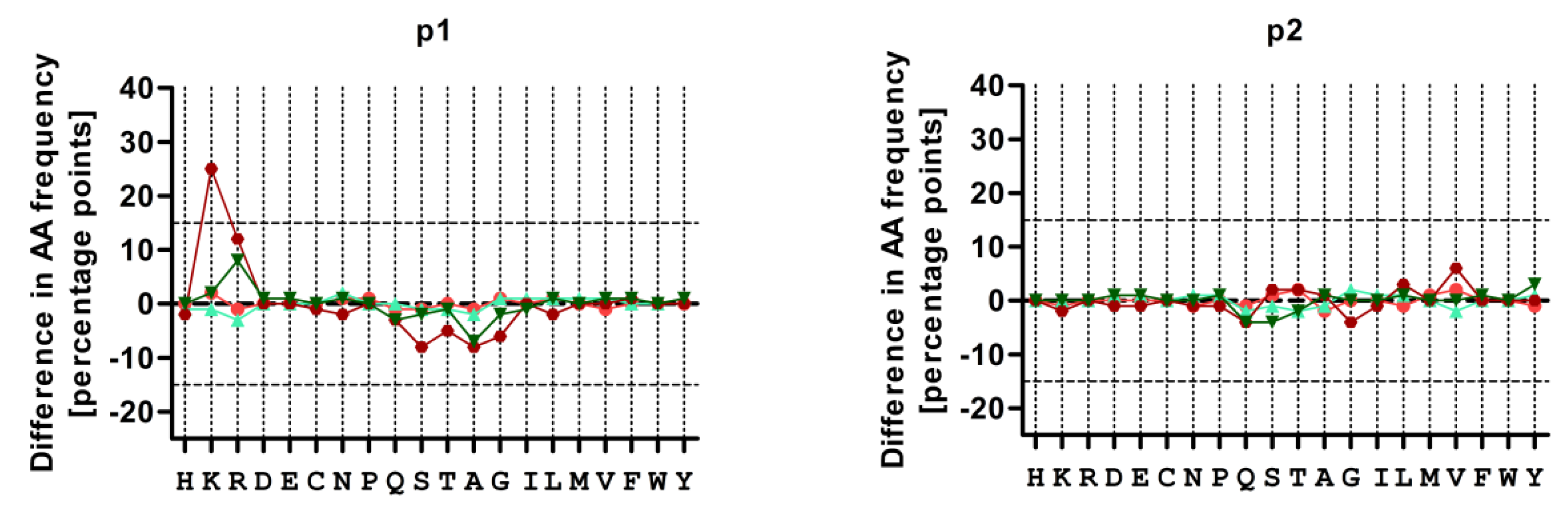
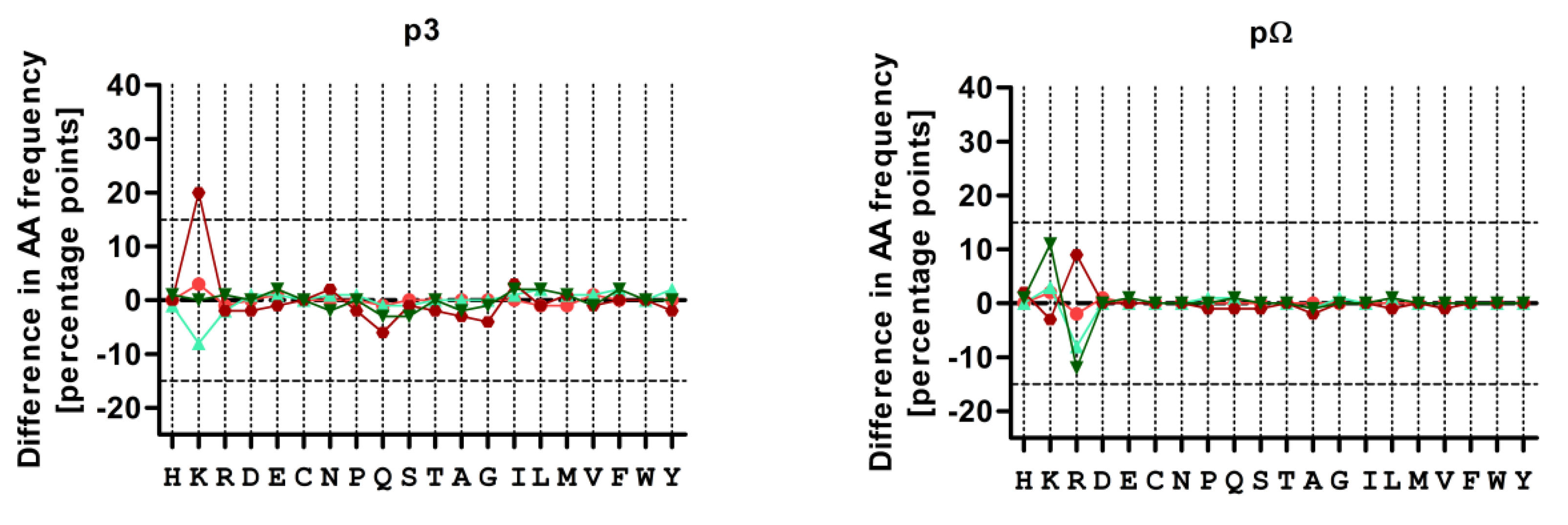
3.4. Treatment with CBZ Alters the Motif of HLA-A*31:01 High Binding Peptides
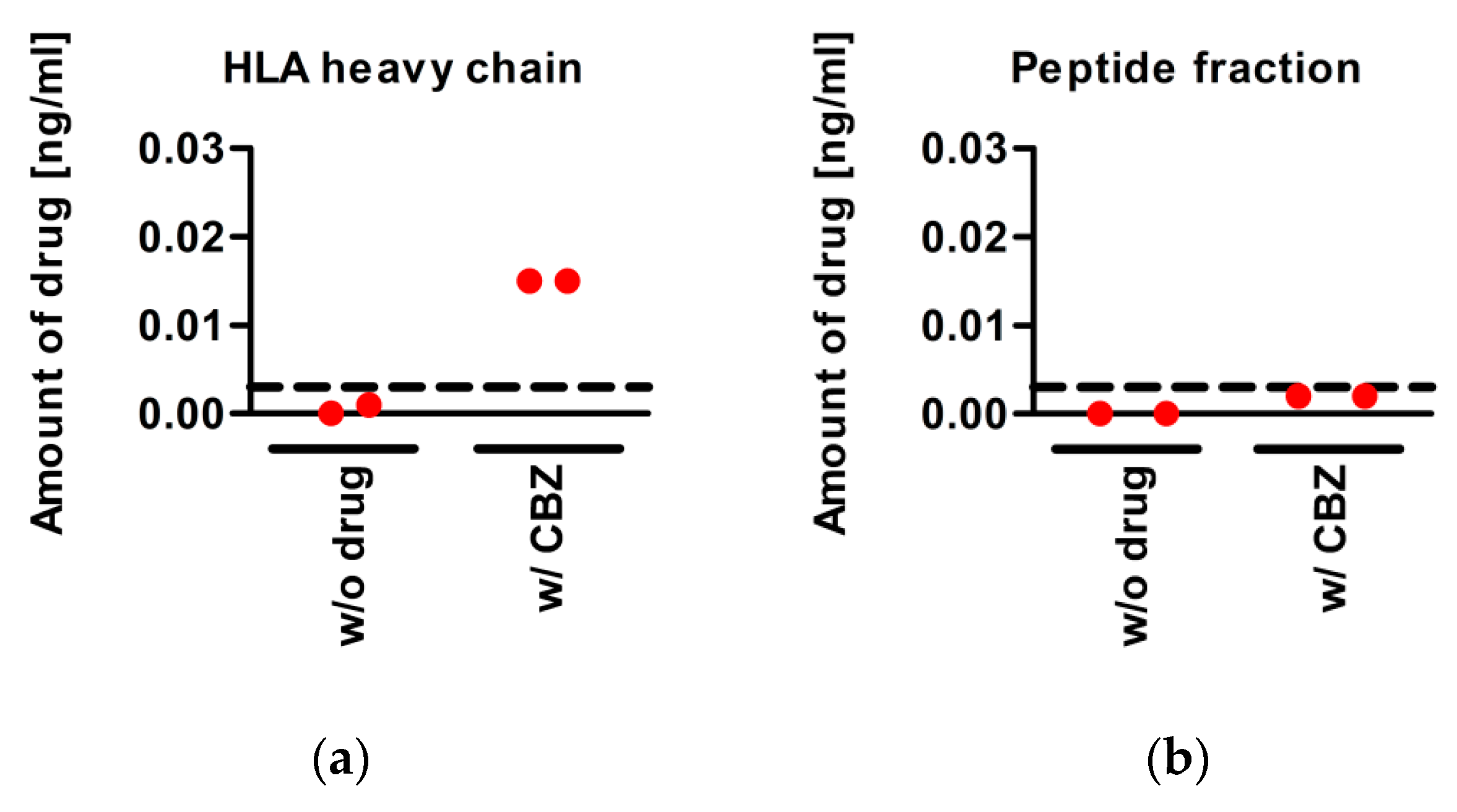
3.5. CBZ Binds to sA*31:01 Heavy Chain
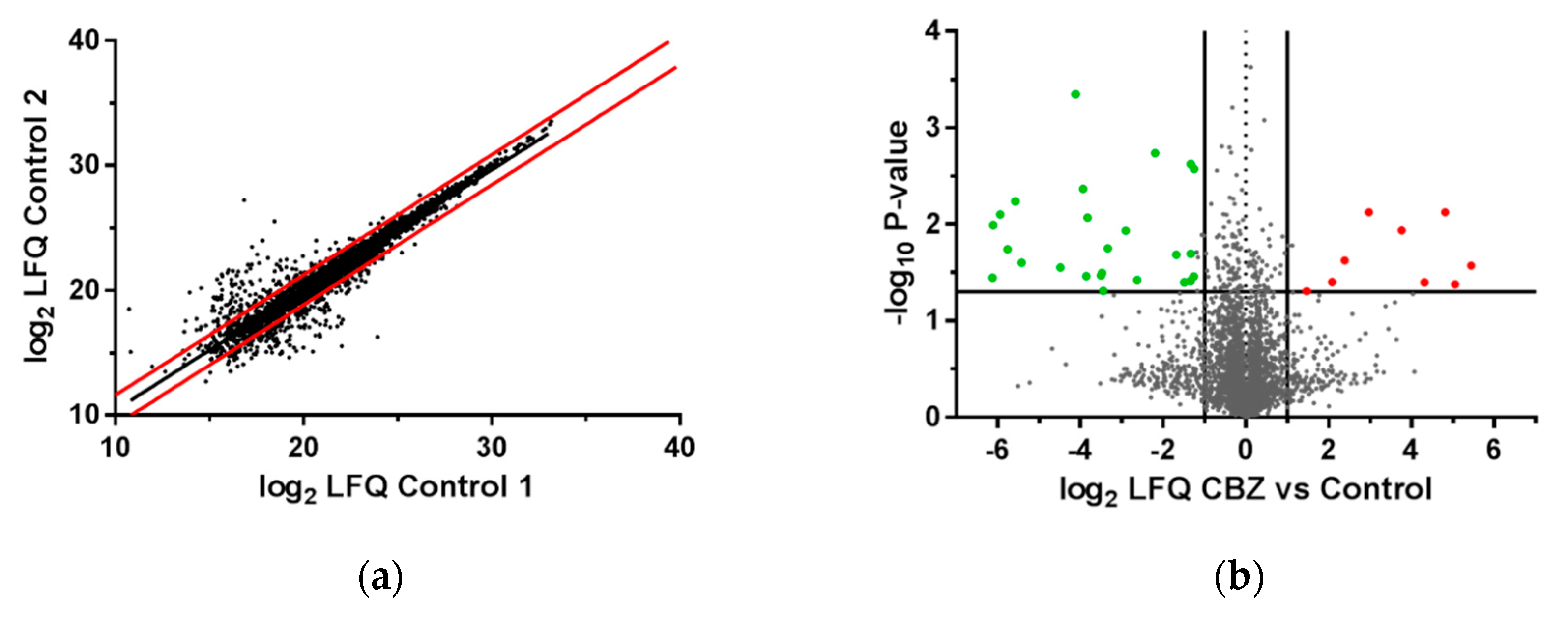
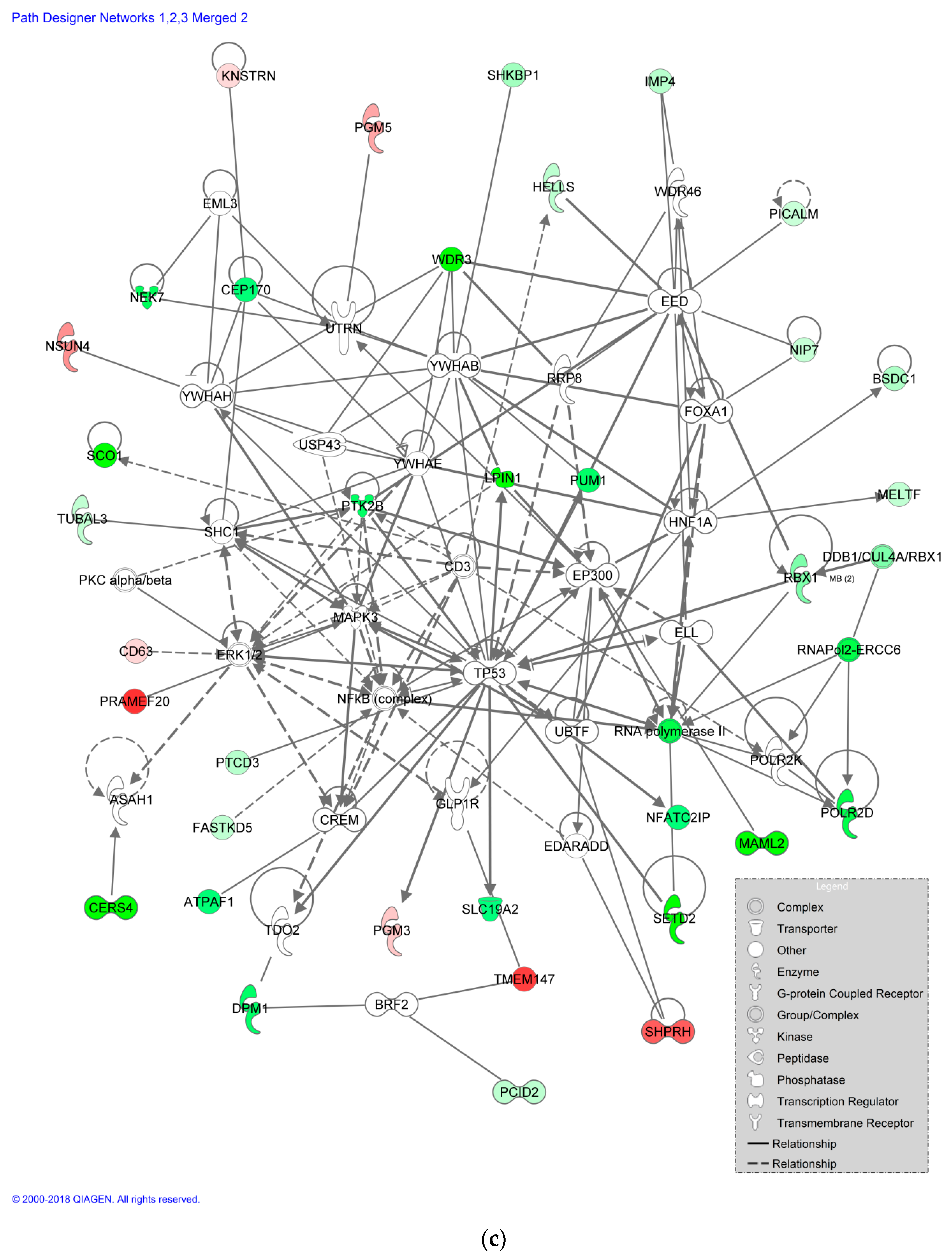
3.6. Impact of CBZ Treatment on Protein Expression of Recombinant B Cells
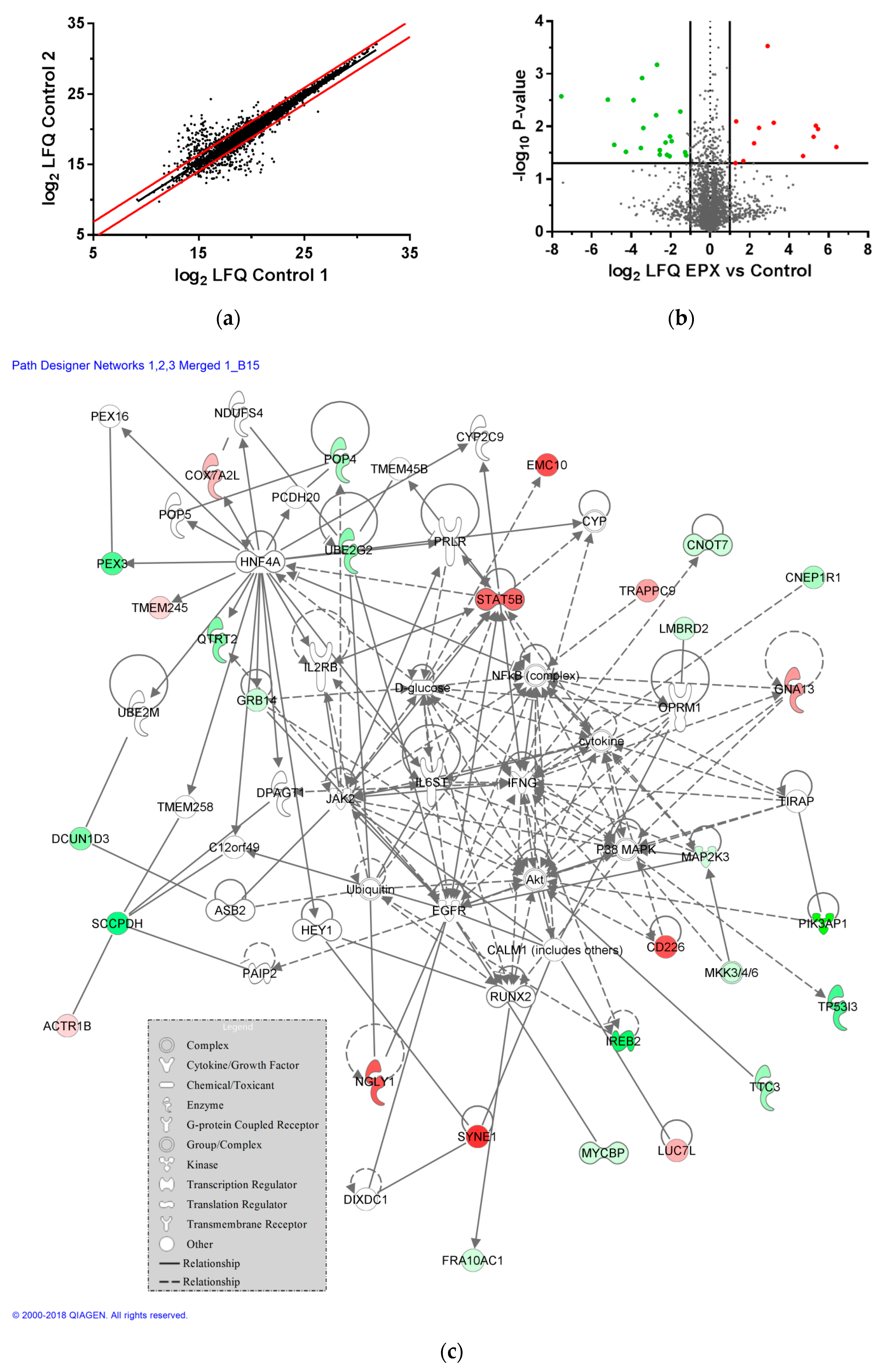
3.7. Impact of EPX Treatment on Protein Expression of Recombinant B Cells
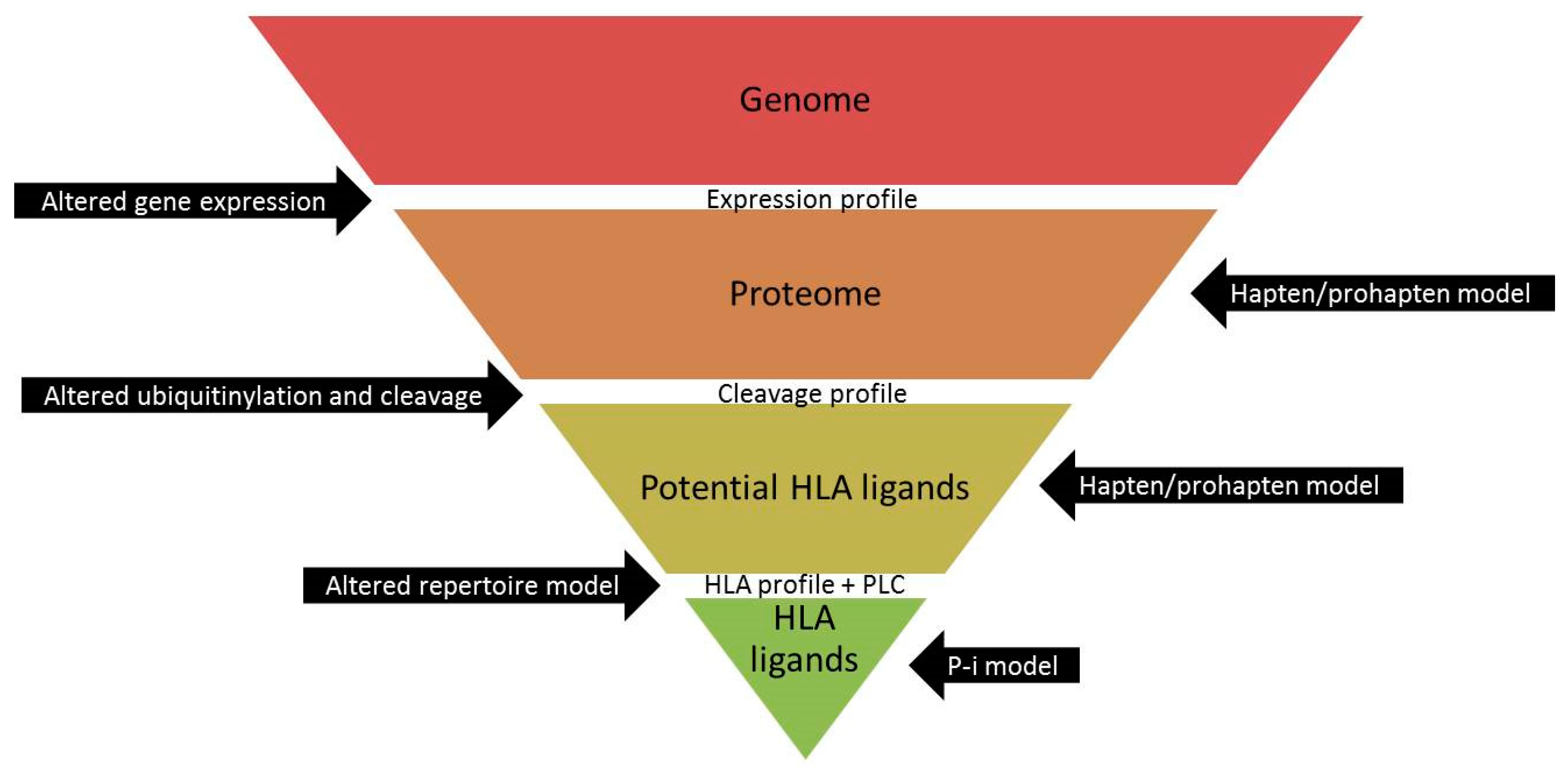
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gomes, E.R.; Demoly, P. Epidemiology of hypersensitivity drug reactions. Curr. Opin. Allergy Clin. Immunol. 2005, 5, 309–316. [Google Scholar] [CrossRef]
- Davies, E.C.; Green, C.F.; Taylor, S.; Williamson, P.R.; Mottram, D.R.; Pirmohamed, M. Adverse drug reactions in hospital in-patients: A prospective analysis of 3695 patient-episodes. PLoS ONE 2009, 4, e4439. [Google Scholar] [CrossRef]
- Pirmohamed, M.; James, S.; Meakin, S.; Green, C.; Scott, A.K.; Walley, T.J.; Farrar, K.; Park, B.K.; Breckenridge, A.M. Adverse drug reactions as cause of admission to hospital: Prospective analysis of 18 820 patients. BMJ 2004, 329, 15–19. [Google Scholar] [CrossRef]
- Mittmann, N.; Knowles, S.R.; Gomez, M.; Fish, J.S.; Cartotto, R.; Shear, N.H. Evaluation of the extent of under-reporting of serious adverse drug reactions: The case of toxic epidermal necrolysis. Drug Saf. 2004, 27, 477–487. [Google Scholar] [CrossRef]
- Hazell, L.; Shakir, S.A. Under-reporting of adverse drug reactions: A systematic review. Drug Saf. 2006, 29, 385–396. [Google Scholar] [CrossRef]
- Alfirevic, A.; Pirmohamed, M. Genomics of adverse drug reactions. Trends Pharmacol. Sci. 2017, 38, 100–109. [Google Scholar] [CrossRef]
- Simper, G.; Celik, A.A.; Kunze-Schumacher, H.; Blasczyk, R.; Bade-Döding, C. Physiology and pathology of drug hypersensitivity: Role of human leukocyte antigens. In Physiology and Pathology of Immunology; IntechOpen: London, UK, 2017. [Google Scholar]
- Rawlins, M.D. Clinical pharmacology. Adverse reactions to drugs. Br. Med. J. 1981, 282, 974–976. [Google Scholar] [CrossRef]
- Pirmohamed, M.; Ostrov, D.A.; Park, B.K. New genetic findings lead the way to a better understanding of fundamental mechanisms of drug hypersensitivity. J. Allergy Clin. Immunol. 2015, 136, 236–244. [Google Scholar] [CrossRef]
- Huyton, T.; Ladas, N.; Schumacher, H.; Blasczyk, R.; Bade-Doeding, C. Pocketcheck: Updating the hla class i peptide specificity roadmap. Tissue Antigens 2012, 80, 239–248. [Google Scholar] [CrossRef]
- Garrett, T.P.; Saper, M.A.; Bjorkman, P.J.; Strominger, J.L.; Wiley, D.C. Specificity pockets for the side chains of peptide antigens in hla-aw68. Nature 1989, 342, 692–696. [Google Scholar] [CrossRef]
- Bade-Doding, C.; Theodossis, A.; Gras, S.; Kjer-Nielsen, L.; Eiz-Vesper, B.; Seltsam, A.; Huyton, T.; Rossjohn, J.; McCluskey, J.; Blasczyk, R. The impact of human leukocyte antigen (hla) micropolymorphism on ligand specificity within the hla-b*41 allotypic family. Haematologica 2010, 96, 110–118. [Google Scholar] [CrossRef]
- Bade-Doeding, C.; DeLuca, D.S.; Seltsam, A.; Blasczyk, R.; Eiz-Vesper, B. Amino acid 95 causes strong alteration of peptide position pomega in hla-b*41 variants. Immunogenetics 2007, 59, 253–259. [Google Scholar] [CrossRef]
- Badrinath, S.; Saunders, P.; Huyton, T.; Aufderbeck, S.; Hiller, O.; Blasczyk, R.; Bade-Doeding, C. Position 156 influences the peptide repertoire and tapasin dependency of human leukocyte antigen b*44 allotypes. Haematologica 2012, 97, 98–106. [Google Scholar] [CrossRef]
- Blees, A.; Januliene, D.; Hofmann, T.; Koller, N.; Schmidt, C.; Trowitzsch, S.; Moeller, A.; Tampe, R. Structure of the human mhc-i peptide-loading complex. Nature 2017, 551, 525–528. [Google Scholar] [CrossRef]
- Aladin, F.; Lautscham, G.; Humphries, E.; Coulson, J.; Blake, N. Targeting tumour cells with defects in the mhc class i antigen processing pathway with cd8+ t cells specific for hydrophobic tap- and tapasin-independent peptides: The requirement for directed access into the er. Cancer Immunol. Immunother. 2007, 56, 1143–1152. [Google Scholar] [CrossRef]
- Coombs, R.R.A.; Gell, P.G.H. Classification of allergic reactions responsible for drug hypersensitivity reactions. In Clinical Aspects of Immunology, 2nd ed.; Coombs, R.R.A., Gell, P.G.H., Eds.; Davis: Philadelphia, PA, USA, 1968; pp. 575–596. [Google Scholar]
- Pichler, W.J.; Hausmann, O. Classification of drug hypersensitivity into allergic, p-i, and pseudo-allergic forms. Int. Arch. Allergy Immunol. 2016, 171, 166–179. [Google Scholar] [CrossRef]
- Faulkner, L.; Meng, X.; Park, B.K.; Naisbitt, D.J. The importance of hapten-protein complex formation in the development of drug allergy. Curr. Opin. Allergy Clin. Immunol. 2014, 14, 293–300. [Google Scholar] [CrossRef]
- Adam, J.; Pichler, W.J.; Yerly, D. Delayed drug hypersensitivity: Models of t-cell stimulation. Br. J. Clin. Pharmacol. 2011, 71, 701–707. [Google Scholar] [CrossRef]
- Naisbitt, D.J.; Farrell, J.; Gordon, S.F.; Maggs, J.L.; Burkhart, C.; Pichler, W.J.; Pirmohamed, M.; Park, B.K. Covalent binding of the nitroso metabolite of sulfamethoxazole leads to toxicity and major histocompatibility complex-restricted antigen presentation. Mol. Pharmacol. 2002, 62, 628–637. [Google Scholar] [CrossRef]
- Ariza, A.; Mayorga, C.; Fernandez, T.D.; Barbero, N.; Martin-Serrano, A.; Perez-Sala, D.; Sanchez-Gomez, F.J.; Blanca, M.; Torres, M.J.; Montanez, M.I. Hypersensitivity reactions to beta-lactams: Relevance of hapten-protein conjugates. J. Investig. Allergol. Clin. Immunol. 2015, 25, 12–25. [Google Scholar]
- Pichler, W.J.; Beeler, A.; Keller, M.; Lerch, M.; Posadas, S.; Schmid, D.; Spanou, Z.; Zawodniak, A.; Gerber, B. Pharmacological interaction of drugs with immune receptors: The p-i concept. Allergol. Int. 2006, 55, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Illing, P.T.; Vivian, J.P.; Dudek, N.L.; Kostenko, L.; Chen, Z.; Bharadwaj, M.; Miles, J.J.; Kjer-Nielsen, L.; Gras, S.; Williamson, N.A.; et al. Immune self-reactivity triggered by drug-modified hla-peptide repertoire. Nature 2012, 486, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Ostrov, D.A.; Grant, B.J.; Pompeu, Y.A.; Sidney, J.; Harndahl, M.; Southwood, S.; Oseroff, C.; Lu, S.; Jakoncic, J.; de Oliveira, C.A.; et al. Drug hypersensitivity caused by alteration of the mhc-presented self-peptide repertoire. Proc. Natl. Acad. Sci. USA 2012, 109, 9959–9964. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, W.A.; Purcell, A.W.; Mifsud, N.A.; Ely, L.K.; Williams, D.S.; Chang, L.; Gorman, J.J.; Clements, C.S.; Kjer-Nielsen, L.; Koelle, D.M.; et al. A naturally selected dimorphism within the hla-b44 supertype alters class i structure, peptide repertoire, and t cell recognition. J. Exp. Med. 2003, 198, 679–691. [Google Scholar] [CrossRef]
- Shorvon, S.D. Drug treatment of epilepsy in the century of the ilae: The second 50 years, 1959–2009. Epilepsia 2009, 50, 93–130. [Google Scholar] [CrossRef]
- Marson, A.G.; Al-Kharusi, A.M.; Alwaidh, M.; Appleton, R.; Baker, G.A.; Chadwick, D.W.; Cramp, C.; Cockerell, O.C.; Cooper, P.N.; Doughty, J.; et al. The sanad study of effectiveness of carbamazepine, gabapentin, lamotrigine, oxcarbazepine, or topiramate for treatment of partial epilepsy: An unblinded randomised controlled trial. Lancet 2007, 369, 1000–1015. [Google Scholar] [CrossRef]
- Di Stefano, G.; La Cesa, S.; Truini, A.; Cruccu, G. Natural history and outcome of 200 outpatients with classical trigeminal neuralgia treated with carbamazepine or oxcarbazepine in a tertiary centre for neuropathic pain. J. Headache Pain 2014, 15, 34. [Google Scholar] [CrossRef]
- Di Stefano, G.; Truini, A.; Cruccu, G. Current and innovative pharmacological options to treat typical and atypical trigeminal neuralgia. Drugs 2018, 78, 1433–1442. [Google Scholar] [CrossRef]
- Simpson, J.R. “Collagen disease” due to carbamazepine (tegretol). Br. Med. J. 1966, 2, 1434. [Google Scholar] [CrossRef][Green Version]
- Gayford, J.J.; Redpath, T.H. The side-effects of carbamazepine. Proc. R. Soc. Med. 1969, 62, 615–616. [Google Scholar] [CrossRef]
- Yip, V.L.M.; Meng, X.; Maggs, J.L.; Jenkins, R.E.; Marlot, P.T.; Marson, A.G.; Park, B.K.; Pirmohamed, M. Mass spectrometric characterization of circulating covalent protein adducts derived from epoxide metabolites of carbamazepine in patients. Chem. Res. Toxicol. 2017, 30, 1419–1435. [Google Scholar] [CrossRef] [PubMed]
- Kardaun, S.H.; Sekula, P.; Valeyrie-Allanore, L.; Liss, Y.; Chu, C.Y.; Creamer, D.; Sidoroff, A.; Naldi, L.; Mockenhaupt, M.; Roujeau, J.C.; et al. Drug reaction with eosinophilia and systemic symptoms (dress): An original multisystem adverse drug reaction. Results from the prospective regiscar study. Br. J. Dermatol. 2013, 169, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Mockenhaupt, M. Stevens-John syndrome and toxic epidermal necrolysis: Clinical patterns, diagnostic considerations, etiology, and therapeutic management. Semin. Cutan. Med. Surg. 2014, 33, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Demoly, P.; Adkinson, N.F.; Brockow, K.; Castells, M.; Chiriac, A.M.; Greenberger, P.A.; Khan, D.A.; Lang, D.M.; Park, H.S.; Pichler, W.; et al. International consensus on drug allergy. Allergy 2014, 69, 420–437. [Google Scholar] [CrossRef]
- Chung, W.H.; Hung, S.I.; Hong, H.S.; Hsih, M.S.; Yang, L.C.; Ho, H.C.; Wu, J.Y.; Chen, Y.T. Medical genetics: A marker for Stevens-Johnson syndrome. Nature 2004, 428, 486. [Google Scholar] [CrossRef]
- Yip, V.L.; Pirmohamed, M. The hla-a*31:01 allele: Influence on carbamazepine treatment. Pharmgenom. Pers. Med. 2017, 10, 29–38. [Google Scholar] [CrossRef]
- Alfirevic, A.; Jorgensen, A.L.; Williamson, P.R.; Chadwick, D.W.; Park, B.K.; Pirmohamed, M. Hla-b locus in caucasian patients with carbamazepine hypersensitivity. Pharmacogenomics 2006, 7, 813–818. [Google Scholar] [CrossRef]
- Hung, S.I.; Chung, W.H.; Jee, S.H.; Chen, W.C.; Chang, Y.T.; Lee, W.R.; Hu, S.L.; Wu, M.T.; Chen, G.S.; Wong, T.W.; et al. Genetic susceptibility to carbamazepine-induced cutaneous adverse drug reactions. Pharmacogenet. Genom. 2006, 16, 297–306. [Google Scholar] [CrossRef]
- Locharernkul, C.; Loplumlert, J.; Limotai, C.; Korkij, W.; Desudchit, T.; Tongkobpetch, S.; Kangwanshiratada, O.; Hirankarn, N.; Suphapeetiporn, K.; Shotelersuk, V. Carbamazepine and phenytoin induced Stevens-Johnson syndrome is associated with hla-b*1502 allele in Thai population. Epilepsia 2008, 49, 2087–2091. [Google Scholar] [CrossRef]
- Mehta, T.Y.; Prajapati, L.M.; Mittal, B.; Joshi, C.G.; Sheth, J.J.; Patel, D.B.; Dave, D.M.; Goyal, R.K. Association of hla-b*1502 allele and carbamazepine-induced Stevens-Johnson syndrome among Indians. Indian J. Dermatol. Venereol. Leprol. 2009, 75, 579–582. [Google Scholar] [CrossRef]
- Tassaneeyakul, W.; Tiamkao, S.; Jantararoungtong, T.; Chen, P.; Lin, S.Y.; Chen, W.H.; Konyoung, P.; Khunarkornsiri, U.; Auvichayapat, N.; Pavakul, K.; et al. Association between hla-b*1502 and carbamazepine-induced severe cutaneous adverse drug reactions in a Thai population. Epilepsia 2010, 51, 926–930. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Too, C.L.; Murad, S.; Hussein, S.H. Association of hla-b*1502 allele with carbamazepine-induced toxic epidermal necrolysis and Stevens-Johnson syndrome in the multi-ethnic Malaysian population. Int. J. Dermatol. 2011, 50, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Then, S.M.; Rani, Z.Z.; Raymond, A.A.; Ratnaningrum, S.; Jamal, R. Frequency of the hla-b*1502 allele contributing to carbamazepine-induced hypersensitivity reactions in a cohort of Malaysian epilepsy patients. Asian Pac. J. Allergy Immunol. 2011, 29, 290–293. [Google Scholar] [PubMed]
- Ferrell, P.B., Jr.; McLeod, H.L. Carbamazepine, hla-b*1502 and risk of Stevens-Johnson syndrome and toxic epidermal necrolysis: US FDA recommendations. Pharmacogenomics 2008, 9, 1543–1546. [Google Scholar] [CrossRef] [PubMed]
- McCormack, M.; Alfirevic, A.; Bourgeois, S.; Farrell, J.J.; Kasperaviciute, D.; Carrington, M.; Sills, G.J.; Marson, T.; Jia, X.; de Bakker, P.I.; et al. Hla-a*3101 and carbamazepine-induced hypersensitivity reactions in Europeans. N. Engl. J. Med. 2011, 364, 1134–1143. [Google Scholar] [CrossRef]
- Ozeki, T.; Mushiroda, T.; Yowang, A.; Takahashi, A.; Kubo, M.; Shirakata, Y.; Ikezawa, Z.; Iijima, M.; Shiohara, T.; Hashimoto, K.; et al. Genome-wide association study identifies hla-a*3101 allele as a genetic risk factor for carbamazepine-induced cutaneous adverse drug reactions in Japanese population. Hum. Mol. Genet. 2011, 20, 1034–1041. [Google Scholar] [CrossRef]
- Kim, S.H.; Lee, K.W.; Song, W.J.; Kim, S.H.; Jee, Y.K.; Lee, S.M.; Kang, H.R.; Park, H.W.; Cho, S.H.; Park, S.H.; et al. Carbamazepine-induced severe cutaneous adverse reactions and hla genotypes in Koreans. Epilepsy Res. 2011, 97, 190–197. [Google Scholar] [CrossRef]
- Yang, C.W.; Hung, S.I.; Juo, C.G.; Lin, Y.P.; Fang, W.H.; Lu, I.H.; Chen, S.T.; Chen, Y.T. Hla-b*1502-bound peptides: Implications for the pathogenesis of carbamazepine-induced Stevens-Johnson syndrome. J. Allergy Clin. Immunol. 2007, 120, 870–877. [Google Scholar] [CrossRef]
- Simper, G.S.; Ho, G.T.; Celik, A.A.; Huyton, T.; Kuhn, J.; Kunze-Schumacher, H.; Blasczyk, R.; Bade-Doding, C. Carbamazepine-mediated adverse drug reactions: Cbz-10,11-epoxide but not carbamazepine induces the alteration of peptides presented by hla-b *15:02. J. Immunol. Res. 2018, 2018, 5086503. [Google Scholar] [CrossRef]
- Abelin, J.G.; Keskin, D.B.; Sarkizova, S.; Hartigan, C.R.; Zhang, W.; Sidney, J.; Stevens, J.; Lane, W.; Zhang, G.L.; Eisenhaure, T.M.; et al. Mass spectrometry profiling of hla-associated peptidomes in mono-allelic cells enables more accurate epitope prediction. Immunity 2017, 46, 315–326. [Google Scholar] [CrossRef]
- Ko, T.M.; Chung, W.H.; Wei, C.Y.; Shih, H.Y.; Chen, J.K.; Lin, C.H.; Chen, Y.T.; Hung, S.I. Shared and restricted t-cell receptor use is crucial for carbamazepine-induced Stevens-Johnson syndrome. J. Allergy Clin. Immunol. 2011, 128, 1266–1276. [Google Scholar] [CrossRef] [PubMed]
- Thorn, C.F.; Leckband, S.G.; Kelsoe, J.; Leeder, J.S.; Muller, D.J.; Klein, T.E.; Altman, R.B. Pharmgkb summary: Carbamazepine pathway. Pharmacogenet. Genom. 2011, 21, 906–910. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.Y.; Chung, W.H.; Huang, H.W.; Chen, Y.T.; Hung, S.I. Direct interaction between hla-b and carbamazepine activates t cells in patients with Stevens-Johnson syndrome. J. Allergy Clin. Immunol. 2012, 129, 1562–1569. [Google Scholar] [CrossRef] [PubMed]
- He, X.J.; Jian, L.Y.; He, X.L.; Tang, M.; Wu, Y.; Xu, Y.Y.; Sun, X.J.; Zhao, L.M. Association of abcb1, cyp3a4, ephx1, fas, scn1a, mica, and bag6 polymorphisms with the risk of carbamazepine-induced Stevens-Johnson syndrome/toxic epidermal necrolysis in Chinese han patients with epilepsy. Epilepsia 2014, 55, 1301–1306. [Google Scholar] [CrossRef]
- Lichtenfels, M.; Farrell, J.; Ogese, M.O.; Bell, C.C.; Eckle, S.; McCluskey, J.; Park, B.K.; Alfirevic, A.; Naisbitt, D.J.; Pirmohamed, M. Hla restriction of carbamazepine-specific t-cell clones from an hla-a*31:01-positive hypersensitive patient. Chem. Res. Toxicol. 2014, 27, 175–177. [Google Scholar] [CrossRef]
- DeMars, R.; Rudersdorf, R.; Chang, C.; Petersen, J.; Strandtmann, J.; Korn, N.; Sidwell, B.; Orr, H.T. Mutations that impair a posttranscriptional step in expression of hla-a and -b antigens. Proc. Natl. Acad. Sci. USA 1985, 82, 8183–8187. [Google Scholar] [CrossRef]
- Kavathas, P.; Bach, F.H.; DeMars, R. Gamma ray-induced loss of expression of hla and glyoxalase i alleles in lymphoblastoid cells. Proc. Natl. Acad. Sci. USA 1980, 77, 4251–4255. [Google Scholar] [CrossRef]
- Graham, F.L.; Smiley, J.; Russell, W.C.; Nairn, R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 1977, 36, 59–74. [Google Scholar] [CrossRef]
- Kunze-Schumacher, H. Analyse des Peptidrepertoires von Hla-A*31:01 und Hla-B*15:02: Auswirkungen Bei Der Hla-Assoziierten Arzneimittelunverträglichkeit. Bachelor Thesis, Hannover Medical School, Hannover, Germany, 2014. [Google Scholar]
- Bade-Doeding, C.; Cano, P.; Huyton, T.; Badrinath, S.; Eiz-Vesper, B.; Hiller, O.; Blasczyk, R. Mismatches outside exons 2 and 3 do not alter the peptide motif of the allele group b*44:02p. Hum. Immunol. 2011, 72, 1039–1044. [Google Scholar] [CrossRef]
- Celik, A.A.; Simper, G.S.; Hiemisch, W.; Blasczyk, R.; Bade-Doding, C. Hla-g peptide preferences change in transformed cells: Impact on the binding motif. Immunogenetics 2018, 70, 485–494. [Google Scholar] [CrossRef]
- Ho, G.T.; Heinen, F.J.; Huyton, T.; Blasczyk, R.; Bade-Doding, C. Hla-f*01:01 presents peptides with n-terminal flexibility and a preferred length of 16 residues. Immunogenetics 2019, 71, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [PubMed]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods. 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Junemann, J.; Birgin, G.; Erdmann, J.; Schroder, A.; Just, I.; Gerhard, R.; Pich, A. Toxin a of the nosocomial pathogen clostridium difficile induces primary effects in the proteome of hep-2 cells. Proteom. Clin. Appl. 2017, 11, 1600031. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. Maxquant enables high peptide identification rates, individualized p.P.B.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. 1d and 2d annotation enrichment: A statistical method integrating quantitative proteomics with complementary high-throughput data. BMC Bioinform. 2012, 13, S12. [Google Scholar] [CrossRef]
- Consortium, T.U. Uniprot: The universal protein knowledgebase. Nucl. Acids Res. 2018, 46, 2699. [Google Scholar]
- Krämer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Kim, S.; Chung, J.K.; Im, S.H.; Jeong, J.M.; Lee, D.S.; Kim, D.G.; Jung, H.W.; Lee, M.C. 11c-methionine pet as a prognostic marker in patients with glioma: Comparison with 18f-fdg pet. Eur. J. Nucl. Med. Mol. Imaging 2005, 32, 52–59. [Google Scholar] [CrossRef]
- Wearsch, P.A.; Cresswell, P. The quality control of mhc class i peptide loading. Curr. Opin. Cell Biol. 2008, 20, 624–631. [Google Scholar] [CrossRef]
- Pichler, W.J. Delayed drug hypersensitivity reactions. Ann. Intern. Med. 2003, 139, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Ambrósio, A.F.; Soares-Da-Silva, P.; Carvalho, C.M.; Carvalho, A.P. Mechanisms of action of carbamazepine and its derivatives, oxcarbazepine, bia 2-093, and bia 2-024. Neurochem. Res. 2002, 27, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.; Sato, A. The hla system. First of two parts. N. Engl. J. Med. 2000, 343, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Unk, I.; Hajdu, I.; Fatyol, K.; Szakal, B.; Blastyak, A.; Bermudez, V.; Hurwitz, J.; Prakash, L.; Prakash, S.; Haracska, L. Human shprh is a ubiquitin ligase for mms2-ubc13-dependent polyubiquitylation of proliferating cell nuclear antigen. Proc. Natl. Acad. Sci. USA 2006, 103, 18107–18112. [Google Scholar] [CrossRef]
- Zhu, H.; Wang, J.; Yin, J.; Lu, B.; Yang, Q.; Wan, Y.; Jia, C. Downregulation of prame suppresses proliferation and promotes apoptosis in hepatocellular carcinoma through the activation of p53 mediated pathway. Cell Physiol. Biochem. 2018, 45, 1121–1135. [Google Scholar] [CrossRef]
- Wu, L.; Sun, T.; Kobayashi, K.; Gao, P.; Griffin, J.D. Identification of a family of mastermind-like transcriptional coactivators for mammalian notch receptors. Mol. Cell Biol. 2002, 22, 7688–7700. [Google Scholar] [CrossRef]
- Camara, Y.; Asin-Cayuela, J.; Park, C.B.; Metodiev, M.D.; Shi, Y.; Ruzzenente, B.; Kukat, C.; Habermann, B.; Wibom, R.; Hultenby, K.; et al. Mterf4 regulates translation by targeting the methyltransferase nsun4 to the mammalian mitochondrial ribosome. Cell Metab. 2011, 13, 527–539. [Google Scholar] [CrossRef]
- Han, G.S.; Wu, W.I.; Carman, G.M. The saccharomyces cerevisiae lipin homolog is a mg2+-dependent phosphatidate phosphatase enzyme. J. Biol. Chem. 2006, 281, 9210–9218. [Google Scholar] [CrossRef]
- Miyazaki, T.; Takaoka, A.; Nogueira, L.; Dikic, I.; Fujii, H.; Tsujino, S.; Mitani, Y.; Maeda, M.; Schlessinger, J.; Taniguchi, T. Pyk2 is a downstream mediator of the il-2 receptor-coupled jak signaling pathway. Genes Dev. 1998, 12, 770–775. [Google Scholar] [CrossRef]
- Mbimba, T.; Hussein, N.J.; Najeed, A.; Safadi, F.F. Trappc9: Novel insights into its trafficking and signaling pathways in health and disease (review). Int. J. Mol. Med. 2018, 42, 2991–2997. [Google Scholar] [CrossRef]
- Rajgor, D.; Shanahan, C.M. Nesprins: From the nuclear envelope and beyond. Expert Rev. Mol. Med. 2013, 15, e5. [Google Scholar] [CrossRef] [PubMed]
- Tahara-Hanaoka, S.; Shibuya, K.; Onoda, Y.; Zhang, H.; Yamazaki, S.; Miyamoto, A.; Honda, S.; Lanier, L.L.; Shibuya, A. Functional characterization of dnam-1 (cd226) interaction with its ligands pvr (cd155) and nectin-2 (prr-2/cd112). Int. Immunol. 2004, 16, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Grimley, P.M.; Dong, F.; Rui, H. Stat5a and stat5b: Fraternal twins of signal transduction and transcriptional activation. Cytokine Growth Factor Rev. 1999, 10, 131–157. [Google Scholar] [CrossRef]
- Hamerman, J.A.; Pottle, J.; Ni, M.; He, Y.; Zhang, Z.Y.; Buckner, J.H. Negative regulation of tlr signaling in myeloid cells–Implications for autoimmune diseases. Immunol. Rev. 2016, 269, 212–227. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fraction No. | Acetonitrile (%) | Triethylamine (%) |
|---|---|---|
| 1 | 7.5 | 0.1 |
| 2 | 12.5 | 0.1 |
| 3 | 15 | 0.1 |
| 4 | 17.5 | 0.1 |
| 5 | 50 | 0.1 |
| Protein Name | Gene Code | log2 Regulation | p-Value |
|---|---|---|---|
| PRAME family member 20/21 | PRAMEF20 | 5.44 | 0.027 |
| Transmembrane protein 147 | TMEM147 | 5.05 | 0.042 |
| Small EDRK-rich factor 2 | SERF2 | 4.81 | 0.008 |
| E3 ubiquitin-protein ligase SHPRH | SHPRH | 4.31 | 0.040 |
| Zinc finger protein Helios | IKZF2;ZNFN1A2 | 3.76 | 0.012 |
| 5-methylcytosine rRNA methyltransferase | NSUN4 | 2.97 | 0.007 |
| Phosphoglucomutase-like protein 5 | PGM5 | 2.39 | 0.024 |
| Protein kinase C-binding protein 1 | ZMYND8 | 2.08 | 0.040 |
| Phosphoacetylglucosamine mutase | PGM3 | 1.47 | 0.049 |
| Protein Name | Gene Code | log2 Regulation | p-Value |
|---|---|---|---|
| Phosphatidate phosphatase LPIN1 | LPIN1 | −6.12 | 0.036 |
| Ceramide synthase 4 | CERS4 | −6.11 | 0.010 |
| Histone-lysine N-methyltransferase SETD2 | SETD2 | −5.94 | 0.008 |
| Mastermind-like protein 2 | MAML2 | −5.76 | 0.018 |
| Protein SCO1 homolog, mitochondrial | SCO1 | −5.58 | 0.006 |
| WD repeat-containing protein 3 | WDR3 | −5.42 | 0.025 |
| DNA-directed RNA polymerase II subunit RPB4 | POLR2D | −4.49 | 0.028 |
| Serine/threonine-protein kinase Nek7 | NEK7 | −4.12 | <0.001 |
| Protein-tyrosine kinase 2-beta | PTK2B | −3.94 | 0.004 |
| Dolichol-phosphate mannosyltransferase subunit 1 | DPM1 | −3.86 | 0.034 |
| Protein Name | Gene Code | log2 Regulation | p-Value |
|---|---|---|---|
| Nesprin-1 | SYNE1 | 6.39 | 0.025 |
| ER membrane protein complex subunit 10 | EMC10 | 5.46 | 0.011 |
| CD226 antigen | CD226 | 5.35 | 0.010 |
| Peptide-N(4)-(N-acetyl-beta-glucosaminyl)asparagine amidase | NGLY1 | 5.24 | 0.016 |
| Signal transducer and activator of transcription 5B | STAT5B | 4.70 | 0.036 |
| Guanine nucleotide-binding protein subunit alpha-13 | GNA13 | 3.22 | 0.008 |
| Trafficking protein particle complex subunit 9 | TRAPPC9 | 2.91 | <0.001 |
| Putative RNA-binding protein Luc7-like 1 | LUC7L | 2.47 | 0.011 |
| Cytochrome c oxidase subunit 7A-related protein, mitochondrial | COX7A2L | 2.23 | 0.021 |
| ESF1 homolog | ESF1 | 1.68 | 0.045 |
| Protein Name | Gene Code | log2 Regulation | p-Value |
|---|---|---|---|
| Phosphoinositide 3-kinase adapter protein 1 | PIK3AP1 | −7.53 | 0.003 |
| Spermidine/spermine N(1)-acetyltransferase-like protein1 | SATL1 | −5.18 | 0.003 |
| Iron-responsive element-binding protein 2 | IREB2 | −4.85 | 0.022 |
| Protein FAM177A1 | FAM177A1 | −4.26 | 0.030 |
| Saccharopine dehydrogenase-like oxidoreductase | SCCPDH | −3.88 | 0.003 |
| Methylsterol monooxygenase 1 | MSMO1 | −3.51 | 0.026 |
| Quinone oxidoreductase PIG3 | TP53I3 | −3.45 | 0.001 |
| Peroxisomal biogenesis factor 3 | PEX3 | −3.38 | 0.011 |
| DCN1-like protein 3 | DCUN1D3 | −2.73 | 0.006 |
| Queuine tRNA-ribosyltransferase subunit | QTRTD1 | −2.69 | 0.001 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simper, G.S.; Gräser, L.S.; Celik, A.A.; Kuhn, J.; Kunze-Schumacher, H.; Hò, G.-G.T.; Blasczyk, R.; Pich, A.; Bade-Doeding, C. The Mechanistic Differences in HLA-Associated Carbamazepine Hypersensitivity. Pharmaceutics 2019, 11, 536. https://doi.org/10.3390/pharmaceutics11100536
Simper GS, Gräser LS, Celik AA, Kuhn J, Kunze-Schumacher H, Hò G-GT, Blasczyk R, Pich A, Bade-Doeding C. The Mechanistic Differences in HLA-Associated Carbamazepine Hypersensitivity. Pharmaceutics. 2019; 11(10):536. https://doi.org/10.3390/pharmaceutics11100536
Chicago/Turabian StyleSimper, Gwendolin S., Lareen S. Gräser, Alexander A. Celik, Joachim Kuhn, Heike Kunze-Schumacher, Gia-Gia T. Hò, Rainer Blasczyk, Andreas Pich, and Christina Bade-Doeding. 2019. "The Mechanistic Differences in HLA-Associated Carbamazepine Hypersensitivity" Pharmaceutics 11, no. 10: 536. https://doi.org/10.3390/pharmaceutics11100536
APA StyleSimper, G. S., Gräser, L. S., Celik, A. A., Kuhn, J., Kunze-Schumacher, H., Hò, G.-G. T., Blasczyk, R., Pich, A., & Bade-Doeding, C. (2019). The Mechanistic Differences in HLA-Associated Carbamazepine Hypersensitivity. Pharmaceutics, 11(10), 536. https://doi.org/10.3390/pharmaceutics11100536