Enhanced In Vitro Antitumor Activity of GnRH-III-Daunorubicin Bioconjugates Influenced by Sequence Modification

 ,
,  ,
, 

Abstract
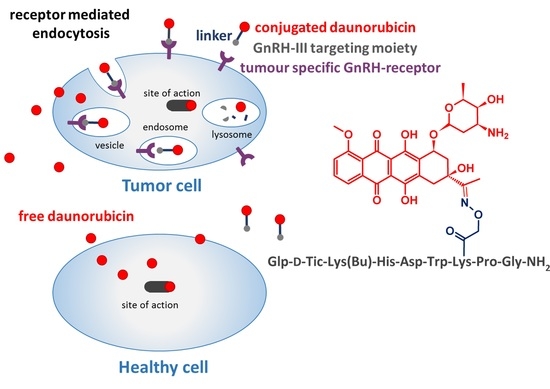
1. Introduction
2. Materials and Methods
2.1. Chemical Reagents
2.2. Synthesis of Oxime Linked GnRH-III-[8Lys(Dau=Aoa)] Bioconjugates
2.3. RP-HPLC
2.4. Mass Spectrometry
2.5. Degradation of GnRH-III Bioconjugates in Rat Liver Lysosomal Homogenate
2.6. Stability in Human Plasma
2.7. Cell Culturing
2.8. In Vitro Cytostatic Effect
2.9. Cellular Uptake Determination by Flow Cytometry
2.10. Confocal Microscopy Imaging
2.11. Western Blotting
2.12. Radioligand Binding Studies
2.13. Statistical Analysis
3. Results
3.1. Synthesis and Characterization of Oxime Bond-Linked GnRH-III-Dau Bioconjugates
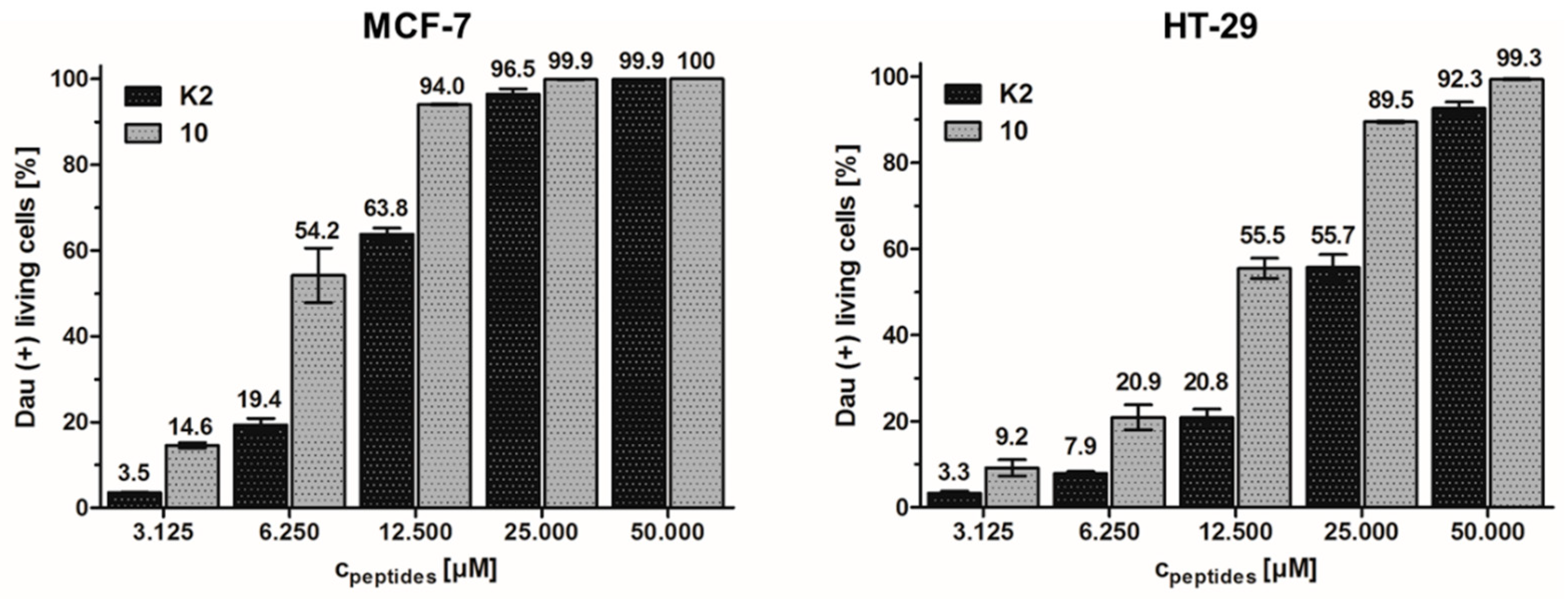
3.2. In Vitro Cytostatic Effect of the Bioconjugates
3.3. Stability in Human Plasma
3.4. Degradation of GnRH-III Bioconjugates in Rat Liver Lysosomal Homogenate
3.5. Cellular Uptake Determination by Flow Cytometry
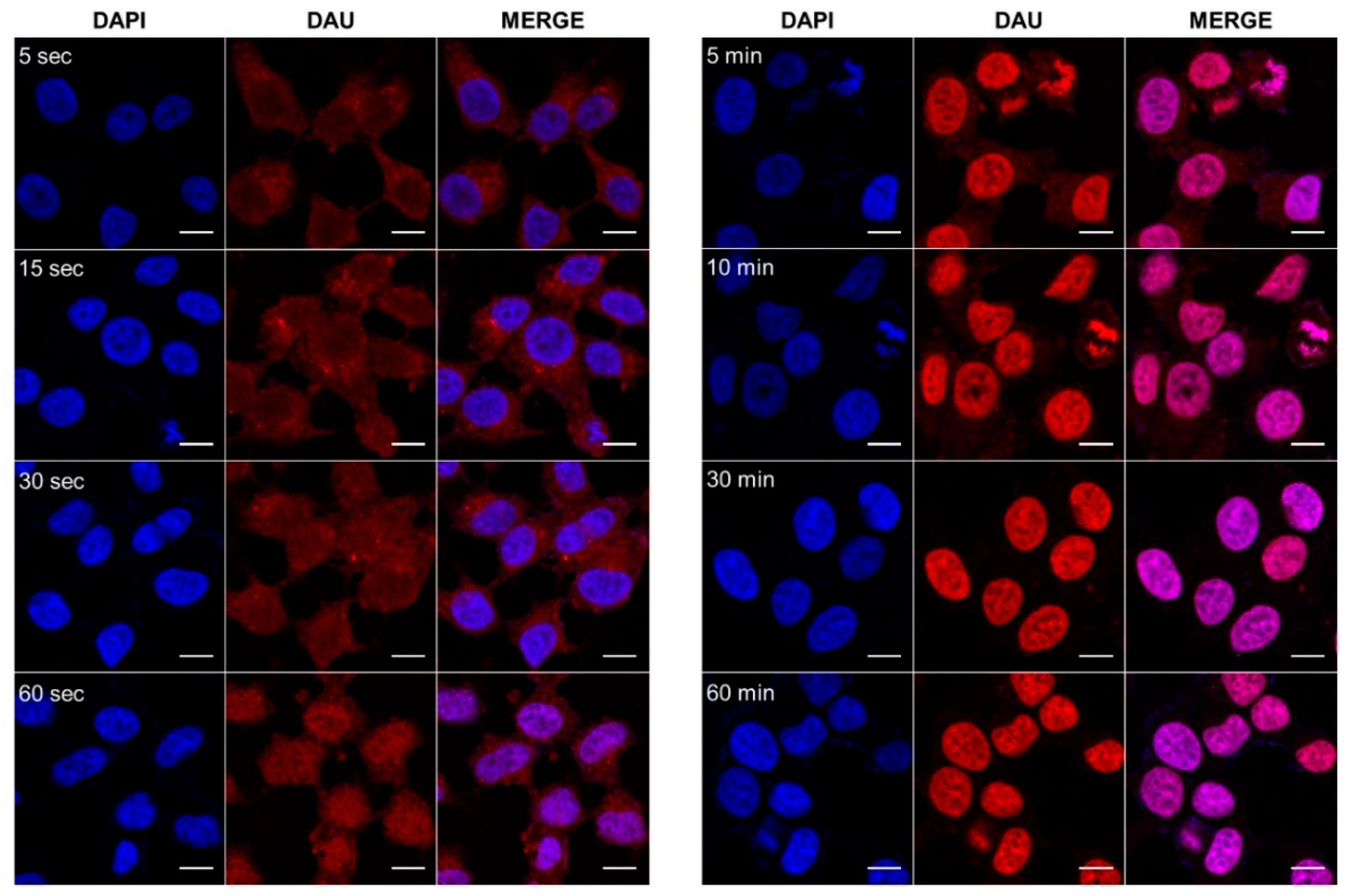
3.6. Cellular Uptake Determination by Confocal Microscopy Imaging
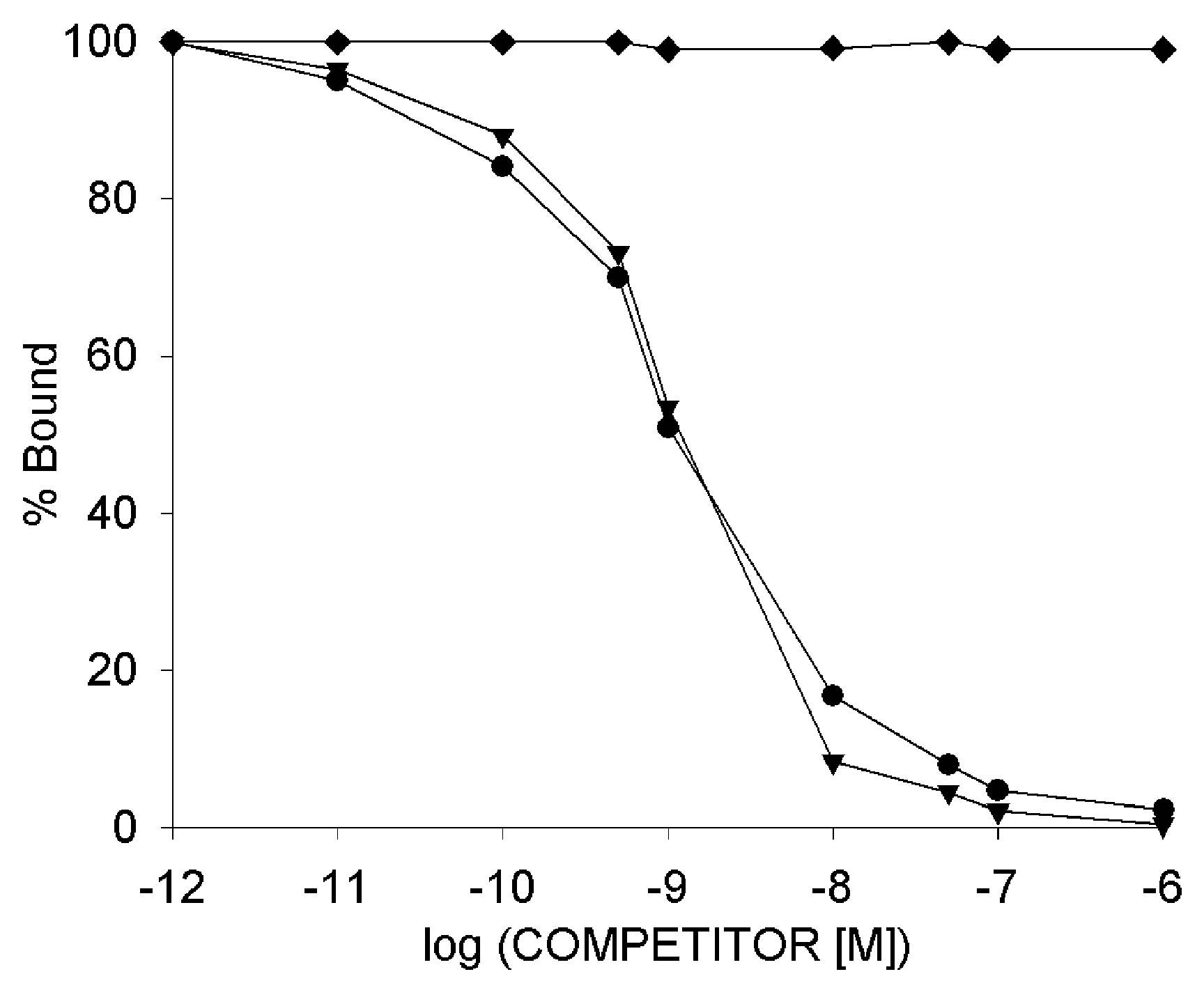
3.7. Radioligand Binding Studies
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Hagimori, M.; Fuchigami, Y.; Kawakami, S. Peptide-Based Cancer-Targeted DDS and Molecular Imaging. Chem. Pharm. Bull. 2017, 65, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Raibaut, L.; Mahdi, O.E.; Melnyk, O. Solid Phase Protein Chemical Synthesis. In Protein Ligation and Total Synthesis II; Topics in Current Chemistry; Springer: Cham, Switzerland, 2014; pp. 103–154. ISBN 978-3-319-19188-1. [Google Scholar]
- Malins, L.R.; Payne, R.J. Recent extensions to native chemical ligation for the chemical synthesis of peptides and proteins. Curr. Opin. Chem. Biol. 2014, 22, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Tugyi, R.; Uray, K.; Iván, D.; Fellinger, E.; Perkins, A.; Hudecz, F. Partial d-amino acid substitution: Improved enzymatic stability and preserved Ab recognition of a MUC2 epitope peptide. Proc. Natl. Acad. Sci. USA 2005, 102, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Gentilucci, L.; De Marco, R.; Cerisoli, L. Chemical Modifications Designed to Improve Peptide Stability: Incorporation of Non-Natural Amino Acids, Pseudo-Peptide Bonds, and Cyclization. Available online: http://www.eurekaselect.com/72674/article (accessed on 20 March 2018).
- Soto, C.; Adessi, C. Converting a Peptide into a Drug: Strategies to Improve Stability and Bioavailability. Available online: http://www.eurekaselect.com/64127/article (accessed on 20 March 2018).
- Coy, D.H.; Vilchez-Martinez, J.A.; Coy, E.J.; Schally, A.V. Analogs of luteinizing hormone-releasing hormone with increased biological activity produced by d-amino acid substitutions in position 6. J. Med. Chem. 1976, 19, 423–425. [Google Scholar] [CrossRef] [PubMed]
- Bajusz, S.; Csernus, V.J.; Janaky, T.; Bokser, L.; Fekete, M.; Schally, A.V. New antagonists of LHRH. II. Inhibition and potentiation of LHRH by closely related analogues. Int. J. Pept. Protein Res. 1988, 32, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Drugs@FDA: FDA Approved Drug Products. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event = overview.process&applno = 022437 (accessed on 19 April 2018).
- Drugs@FDA: FDA Approved Drug Products. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event = overview.process&ApplNo = 021197 (accessed on 19 April 2018).
- Mezo, G. Peptide and protein based pharmaceuticals. In Amino Acids, Peptides and Proteins Vol. 38; Farkas, E., Ryadnov, M., Eds.; RCS Publishing: Cambridge, UK, 2013; pp. 203–252. [Google Scholar]
- Nair, R.M.; Schally, A.V. Structure of a hypothalamic peptide possessing gonadotropin-releasing activity. Int. J. Pept. Protein Res. 1972, 4, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Ling, N.; Rivier, J.; Burgus, R.; Guillemin, R. Direct sequence determination of ovine luteinizing hormone releasing factor by mass spectrometry. Biochemistry 1973, 12, 5305–5310. [Google Scholar] [CrossRef] [PubMed]
- Padula, A.M. GnRH analogues--agonists and antagonists. Anim. Reprod. Sci. 2005, 88, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Mezo, G.; Manea, M. Luteinizing hormone-releasing hormone antagonists. Expert Opin. Ther. Pat. 2009, 19, 1771–1785. [Google Scholar] [CrossRef] [PubMed]
- Nagy, A.; Schally, A.V. Targeting of Cytotoxic Luteinizing Hormone-Releasing Hormone Analogs to Breast, Ovarian, Endometrial, and Prostate Cancers. Biol. Reprod. 2005, 73, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Nagy, A.; Schally, A.V.; Armatis, P.; Szepeshazi, K.; Halmos, G.; Kovacs, M.; Zarandi, M.; Groot, K.; Miyazaki, M.; Jungwirth, A.; et al. Cytotoxic analogs of luteinizing hormone-releasing hormone containing doxorubicin or 2-pyrrolinodoxorubicin, a derivative 500-1000 times more potent. Proc. Natl. Acad. Sci. USA 1996, 93, 7269–7273. [Google Scholar] [CrossRef] [PubMed]
- Nagy, A.; Schally, A.V. Cytotoxic analogs of luteinizing hormone-releasing hormone (LHRH): A new approach to targeted chemotherapy. Drugs Future 2002, 27, 359–370. [Google Scholar] [CrossRef]
- Westphalen, S.; Kotulla, G.; Kaiser, F.; Krauss, W.; Werning, G.; Elsasser, H.P.; Nagy, A.; Schulz, K.D.; Grundker, C.; Schally, A.V.; et al. Receptor mediated antiproliferative effects of the cytotoxic LHRH agonist AN-152 in human ovarian and endometrial cancer cell lines. Int. J. Oncol. 2000, 17, 1063–1069. [Google Scholar] [CrossRef] [PubMed]
- Emons, G.; Kaufmann, M.; Gorchev, G.; Tsekova, V.; Gründker, C.; Günthert, A.R.; Hanker, L.C.; Velikova, M.; Sindermann, H.; Engel, J.; et al. Dose escalation and pharmacokinetic study of AEZS-108 (AN-152), an LHRH agonist linked to doxorubicin, in women with LHRH receptor-positive tumors. Gynecol. Oncol. 2010, 119, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Engel, J.; Emons, G.; Pinski, J.; Schally, A.V. AEZS-108: A targeted cytotoxic analog of LHRH for the treatment of cancers positive for LHRH receptors. Expert Opin. Investig. Drugs 2012, 21, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Schally, A.V. New approaches to the therapy of various tumors based on peptide analogues. Horm. Metab. Res. 2008, 40, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Schally, A.V.; Nagy, A. Chemotherapy targeted to cancers through tumoral hormone receptors. Trends Endocrinol. Metab. 2004, 15, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Mezo, G.; Manea, M. Receptor-mediated tumor targeting based on peptide hormones. Expert Opin. Drug Deliv. 2010, 7, 79–96. [Google Scholar] [CrossRef] [PubMed]
- Aeterna Zentaris Announces that ZoptEC Phase 3 Clinical Study of Zoptrex™ Did Not Achieve its Primary Endpoint | Æterna Zentaris Investor Center. Available online: http://ir.aezsinc.com/press-release/aeternazentaris/aeterna-zentaris-announces-zoptec-phase-3-clinical-study-zoptrex-did (accessed on 21 October 2017).
- Nagy, A.; Plonowski, A.; Schally, A.V. Stability of cytotoxic luteinizing hormone-releasing hormone conjugate (AN-152) containing doxorubicin 14-O-hemiglutarate in mouse and human serum in vitro: Implications for the design of preclinical studies. Proc. Natl. Acad. Sci. USA 2000, 97, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Szabó, I.; Manea, M.; Orbán, E.; Csámpai, A.; Bosze, S.; Szabó, R.; Tejeda, M.; Gaál, D.; Kapuvári, B.; Przybylski, M.; et al. Development of an oxime bond containing daunorubicin-gonadotropin-releasing hormone-III conjugate as a potential anticancer drug. Bioconjug. Chem. 2009, 20, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Orbán, E.; Mezo, G.; Schlage, P.; Csík, G.; Kulić, Z.; Ansorge, P.; Fellinger, E.; Möller, H.M.; Manea, M. In vitro degradation and antitumor activity of oxime bond-linked daunorubicin-GnRH-III bioconjugates and DNA-binding properties of daunorubicin-amino acid metabolites. Amino Acids 2011, 41, 469–483. [Google Scholar] [CrossRef] [PubMed]
- Schlage, P.; Mezo, G.; Orbán, E.; Bosze, S.; Manea, M. Anthracycline-GnRH derivative bioconjugates with different linkages: Synthesis, in vitro drug release and cytostatic effect. J. Control. Release 2011, 156, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Szabó, I.; Bősze, S.; Orbán, E.; Sipos, É.; Halmos, G.; Kovács, M.; Mező, G. Comparative in vitro biological evaluation of daunorubicin containing GnRH-I and GnRH-II conjugates developed for tumor targeting. J. Pept. Sci. 2015, 21, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, M.; Seprodi, J.; Koppan, M.; Horvath, J.E.; Vincze, B.; Teplan, I.; Flerko, B. Lamprey gonadotropin hormone-releasing hormone-III has no selective follicle-stimulating hormone-releasing effect in rats. J. Neuroendocrinol. 2002, 14, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Hegedüs, R.; Pauschert, A.; Orbán, E.; Szabó, I.; Andreu, D.; Marquardt, A.; Mező, G.; Manea, M. Modification of daunorubicin-GnRH-III bioconjugates with oligoethylene glycol derivatives to improve solubility and bioavailability for targeted cancer chemotherapy. Biopolymers 2015, 104, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Hegedüs, R.; Manea, M.; Orbán, E.; Szabó, I.; Kiss, É.; Sipos, É.; Halmos, G.; Mező, G. Enhanced cellular uptake and in vitro antitumor activity of short-chain fatty acid acylated daunorubicin–GnRH-III bioconjugates. Eur. J. Med. Chem. 2012, 56, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Manea, M.; Leurs, U.; Orbán, E.; Baranyai, Z.; Öhlschläger, P.; Marquardt, A.; Schulcz, Á.; Tejeda, M.; Kapuvári, B.; Tóvári, J.; et al. Enhanced enzymatic stability and antitumor activity of daunorubicin-GnRH-III bioconjugates modified in position 4. Bioconjug. Chem. 2011, 22, 1320–1329. [Google Scholar] [CrossRef] [PubMed]
- Schreier, V.N.; Mező, G.; Orbán, E.; Dürr, C.; Marquardt, A.; Manea, M. Synthesis, enzymatic stability and in vitro cytostatic effect of Daunorubicin-GnRH-III derivative dimers. Bioorg. Med. Chem. Lett. 2013, 23, 2145–2150. [Google Scholar] [CrossRef] [PubMed]
- Kapuvári, B.; Hegedüs, R.; Schulcz, Á.; Manea, M.; Tóvári, J.; Gacs, A.; Vincze, B.; Mező, G. Improved in vivo antitumor effect of a daunorubicin—GnRH-III bioconjugate modified by apoptosis inducing agent butyric acid on colorectal carcinoma bearing mice. Investig. New Drugs 2016, 34, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Pappa, E.V.; Zompra, A.A.; Diamantopoulou, Z.; Spyranti, Z.; Pairas, G.; Lamari, F.N.; Katsoris, P.; Spyroulias, G.A.; Cordopatis, P. Structure-activity studies of lGnRH-III through rational amino acid substitution and NMR conformational studies. Biopolymers 2012, 98, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Mezö, G.; Szabó, I.; Kertész, I.; Hegedüs, R.; Orbán, E.; Leurs, U.; Bösze, S.; Halmos, G.; Manea, M. Efficient synthesis of an (aminooxy) acetylated-somatostatin derivative using (aminooxy)acetic acid as a “carbonyl capture” reagent. J. Pept. Sci. 2011, 17, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Halmos, G.; Arencibia, J.M.; Schally, A.V.; Davis, R.; Bostwick, D.G. High incidence of receptors for luteinizing hormone-releasing hormone (LHRH) and LHRH receptor gene expression in human prostate cancers. J. Urol. 2000, 163, 623–629. [Google Scholar] [CrossRef]
- Rozsa, B.; Nadji, M.; Schally, A.V.; Dezso, B.; Flasko, T.; Toth, G.; Mile, M.; Block, N.L.; Halmos, G. Receptors for luteinizing hormone-releasing hormone (LHRH) in benign prostatic hyperplasia (BPH) as potential molecular targets for therapy with LHRH antagonist cetrorelix. Prostate 2011, 71, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.; Biri-Kovács, B.; Szeder, B.; Farkas, V.; Buday, L.; Szabó, Z.; Halmos, G.; Mező, G. Synthesis and in vitro biochemical evaluation of oxime bond-linked daunorubicin-GnRH-III conjugates developed for targeted drug delivery. Beilstein J. Org. Chem. 2018, 14, 756–771. [Google Scholar] [CrossRef] [PubMed]
- Halmos, G.; Wittliff, J.L.; Schally, A.V. Characterization of bombesin/gastrin-releasing peptide receptors in human breast cancer and their relationship to steroid receptor expression. Cancer Res. 1995, 55, 280–287. [Google Scholar] [PubMed]
- Hunter, W.M.; Greenwood, F.C. Preparation of Iodine-131 Labelled Human Growth Hormone of High Specific Activity. Nature 1962, 194, 495–496. [Google Scholar] [CrossRef] [PubMed]
- Biostatistical Analysis. Available online: https://www.pearson.com/us/higher-education/product/Zar-Biostatistical-Analysis-5th-Edition/9780131008465.html (accessed on 20 October 2018).
- R: The R Project for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 20 October 2018).
- GNRHR—Gonadotropin-Releasing Hormone Receptor—Homo sapiens (Human)—GNRHR Gene & Protein. Available online: https://www.uniprot.org/uniprot/P30968 (accessed on 20 October 2018).
- Neill, J.D.; Sellers, J.C.; Musgrove, L.C.; Duck, L.W. Epitope-tagged gonadotropin-releasing hormone receptors heterologously-expressed in mammalian (COS-1) and insect (Sf9) cells1Note: Supported by NIH Research Grant DK45519 to J.D.N.12Note: Presented as preliminary reports at the 1995 Endocrine Society Meetings (Washington, DC), and at the 1995 Society for Neuroscience Meetings (San Diego, CA). Mol. Cell. Endocrinol. 1997, 127, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Gangadharan, S.; Karande, A.A. Modulation of Proliferation by Gonadotropin-Releasing Hormone Receptors in Breast Cancer Cells. Biomed. Res. J. 2014, 1, 71–85. [Google Scholar]
- Lajkó, E.; Spring, S.; Hegedüs, R.; Biri-Kovács, B.; Ingebrandt, S.; Mező, G.; Kőhidai, L. Comparative cell biological study of in vitro antitumor and antimetastatic activity on melanoma cells of GnRH-III-containing conjugates modified with short-chain fatty acids. Beilstein J. Org. Chem. 2018, 14, 2495–2509. [Google Scholar] [CrossRef] [PubMed]
- Cancer. Available online: http://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 1 August 2018).
- Miller, W.R.; Scott, W.N.; Morris, R.; Fraser, H.M.; Sharpe, R.M. Growth of human breast cancer cells inhibited by a luteinizing hormone-releasing hormone agonist. Nature 1985, 313, 231–233. [Google Scholar] [CrossRef] [PubMed]
- Limonta, P.; Montagnani Marelli, M.; Mai, S.; Motta, M.; Martini, L.; Moretti, R.M. GnRH receptors in cancer: From cell biology to novel targeted therapeutic strategies. Endocr. Rev. 2012, 33, 784–811. [Google Scholar] [CrossRef] [PubMed]
- Sealfon, S.C.; Weinstein, H.; Millar, R.P. Molecular Mechanisms of Ligand Interaction with the Gonadotropin-Releasing Hormone Receptor. Endocr. Rev. 1997, 18, 180–205. [Google Scholar] [CrossRef] [PubMed]
- Millar, R.P. GnRHs and GnRH receptors. Anim. Reprod. Sci. 2005, 88, 5–28. [Google Scholar] [CrossRef] [PubMed]
- Mezö, I.; Lovas, S.; Pályi, I.; Vincze, B.; Kálnay, A.; Turi, G.; Vadász, Z.; Seprödi, J.; Idei, M.; Tóth, G.; et al. Synthesis of gonadotropin-releasing hormone III analogs. Structure-antitumor activity relationships. J. Med. Chem. 1997, 40, 3353–3358. [Google Scholar] [CrossRef] [PubMed]
- Fujino, M.; Shinagawa, S.; Yamazaki, I.; Kobayashi, S.; Obayashi, M.; Fukuda, T.; Nakayama, R.; White, W.F.; Rippel, R.H. [Des-Gly-NH210, pro-ethylamide9]-LH-RH: A highly potent analog of luteinizing hormone releasing hormone. Arch. Biochem. Biophys. 1973, 154, 488–489. [Google Scholar] [CrossRef]
- Chenault, J.R.; Kratzer, D.D.; Rzepkowski, R.A.; Goodwin, M.C. LH and FSH response of Holstein heifers to fertirelin acetate, gonadorelin and buserelin. Theriogenology 1990, 34, 81–98. [Google Scholar] [CrossRef]
- Kovács, M.; Vincze, B.; Horváth, J.E.; Seprodi, J. Structure-activity study on the LH- and FSH-releasing and anticancer effects of gonadotropin-releasing hormone (GnRH)-III analogs. Peptides 2007, 28, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Zompra, A.A.; Magafa, V.; Lamari, F.N.; Nikolopoulou, A.; Nock, B.; Maina, T.; Spyroulias, G.A.; Karamanos, N.K.; Cordopatis, P. GnRH analogues containing conformationally restricted amino acids in positions 3 and 6: Differential impact on pituitary binding affinity and direct antiproliferative effect on breast cancer cells†. J. Pept. Res. 2005, 66, 57–64. [Google Scholar] [CrossRef]
- Orbán, E.; Manea, M.; Marquadt, A.; Bánóczi, Z.; Csík, G.; Fellinger, E.; Bosze, S.; Hudecz, F. A new daunomycin-peptide conjugate: Synthesis, characterization and the effect on the protein expression profile of HL-60 cells in vitro. Bioconjug. Chem. 2011, 22, 2154–2165. [Google Scholar] [CrossRef] [PubMed]
- Coley, H.M.; Amos, W.B.; Twentyman, P.R.; Workman, P. Examination by laser scanning confocal fluorescence imaging microscopy of the subcellular localisation of anthracyclines in parent and multidrug resistant cell lines. Br. J. Cancer 1993, 67, 1316–1323. [Google Scholar] [CrossRef] [PubMed]
- Gründker, C.; Föst, C.; Fister, S.; Nolte, N.; Günthert, A.R.; Emons, G. Gonadotropin-releasing hormone type II antagonist induces apoptosis in MCF-7 and triple-negative MDA-MB-231 human breast cancer cells in vitro and in vivo. Breast Cancer Res. 2010, 12, R49. [Google Scholar] [CrossRef] [PubMed]
- Vincze, B.; Pályi, I.; Daubner, D.; Kremmer, T.; Számel, I.; Bodrogi, I.; Sugár, J.; Seprödi, J.; Mezö, I.; Teplán, I. Influence of luteinizing hormone-releasing hormone agonists on human mammary carcinoma cell lines and their xenografts. J. Steroid Biochem. Mol. Biol. 1991, 38, 119–126. [Google Scholar] [CrossRef]
- Gründker, C.; Emons, G. The Role of Gonadotropin-Releasing Hormone in Cancer Cell Proliferation and Metastasis. Front. Endocrinol. 2017, 8, 187. [Google Scholar] [CrossRef] [PubMed]
- Giovannelli, P.; Di Donato, M.; Galasso, G.; Di Zazzo, E.; Bilancio, A.; Migliaccio, A. The Androgen Receptor in Breast Cancer. Front. Endocrinol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | [8Lys(Dau=Aoa)]-GnRH-III Compound | Purity [%] | RP-HPLC Rt [min] a | ESI-MS MWcal/MWexp [g/mol] b |
|---|---|---|---|---|
| K1 | ≥97 | 21.37 | 1841.89/1841.66 | |
| 1 | [3d-Trp] | ≥98 | 21.43 | 1841.89/1841.65 |
| 2 | [3d-Tic] | ≥98 | 21.22 | 1814.86/1814.65 |
| 3 | [2ΔHis, 3d-Tic] | ≥95 | 22.98 | 1677.72/1677.54 |
| 4 | [3d-Tic, 7d-Trp] | >97 | 21.58 | 1814.86/1814.62 |
| 5 | [2ΔHis, 3d-Tic, 7d-Trp] | ≥95 | 23.10 | 1677.72/1677.53 |
| 6 | [6Asp(OMe)] | ≥95 | 21.85 | 1855.91/1855.64 |
| 7 | [10ΔGly-NHEt] | ≥97 | 21.18 | 1812.88/1812.82 |
| K2 | [4Lys(Bu)] | ≥99 | 22.41 | 1953.07/1952.79 |
| 8 | [3d-Trp, 4Lys(Bu)] | ≥98 | 22.83 | 1953.07/1952.65 |
| 9 | [3d-Tic, 4Lys(Bu)] | ≥97 | 22.57 | 1926.05/1925.73 |
| 10 | [2ΔHis, 3d-Tic, 4Lys(Bu)] | ≥96 | 24.27 | 1788.91/1788.64 |
| 11 | [3d-Tic, 4Lys(Bu), 7d-Trp] | ≥96 | 22.77 | 1926.05/1925.81 |
| 12 | [2ΔHis, 3d-Tic, 4Lys(Bu), 7d-Trp] | ≥98 | 24.27 | 1788.91/1788.68 |
| 13 | [4Lys(Bu), 6Asp(OMe)] | ≥98 | 22.92 | 1967.10/1966.68 |
| 14 | [4Lys(Bu), 10ΔGly-NHEt] | ≥97 | 23.17 | 1924.07/1923.72 |
| Code | [8Lys(Dau=Aoa)]-GnRH-III Compound | HT-29 IC50 [µM] | MCF-7 IC50 [µM] | MDA-MB-231 IC50 [µM] |
|---|---|---|---|---|
| K1 | 13.89 ± 3.62 | 2.54 ± 0.67 | 8.22 ± 0.13 | |
| 1 | [3d-Trp] | 15.25 ± 2.51 | 3.60 ± 0.28 | n.d. |
| 2 | [3d-Tic] | 8.75 ± 0.86 | 2.89 ± 0.62 | n.d. |
| 3 | [2ΔHis, 3d-Tic] | 10.32 ± 1.32 | 2.75 ± 0.17 | 9.35 ± 1.93 |
| 4 | [3d-Tic, 7d-Trp] | 15.34 ± 2.63 | 3.42 ± 0.39 | n.d. |
| 5 | [2ΔHis, 3d-Tic, 7d-Trp] | 10.70 ± 0.95 | 1.90 ± 0.58 | 7.88 ± 1.24 |
| 6 | [6Asp(OMe)] | 10.66 ± 1.76 | 4.81 ± 0.72 | n.d. |
| 7 | [10ΔGly-NHEt] | 14.18 ± 3.59 | 4.88 ± 0.01 | 14.33 ± 1.18 |
| K2 | [4Lys(Bu)] | 15.93 ± 0.99 | 2.36 ± 0.07 | 9.00 ± 1.33 |
| 8 | [3d-Trp, 4Lys(Bu)] | 15.03 ± 2.51 | 6.64 ± 1.58 | n.d. |
| 9 | [3d-Tic, 4Lys(Bu)] | 12.73 ± 3.10 | 2.56 ± 0.51 | n.d. |
| 10 | [2ΔHis, 3d-Tic, 4Lys(Bu)] | 3.31 ± 0.90 | 0.14 ± 0.01 | 2.49 ± 0.53 |
| 11 | [3d-Tic, 4Lys(Bu), 7d-Trp,] | 16.83 ± 0.66 | 2.57 ± 0.47 | n.d. |
| 12 | [2ΔHis, 3d-Tic, 4Lys(Bu), 7d-Trp] | 16.55 ± 0.30 | 2.81 ± 0.04 | 8.18 ± 0.18 |
| 13 | [4Lys(Bu), 6Asp(OMe)] | 18.00 ± 0.13 | 3.44 ± 0.51 | n.d. |
| 14 | [4Lys(Bu), 10ΔGly-NHEt] | 17.84 ± 0.08 | 2.23 ± 0.40 | 12.41 ± 2.30 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schuster, S.; Biri-Kovács, B.; Szeder, B.; Buday, L.; Gardi, J.; Szabó, Z.; Halmos, G.; Mező, G. Enhanced In Vitro Antitumor Activity of GnRH-III-Daunorubicin Bioconjugates Influenced by Sequence Modification. Pharmaceutics 2018, 10, 223. https://doi.org/10.3390/pharmaceutics10040223
Schuster S, Biri-Kovács B, Szeder B, Buday L, Gardi J, Szabó Z, Halmos G, Mező G. Enhanced In Vitro Antitumor Activity of GnRH-III-Daunorubicin Bioconjugates Influenced by Sequence Modification. Pharmaceutics. 2018; 10(4):223. https://doi.org/10.3390/pharmaceutics10040223
Chicago/Turabian StyleSchuster, Sabine, Beáta Biri-Kovács, Bálint Szeder, László Buday, János Gardi, Zsuzsanna Szabó, Gábor Halmos, and Gábor Mező. 2018. "Enhanced In Vitro Antitumor Activity of GnRH-III-Daunorubicin Bioconjugates Influenced by Sequence Modification" Pharmaceutics 10, no. 4: 223. https://doi.org/10.3390/pharmaceutics10040223
APA StyleSchuster, S., Biri-Kovács, B., Szeder, B., Buday, L., Gardi, J., Szabó, Z., Halmos, G., & Mező, G. (2018). Enhanced In Vitro Antitumor Activity of GnRH-III-Daunorubicin Bioconjugates Influenced by Sequence Modification. Pharmaceutics, 10(4), 223. https://doi.org/10.3390/pharmaceutics10040223