Exosomes and Exosome-Inspired Vesicles for Targeted Drug Delivery




Abstract
1. Introduction
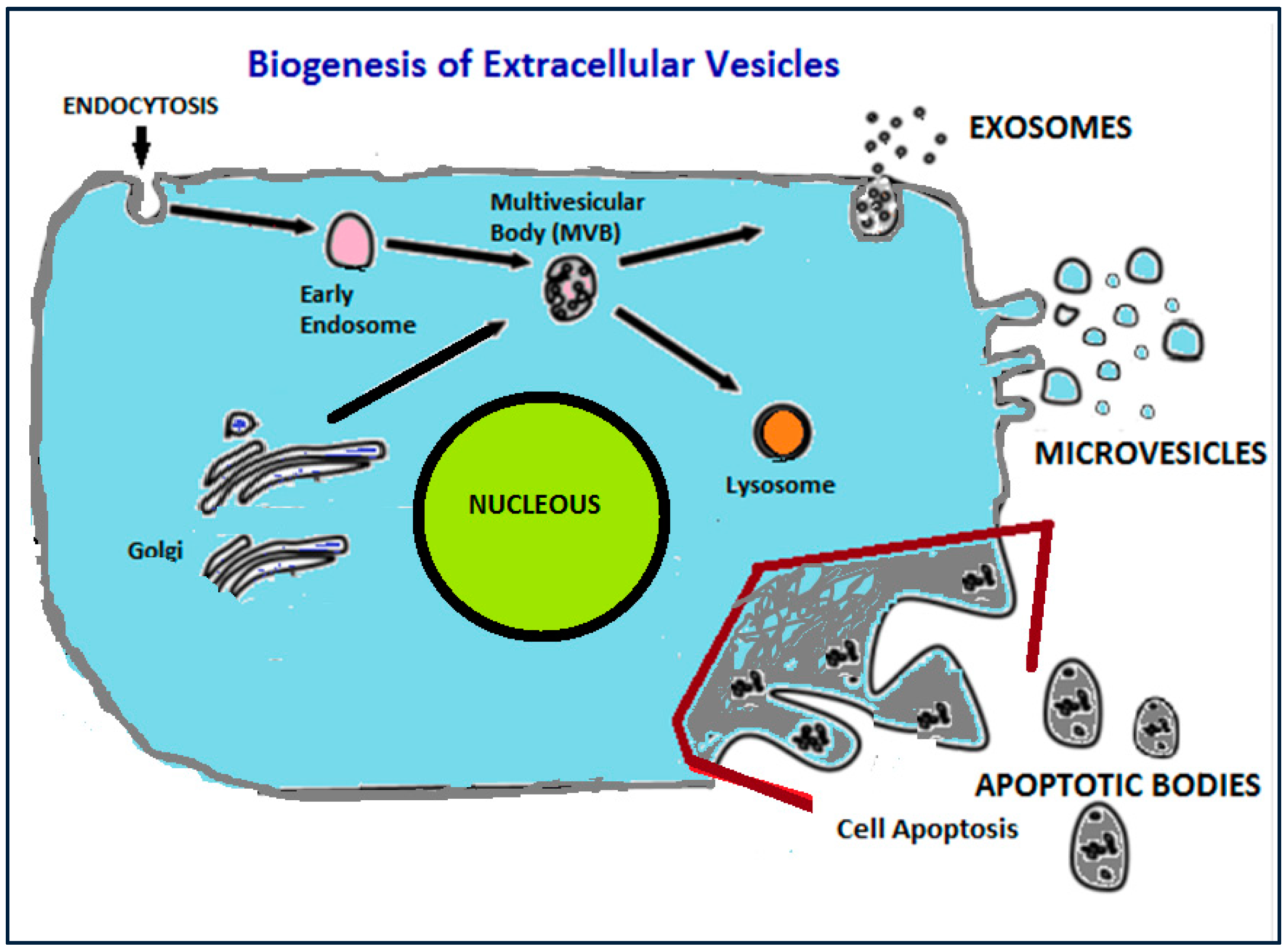
1.1. Definition, Biogenesis and Main Functions of Extracellular Vesicles (EVs) and Exosomes (EXs)
- (i)
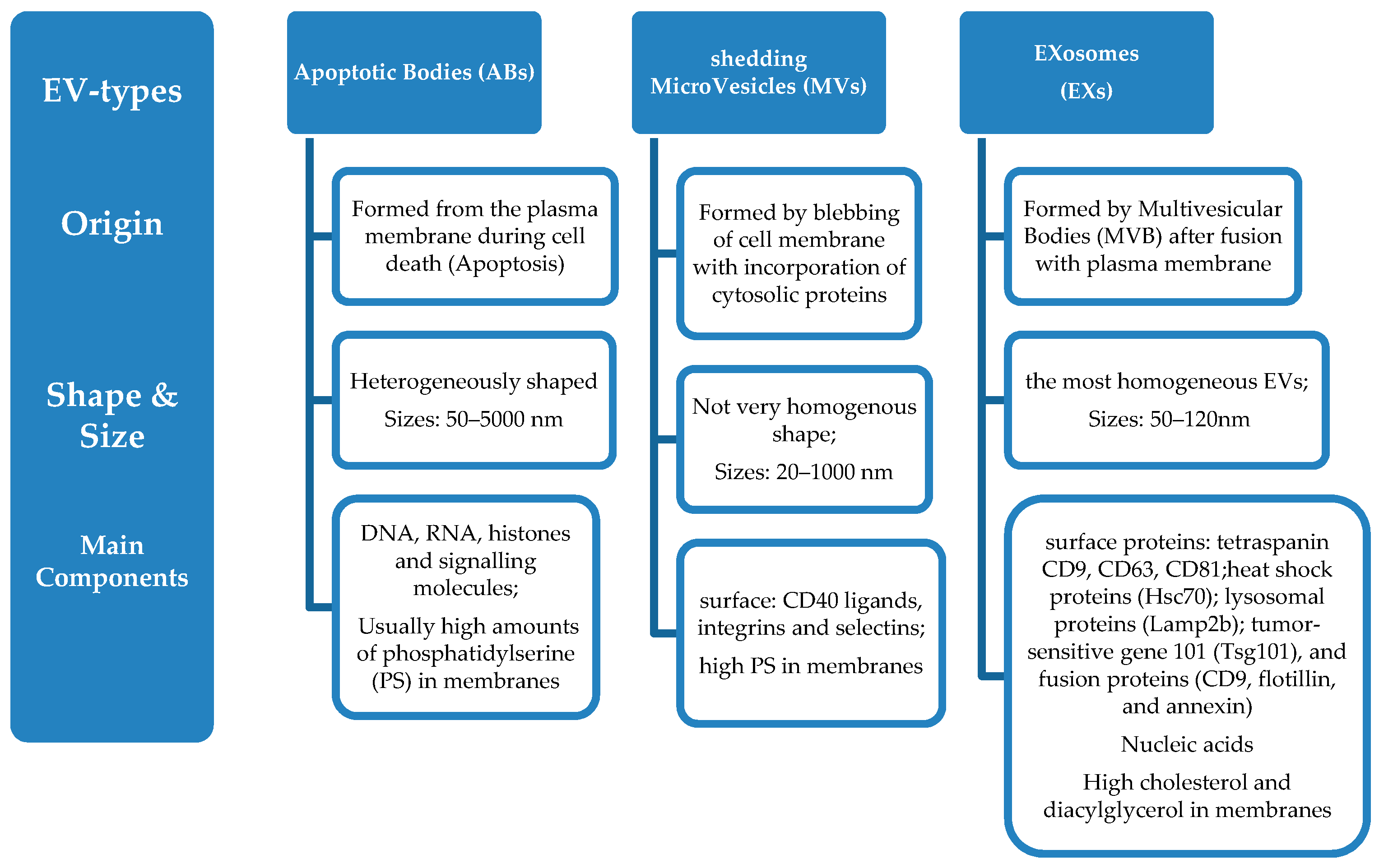
- Apoptotic bodies are released during cell death (apoptosis) and are heterogeneously shaped vesicles with sizes between 50–5000 nm. They are formed from the plasma membrane, and they contain DNA, RNA, histones, and signalling molecules [22]. They usually have high amounts of phosphatidylserine (PS) in their membranes, since the outer membrane of apoptotic cells is enriched in PS.
- (ii)
- Micro vesicles are formed by blebbing of the cell membrane with concurrent incorporation of cytosolic proteins, and their sizes range between 20–1000 nm, depending on the origin cells and the method applied for their isolation from cell media [23]. Their formation can be triggered through Ca2+ influx, phorbol esters, ATP, etc. [24]. Some common biochemical characteristics have been identified between microvesicless from different cells, such as their high membrane levels of PS, and some common surface markers (CD40 ligands, integrins and selectins) [12].
- (iii)
- Finally, exosomes include a more homogeneous (in shape and size) population of vesicles compared to microvesicles, with sizes that range from 50 nm up to 120 nm. Their biogenesis is initiated by inward budding of the plasma membrane which results in the formation of intermediate endosome-vesicles, the multivesicular bodies (MVBs). After that, depending on their composition, MVBs are either degraded after fusion with lysosomes or they fused with the plasma membrane and form exosomess that are released from the cells [25,26,27,28]. Exosomes contain surface proteins unique to the endosomal pathway, which are generally used to characterize exosomes and distinguish them from microvesicles, apoptotic bodies, and other vesicles such as tetraspanin CD9, CD63, CD81 [29], heat shock proteins (Hsc70), lysosomal proteins (Lamp2b), the tumor-sensitive gene 101 (Tsg101), and fusion proteins (CD9, flotillin, and annexin) [30], and incorporate nucleic acids, cytosolic proteins, and receptors. Their lipid composition differs from other extracellular vesicle-types, since they are rich in cholesterol and diacylglycerol. Exosomes are generally considered as transporters of miRNA that regulate specific intracellular mRNA activity [31].
1.2. Current Bottlenecks in Nanoparticle-Assisted Targeted Drug Delivery and Liposomes
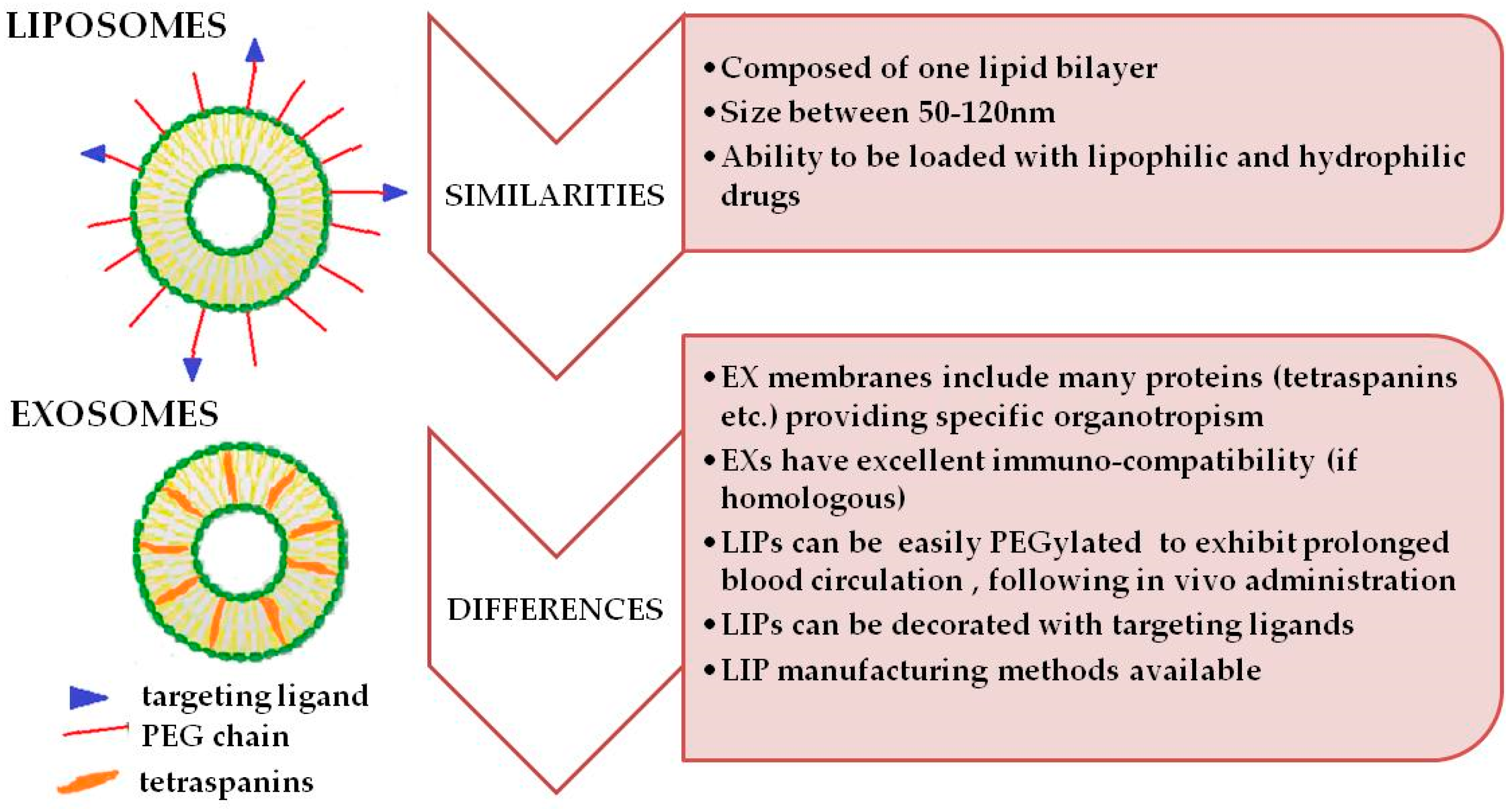
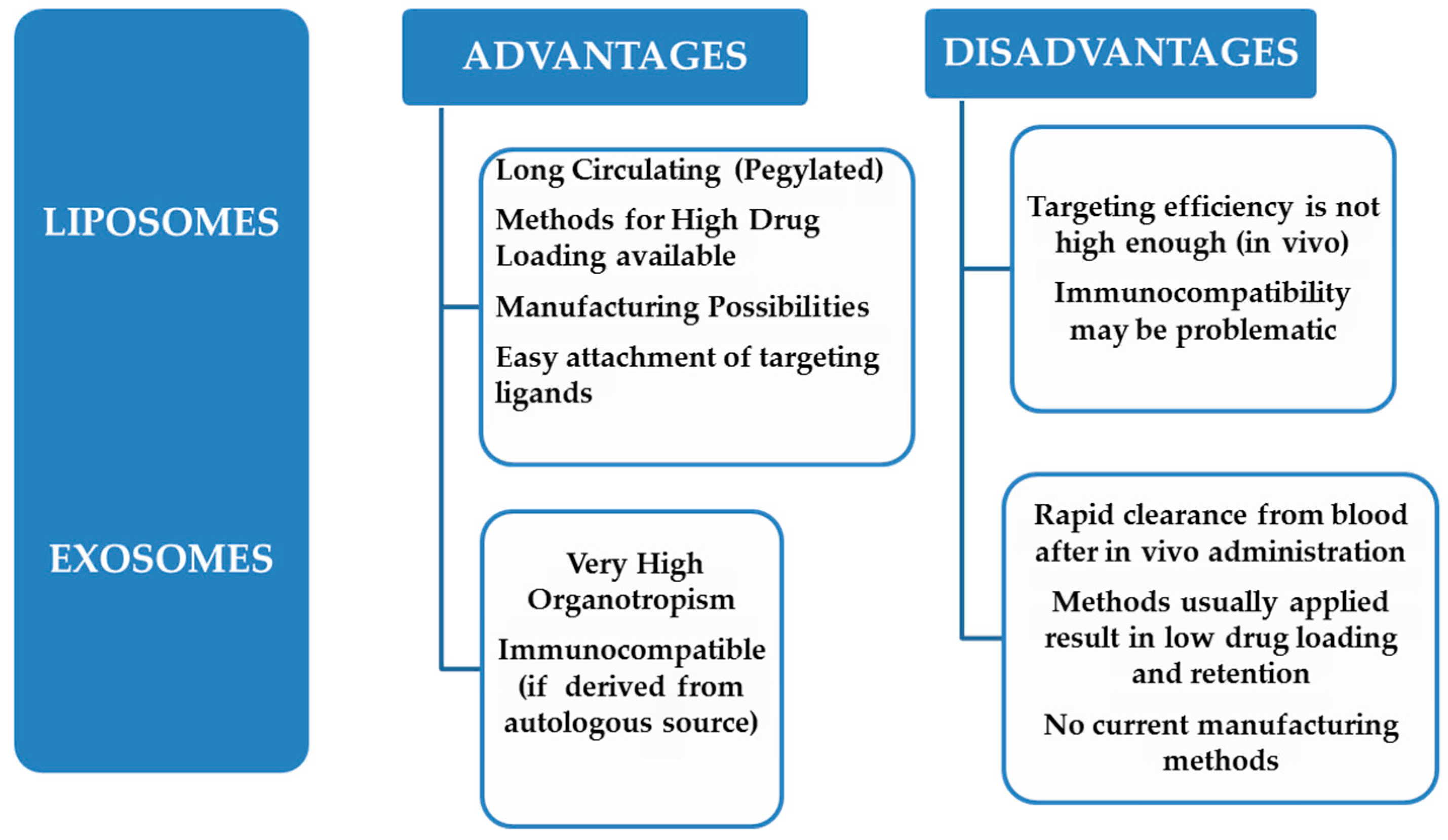
2. Similarities and Differences between Exosomes and Liposomes
3. Sources, Methods of Isolation and In Vivo Clearance of Unmodified Exosomes
3.1. Sources of Exosomes
3.2. Isolation Methods
3.2.1. Ultracentrifugation
3.2.2. Immunoaffinity
3.2.3. Other Size-Based Isolation Methods
3.2.4. Precipitation
3.2.5. Yield of Common Isolation Methodologies
3.2.6. Microfluidic Methods for EX/EV Purification
3.3. In Vivo Clearance of Unmodified Exosomes
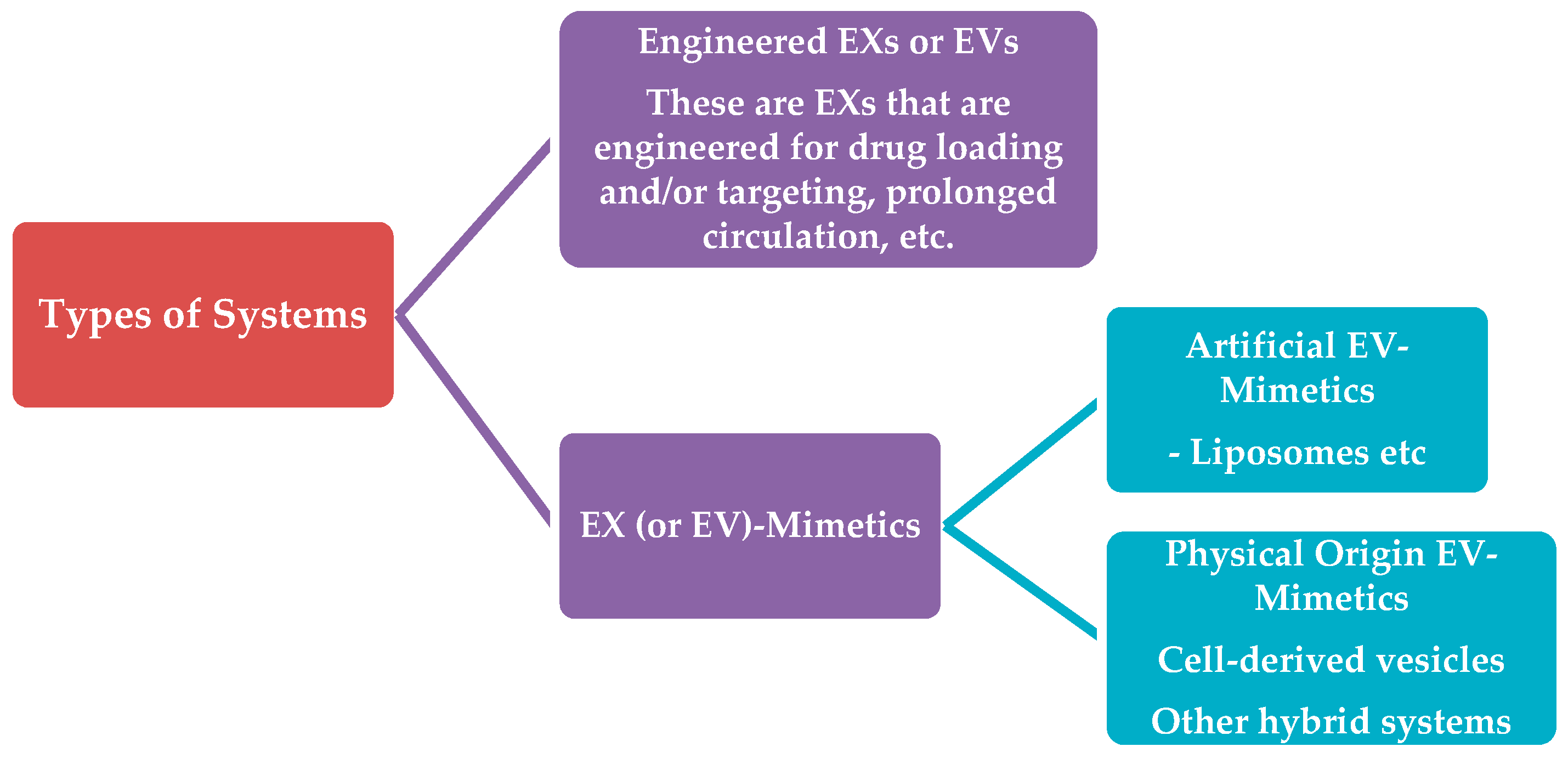
4. Types of Systems
- The efficient loading of drugs and/or
- Surface-modification/attachment of molecules on their surfaces; such modifications may be required when the in vivo fate (stability and/or pharmacokinetic/biodistribution profile) of the exosomes is not considered to be adequate for the planned drug delivery and/or targeting application. In fact, as analyzed above, in most cases, the clearance of unmodified exosomes after their in vivo administration (especially if iv-injection is used) is rapid, posing a problem for their applicability as targeted drug carriers.
- Artificial Exosome-mimetics, when the starting material is of synthetic or semi-synthetic origin (this category also includes lipids extracted from natural sources, such as cells or extracellular vesicless). In most cases, artificial exosome-mimetics are actually liposomes with or without specific proteins in their membrane (which are inspired from specific types of exosomes with high organotropism).
- Physical-origin exosome-mimetics, when the starting material may be derived from other types of cellular components excluding extracellular vesicles, such as whole cells (in this case the vesicles are names “cellular vesicles”). In this subcategory, again, the starting material is engineered as mentioned above for engineered-exosomes, and the same methodologies apply.
4.1. Engineered-Exosomes or Extracellular Vesicles
4.2. Exosome (or Extracellular Vesicle)-Mimetics
4.2.1. Artificial Extracellular Vesicle-Mimetics
4.2.2. Physical-Origin Extracellular Vesicle-Mimetics
4.2.3. Other Types of Extracellular Vesicle-Mimetic Systems
5. Methods of Preparation and Engineering of Engineered Exosomes and Exosome-Mimetics
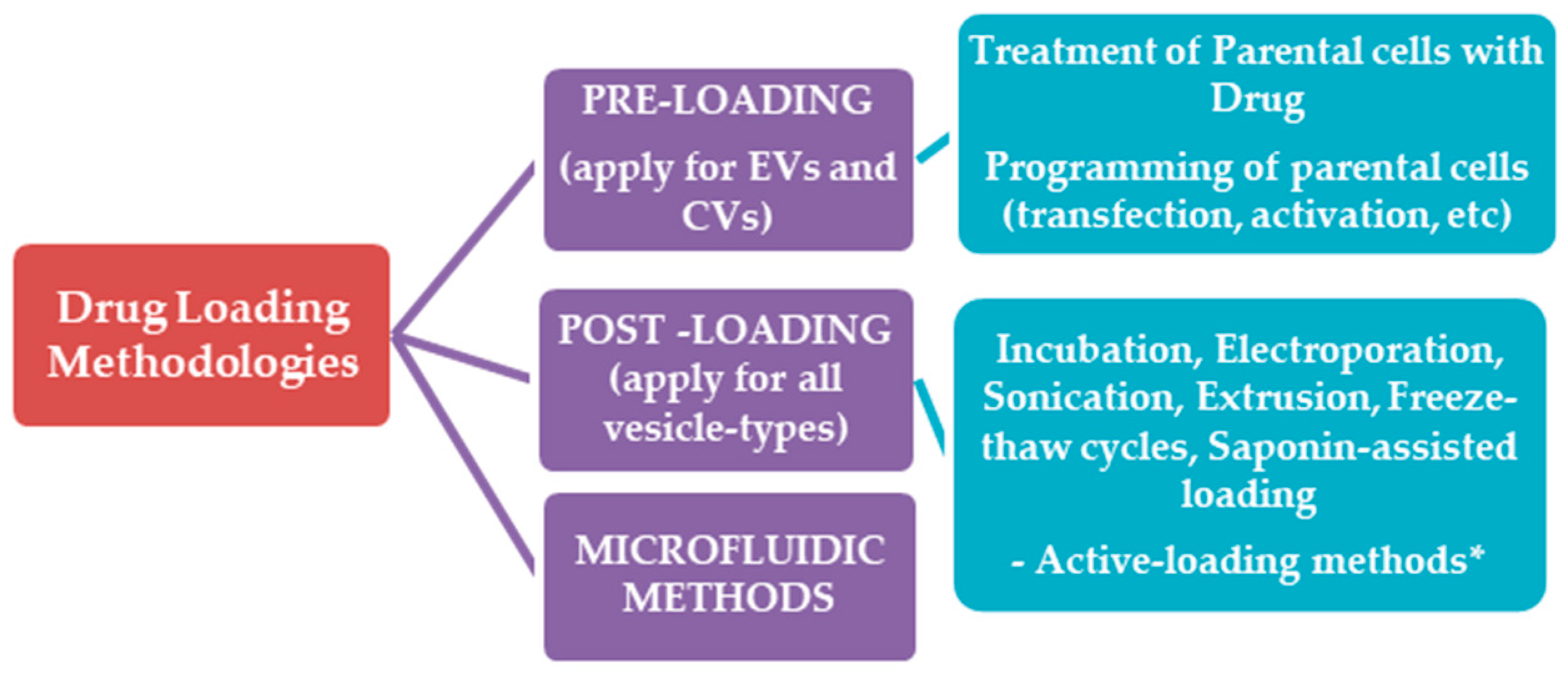
5.1. Drug Loading Methodologies
5.1.1. Pre-Loading Methods
Treatment of Parental Cells with Drugs
Parental Cell Engineering
5.1.2. Post-Loading Methods
Incubation with Drug
Electroporation
Sonication
Extrusion
Freeze/Thaw Cycle Method
Saponin-Assisted Loading
5.1.3. Comparison of Different Loading Methods
5.2. Surface Modification Methods
5.3. Microfluidic Methods for Engineering of Exosomes and Exosome-Mimetics
6. Potential Clinical Applications of EXs and EXs-Mimetics
6.1. Current Status
6.2. Challenges and Future Perspectives
Acknowledgments
Conflicts of Interest
References
- Meldolesi, J. Exosomes and Ectosomes in Intercellular Communication. Curr. Biol. 2018, 28, R435–R444. [Google Scholar] [CrossRef] [PubMed]
- Maia, J.; Caja, S.; Strano Moraes, M.C.; Couto, N.; Costa-Silva, B. Exosome-Based Cell-Cell Communication in the Tumor Microenvironment. Front. Cell Dev. Biol. 2018, 6, 18. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.; Thakur, B.K.; Weiss, J.M.; Kim, H.S.; Peinado, H.; Lyden, D. Extracellular vesicles in cancer: Cell-to-cell mediators of metastasis. Cancer Cell 2016, 30, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Yang, H.; Zhang, X.; Xu, W. The emerging roles of exosomes in tumor-stroma interaction. J. Cancer Res. Clin. Oncol. 2016, 142, 1897–1907. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Paila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, D.; Joshi, N.; Tao, W.; Karp, J.M.; Peer, D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat. Commun. 2018, 9, 1410. [Google Scholar] [CrossRef] [PubMed]
- Akhter, M.H.; Rizwanullah, M.; Ahmad, J.; Ahsan, M.J.; Mujtaba, M.A.; Amin, S. Nanocarriers in advanced drug targeting: Setting novel paradigm in cancer therapeutics. Artif. Cells Nanomed. Biotechnol. 2018, 46, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.M.; Cullis, P.R. Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Belfiore, L.; Saunders, D.N.; Ranson, M.; Thurecht, K.J.; Storm, G.; Vine, K.L. Towards clinical translation of ligand-functionalized liposomes in targeted cancer therapy: Challenges and opportunities. J. Control. Release 2018, 277, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kooijmans, S.A.; Vader, P.; van Dommelen, S.M.; van Solinge, W.W.; Schiffelers, R.M. Exosome mimetics: A novel class of drug delivery systems. Int. J. Nanomed. 2012, 7, 1525–1541. [Google Scholar] [CrossRef]
- Van Dommelen, S.M.; Vader, P.; Lakhal, S.; Kooijmans, S.A.; van Solinge, W.W.; Wood, M.J.; Schiffelers, R.M. Microvesicles and exosomes: Opportunities for cell-derived membrane vesicles in drug delivery. J. Control. Release 2012, 161, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Aryani, A.; Denecke, B. Exosomes as a Nanodelivery System: A Key to the Future of Neuromedicine? Mol. Neurobiol. 2016, 53, 818–834. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, K.B.; Gudbergsson, J.M.; Skov, M.N.; Pilgaard, L.; Moos, T.; Duroux, M. A comprehensive overview of exosomes as drug delivery vehicles—Endogenous nanocarriers for targeted cancer therapy. Biochim. Biophys. Acta 2014, 1846, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Vader, P.; Mol, E.A.; Pasterkamp, G.; Schiffelers, R.M. Extracellular vesicles for drug delivery. Adv. Drug Deliv. Rev. 2016, 106 Pt A, 148–156. [Google Scholar] [CrossRef]
- Yim, N.; Choi, C. Extracellular vesicles as novel carrier for therapeutic molecules. BMB Rep. 2016, 49, 585–586. [Google Scholar] [CrossRef] [PubMed]
- Rufino-Ramos, D.; Albuquerque, P.R.; Carmona, V.; Perfeito, R.; Nobre, R.J.; Pereira de Almeida, L. Extracellular vesicles: Novel promising delivery systems for therapy of brain diseases. J. Control. Release 2017, 262, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Lugli, G.; Cohen, A.M.; Bennett, D.A.; Shah, R.C.; Fields, C.J.; Hernandez, A.G.; Smalheiser, N.R. Plasma exosomal miRNAs in persons with and without Alzheimer disease: Altered expression and prospects for biomarkers. PLoS ONE 2015, 10, e0139233. [Google Scholar] [CrossRef] [PubMed]
- Abels, E.R.; Breakefield, X.O. Introduction to extracellular vesicles: Biogenesis, RNA cargo selection, content, release, and uptake. Cell. Mol. Neurobiol. 2016, 36, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Silverman, J.M.; Reiner, N.E. Exosomes and other microvesicles in infection biology: Organelles with unanticipated phenotypes. Cell Microbiol. 2011, 13, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ellis, T.N.; Kuehn, M.J. Virulence and immunomodulatory roles of bacterial outer membrane vesicles. Microbiol. Mol. Biol. Rev. 2010, 74, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Van der Pol, E.; Boing, A.N.; Harrison, P.; Sturk, A.; Nieuwland, R. Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol. Rev. 2012, 64, 676–705. [Google Scholar] [CrossRef] [PubMed]
- Cocucci, E.; Racchetti, G.; Meldolesi, J. Shedding microvesicles: Artefacts no more. Trends Cell Biol. 2009, 19, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Cocucci, E.; Racchetti, G.; Podini, P.; Meldolesi, L. Enlargeosome traffic: Exocytosis triggered by various signals is followed by endocytosis, membrane shedding or both. Traffic 2007, 8, 742–757. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- György, B.; Szabó, T.G.; Pásztói, M.; Pál, Z.; Misják, P.; Aradi, B.; László, V.; Pállinger, É.; Pap, E.; Kittel, Á.; et al. Membrane vesicles, current state-of-the-art: Emerging role of extracellular vesicles. Cell. Mol. Life Sci. 2011, 68, 2667–2688. [Google Scholar] [CrossRef] [PubMed]
- Fevrier, B.; Raposo, G. Exosomes: Endosomal-derived vesicles shipping extracellular messages. Curr. Opin. Cell Biol. 2004, 16, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.T.; Teng, K.; Wu, C.; Adam, M.; Johnstone, R.M. Electron microscopic evidence for externalization of the transferrin receptor in vesicular form in sheep reticulocytes. J. Cell Biol. 1985, 101, 942–948. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immun. 2002, 2, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Kahlert, C.; Melo, S.A.; Protopopov, A.; Tang, J.; Seth, S.; Koch, M.; Zhang, J.; Weitz, J.; Chin, L.; Futreal, A.; et al. Identification of double-stranded genomic DNA spanning all chromosomes with mutated KRAS and p53 DNA in the serum exosomes of patients with pancreatic cancer. J. Biol. Chem. 2014, 289, 3869–3875. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, J.; Wysoczynski, M.; Hayek, F.; Janowska-Wieczorek, A.; Ratajczak, M.Z. Membrane-derived microvesicles: Important and underappreciated mediators of cell-to-cell communication. Leukemia 2006, 20, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Kanada, M.; Bachmann, M.H.; Hardy, J.W.; Frimannson, D.O.; Bronsart, L.; Wang, A.; Sylvester, M.D.; Schmidt, T.L.; Kaspar, R.L.; Butte, M.J.; et al. Differential fates of biomolecules delivered to target cells via extracellular vesicles. Proc. Natl. Acad. Sci. USA 2015, 112, E1433–E1442. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, J.; Miekus, K.; Kucia, M.; Zhang, J.; Reca, R.; Dvorak, P.; Ratajczak, M.Z. Embryonic stem cell-derived microvesicles reprogram hematopoietic progenitors: Evidence for horizontal transfer of mRNA and protein delivery. Leukemia 2006, 20, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Azmi, A.S.; Bao, B.; Sarkar, F.H. Exosomes in cancer development, metastasis, and drug resistance: A comprehensive review. Cancer Metastasis Rev. 2013, 32, 623–642. [Google Scholar] [CrossRef] [PubMed]
- Buzas, E.I.; György, B.; Nagy, G.; Falus, A.; Gay, S. Emerging role of extracellular vesicles in inflammatory diseases. Nat. Rev. Rheumatol. 2014, 10, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.D.; Morelli, A.E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014, 14, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Antimisiaris, S.G.; Kallinteri, P.; Fatouris, D.G. Chapter 13. Liposomes & Drug Delivery. In Pharmaceutical Manufacturing: Production and Processes; Cox Gad, S., Ed.; John Wiley & Sons, Inc.: New York, NY, USA, 2008; pp. 443–533. [Google Scholar] [CrossRef]
- Deshpande, P.P.; Biswas, S.; Torchilin, V.P. Current trends in the use of liposomes for tumor targeting. Nanomedicine (Lond.) 2013, 8, 1509–1528. [Google Scholar] [CrossRef] [PubMed]
- Rip, J.; Chen, L.; Hartman, R.; van den Heuvel, A.; Reijerkerk, A.; van Kregten, J.; van der Boom, B.; Appeldoorn, C.; de Boer, M.; Maussang, D.; et al. Glutathione PEGylated liposomes: Delivery of cargo across the BBB. J. Drug Target. 2014, 22, 460–467. [Google Scholar] [CrossRef] [PubMed]
- Hua, S.; Marks, E.; Schneider, J.J.; Keely, S. Advances in oral nanodelivery systems for colon targeted drug delivery in IBD. Nanomedicine 2015, 11, 1117–1132. [Google Scholar] [CrossRef] [PubMed]
- Puri, A.; Loomis, K.; Smith, B.; Lee, J.H.; Yavlovich, A.; Heldman, E.; Blumenthal, R. Lipid based nanopart. As pharmac. drug carriers. Crit. Rev. Ther. Drug Carrier Syst. 2009, 26, 523–558. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, E.; Gomes, A.C.; Preto, A.; Cavaco-Paulo, A. Design of liposomal formulations for cell targeting. Colloids Surf. B Biointerfaces 2015, 136, 514–526. [Google Scholar] [CrossRef] [PubMed]
- Antimisiaris, S.; Mourtas, S.; Papadia, K. Targeted si-RNA with liposomes and exosomes (extracellular vesicles): How to unlock the potential. Int. J. Pharm. 2017, 525, 293–312. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, M.; Ajazuddin, D.K.; Tripathi, S.; Saraf, S.; Saraf, S.G.; Antimisiaris, S.G.; Mourtas, S.; Hammarlund-Udenaese, M.; Alexander, A. Recent advancements in liposomes targeting strategies to cross blood-brain barrier (BBB) for the treatment of Alzheimer’s disease. J. Control. Release 2017, 260, 61–77. [Google Scholar] [CrossRef] [PubMed]
- Eloya, J.O.; Petrilli, R.; Trevizana, L.N.F.; Chorilli, M. Immunoliposomes: A review on functionalization strategies and targets for drug delivery. Colloids Surf. B Biointerfaces 2017, 159, 454–467. [Google Scholar] [CrossRef] [PubMed]
- Petrenko, V.A. Landscape Phage: Evolution from Phage Display to Nanobiotechnology. Viruses 2018, 10, 311. [Google Scholar] [CrossRef] [PubMed]
- Mu, L.M.; Ju, R.J.; Liu, R.; Bu, Y.Z.; Zhang, J.Y.; Li, X.Q.; Zeng, F.; Lu, W.L. Dual-functional drug liposomes in treatment of resistant cancers. Adv. Drug Del. Rev. 2017, 115, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Tiwari, A.; Verma, A.; Jain, S.K. Ultrasound-based triggered drug delivery to tumors. Drug Deliv. Transl. Res. 2018, 8, 150–164. [Google Scholar] [CrossRef] [PubMed]
- Zangabad, P.S.; Mirkiani, S.; Shahsavari, S.; Masoudi, B.; Masroor, M.; Hamed, H.; Jafari, Z.; Taghipour, Y.D.; Hashemi, H.; Karimi, M.; et al. Stimulus-responsive liposomes as smart nanoplatforms for drug delivery applications. Nanotechnol. Rev. 2018, 7, 95–122. [Google Scholar] [CrossRef] [PubMed]
- Skouras, A.; Papadia, K.; Mourtas, S.; Klepetsanis, P.; Antimisiaris, S.G. Multifunctional doxorubicin-loaded magnetoliposomes with active and magnetic targeting properties. Eur. J. Pharm. Sci. 2018, 123, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Van der Meel, R.; Fens, M.H.; Vader, P.; van Solinge, W.W.; Eniola-Adefeso, O.; Schiffelers, R.M. Extracellular vesicles as drug delivery systems: Lessons from the liposome field. J. Control. Release 2014, 195, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.T.; Umezaki, K.; Sawada, S.; Mukai, S.; Sasaki, Y.; Harada, N.; Shiku, H.; Akiyoshi, K. Engineering hybrid exosomes by membrane fusion with liposomes. Sci. Rep. 2016, 6, 21933. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, K.B.; Gudbergsson, J.M.; Duroux, M.; Moos, T.; Andresen, T.L.; Simonsen, J.B. On the use of liposome controls in studies investigating the clinical potentialof extracellular vesicle-based drug delivery systems—A commentary. J. Control. Release 2018, 269, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Mathivanan, S.; Simpson, R.J. ExoCarta: A compendium of exosomal proteins and RNA. Proteomics 2009, 9, 4997–5000. [Google Scholar] [CrossRef] [PubMed]
- Marban, E. The secret life of exosomes. What Bees Can Teach Us About Next-Generation Therapeutics. J. Am. Coll. Cardiol. 2018, 71, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Boon, R.A.; Vickers, K.C. Intercellular transport of microRNAs. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Zhang, K.; Wu, S.; Cui, M.; Xu, T. Focus on Mesenchymal Stem Cell-Derived Exosomes: Opportunities and Challenges in Cell-Free Therapy. Stem Cells Int. 2017, 6305295. [Google Scholar] [CrossRef] [PubMed]
- Michael, A.; Bajracharya, S.D.; Yuen, P.S.; Zhou, H.; Star, R.A.; Illei, G.G.; Alevizos, I. Exosomes from human saliva as a source of microRNA biomarkers. Oral Dis. 2010, 16, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; Raposo, G.; Candalh, C.; Boussac, M.; Hershberg, R.; Cerf-Bensussan, N.; Heyman, M. Intestinal epithelial cells secrete exosome-like vesicles. Gastroenterology 2001, 121, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Saunderson, S.C.; Dunn, A.C.; Crocker, P.R.; McLellan, A.D. CD169 mediates the capture of exosomes in spleen and lymph node. Blood 2014, 123, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Record, M.; Subra, C.; Silvente-Poirot, S.; Poirot, M. Exosomes as intercellular signalosomes and pharmacological effectors. Biochem. Pharmacol. 2011, 81, 1171–1182. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, G.; Brody, I. The prostasome: Its secretion and function in man. Biochim. Biophys. Acta 1985, 822, 203–218. [Google Scholar] [CrossRef]
- Pisitkun, T.; Shen, R.-F.; Knepper, M.A. Identification and proteomic profiling of exosomes in human urine. Proc. Natl. Acad. Sci. USA 2004, 101, 13368–13373. [Google Scholar] [CrossRef] [PubMed]
- Asea, A.; Jean-Pierre, C.; Kaur, P.; Rao, P.; Linhares, I.M.; Skupski, D.; Witkin, S.S. Heat shock protein-containing exosomes in mid-trimester amniotic fluids. J. Reprod. Immunol. 2008, 79, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Lässer, C.; Alikhani, V.S.; Ekström, K.; Eldh, M.; Paredes, P.T.; Bossios, A.; Sjöstrand, M.; Gabrielsson, S.; Lötvall, J.; Valadi, H. Human saliva, plasma and breast milk exosomes contain RNA: Uptake by macrophages. J. Transl. Med. 2011, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Nijman, H.W.; Stoorvogel, W.; Liejendekker, R.; Harding, C.V.; Melief, C.J.; Geuze, H.J. B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 1996, 183, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Gutwein, P.; Stoeck, A.; Riedle, S.; Gast, D.; Runz, S.; Condon, T.P.; Marmé, A.; Phong, M.C.; Linderkamp, O.; Skorokhod, A.; et al. Cleavage of L1 in exosomes and apoptotic membrane vesicles released from ovarian carcinoma cells. Clin. Cancer Res. 2005, 11, 2492–2501. [Google Scholar] [CrossRef] [PubMed]
- Paggetti, J.; Haderk, F.; Seiffert, M.; Janji, B.; Distler, U.; Ammerlaan, W.; Kim, Y.J.; Adam, J.; Lichter, P.; Solary, E.; et al. Exosomes released by chronic lymphocytic leukemia cells induce the transition of stromal cells into cancer-associated fibroblasts. Blood 2015, 126, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.A.; Park, H.; Lim, E.H.; Kim, K.H.; Choi, J.S.; Lee, J.H.; Shin, J.W.; Lee, K.W. Exosomes from ovarian cancer cells induce adipose tissue-derived mesenchymal stem cells to acquire the physical and functional characteristics of tumor-supporting myofibroblasts. Gynecol. Oncol. 2011, 123, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Feng, S.; Wang, X.; Long, K.; Luo, Y.; Wang, Y.; Ma, J.; Tang, Q.; Jin, L.; Li, X.; et al. Identification of exosome-like nanoparticle-derived microRNAs from 11 edible fruits and vegetables. PeerJ 2018, 6, e5186. [Google Scholar] [CrossRef] [PubMed]
- Munagala, R.; Aqil, F.; Jeyabalan, J.; Gupta, R.C. Bovine milk-derived exosomes for drug delivery. Cancer Lett. 2016, 371, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Manca, S.; Upadhyaya, B.; Mutai, E.; Desaulniers, A.T.; Cederberg, R.A.; White, B.R.; Zempleni, J. Milk exosomes are bioavailable and distinct microRNA cargos have unique tissue distribution patterns. Sci. Rep. 2018, 8, 11321. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 194, 3.22.1–3.22.29. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Kim, J.; Park, J. Methods to isolate extracellular vesicles for diagnosis. Micro Nano Syst. Lett. 2017, 5, 15. [Google Scholar] [CrossRef]
- Li, P.; Kaslan, M.; Lee, S.H.; Yao, J.; Gao, Z. Progress in Exosome Isolation Techniques. Theranostics 2017, 7, 789–804. [Google Scholar] [CrossRef] [PubMed]
- Khatun, Z.; Bhat, A.; Sharma, S.; Sharma, A. Elucidating diversity of exosomes: Biophysical and molecular characterization methods. Nanomedicine 2016, 11, 2359–2377. [Google Scholar] [CrossRef] [PubMed]
- Higginbotham, J.N.; Zhang, Q.; Jeppesen, D.K.; Scott, A.M.; Manning, H.C.; Ochieng, J.; Franklin, J.L.; Coffey, R.J. Identification and characterization of EGF receptor in individual exosomes by fluorescence-activated vesicle sorting. J. Extracell. Vesicles 2016, 5, 29254. [Google Scholar] [CrossRef] [PubMed]
- Zheringer, E.; Barta, T.; Li, M.; Vlassov, A. Strategies for Isolation of Exosomes. Cold Spring Harb. Protoc. 2015, 2015, 319–323. [Google Scholar] [CrossRef]
- Heinemann, M.L.; Ilmer, M.; Silva, L.P.; Hawke, D.H.; Recio, A.; Vorontsova, M.A.; Vykoukal, J. Benchtop isolation and characterization of functional exosomes by sequential filtration. J. Chromatogr. A 2014, 1371, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Han, C.; Labuz, J.M.; Kim, J.; Kim, J.; Cho, S.; Gho, Y.S.; Takayama, S.; Park, J. High-yield isolation of extracellular vesicles using aqueous two-phase system. Sci. Rep. 2015, 5, 13103. [Google Scholar] [CrossRef] [PubMed]
- Lamparski, H.G.; Metha-Damani, A.; Yao, J.Y.; Patel, S.; Hsu, D.H.; Ruegg, C.; Le Pecq, J.B. Production and characterization of clinical grade exosomes derived from dendritic cells. J. Immunol. Methods 2002, 270, 211–226. [Google Scholar] [CrossRef]
- Tauro, B.J.; Greening, D.W.; Mathias, R.A.; Ji, H.; Mathivanan, S.; Scott, A.M.; Simpson, R.J. Comparison of ultracentrifugation, density gradient separation, and immunoaffinity capture methods for isolating human colon cancer cell line LIM1863-derived exosomes. Methods 2012, 56, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Chen, Y.; Zhang, F.; Zhao, Q.; Zhong, H. Increased anti-tumour activity by exosomes derived from doxorubicin-treated tumour cells via heat stress. Int. J. Hyperth. 2015, 31, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Ouyang, Y.; Wang, Z.; Zhang, R.; Huang, P.H.; Chen, C.; Li, H.; Li, P.; Quinn, D.; Dao, M.; et al. Isolation of exosomes from whole blood by integrating acoustics and microfluidics. Proc. Natl. Acad. Sci. USA 2017, 114, 10584–10589. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Guo, J.; Tian, F.; Yang, N.; Yan, F.; Ding, Y.; Wei, J.; Hu, G.; Nie, G.; Sun, J. Field-Free Isolation of Exosomes from Extracellular Vesicles by Microfluidic Viscoelastic Flows. ACS Nano 2017, 11, 6968–6976. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wu, H.J.; Fine, D.; Schmulen, J.; Hu, Y.; Godin, B.; Zhang, J.X.; Liu, X. Ciliated micropillars for the microfluidic-based isolation of nanoscale lipid vesicles. Lab. Chip 2013, 13, 2879–2882. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.G.; Kong, M.Q.; Zhou, S.; Sheng, Y.F.; Wang, P.; Yu, T.; Inci, F.; Kuo, W.P.; Li, L.J.; Demirci, U.; et al. An integrated double-filtration microfluidic device for isolation, enrichment and quantification of urinary extracellular vesicles for detection of bladder cancer. Sci. Rep. 2017, 7, 46224. [Google Scholar] [CrossRef] [PubMed]
- Smyth, T.; Kullberg, M.; Malik, N.; Smith-Jones, P.; Graner, M.W.; Anchordoquy, T.J. Biodistribution and Delivery Efficiency of Unmodified Tumor-Derived Exosomes. J. Control. Release 2015, 199, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Cataldi, M.; Vigliotti, D.; Mosca, T.; Cammarota, M.; Capone, D. Emerging Role of the Spleen in the Pharmacokinetics of Monoclonal Antibodies, Nanoparticles and Exosomes. Int. J. Mol. Sci. 2017, 18, E1249. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Zhuang, X.; Xiang, X.; Liu, Y.; Zhang, S.; Liu, C.; Barnes, S.; Grizzle, W.; Miller, D.; Zhang, H.G. A novel nanoparticle drug delivery system: The anti-inflammatory activity of curcumin is enhanced whenencapsulated in exosomes. Mol. Ther. 2010, 18, 1606–1614. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.W.; Choi, H.; Jang, S.C.; Yoo, M.Y.; Park, J.Y.; Choi, N.E.; Oh, H.J.; Ha, S.; Lee, Y.S.; Jeong, J.M.; et al. Noninvasive imaging of radiolabeled exosome-mimetic nanovesicle using (99m)Tc-HMPAO. Sci. Rep. 2015, 5, 15636. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Nishikawa, M.; Shinotsuka, H.; Matsui, Y.; Ohara, S.; Imai, T.; Takakura, Y. Visualization and in vivo tracking of the exosomes of murine melanoma B16-BL6 cells in mice after intravenous injection. J. Biotechnol. 2013, 165, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Morishita, M.; Takahashi, Y.; Nishikawa, M.; Sano, K.; Kato, K.; Yamashita, T.; Imai, T.; Saji, H.; Takakura, Y. Quantitative analysis of tissue distribution of the B16BL6-derived exosomes using a streptavidin-lactadherinfusion protein and iodine-125-labeled biotin derivative after intravenous injection in mice. J. Pharm. Sci. 2015, 104, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Takahashi, Y.; Nishikawa, M.; Kato, K.; Morishita, M.; Yamashita, T.; Matsumoto, A.; Charoenviriyakul, C.; Takakura, Y. Macrophage-dependent clearance of systemically administeredB16BL6-derived exosomes from the blood circulation in mice. J. Extracell. Vesicles 2015, 4, 26238. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; Zhao, W.L.; Ye, Y.Y.; Bai, X.C.; Liu, R.Q.; Chang, L.F.; Zhou, Q.; Sui, S.F. Cellular internalization ofexosomes occurs through phagocytosis. Traffic 2010, 11, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3, 24641. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A.; Turkes, A.; Dewitt, S.; Steadman, R.; Mason, M.D.; Hallett, M.B. Adhesion and signaling by Bcell-derived exosomes: The role of integrins. FASEB J. 2004, 18, 977–979. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Singh, S.P.; Elkahloun, A.G.; Wu, W.; Abu-Asab, M.S.; Roberts, D.D. CD47-dependent immunomodulatory and angiogenic activities ofextracellular vesicles produced by T cells. Matrix Biol. 2014, 37, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Long, K.B.; Beatty, G.L. Harnessing the antitumor potential of macrophages for cancer immunotherapy. Oncoimmunology 2013, 2, e26860. [Google Scholar] [CrossRef] [PubMed]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Kooijmans, S.A.; Fliervoet, L.A.; van der Meel, R.; Fens, M.H.; Heijnen, H.F.; van Bergen En Henegouwen, P.M.P.; Vader, P.; Schiffelers, R.M. PEGylated and targeted extracellular vesicles display enhanced cell specificity and circulation time. J. Control. Release 2016, 224, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Pascucci, L.; Cocce, V.; Bonomi, A.; Ami, D.; Ceccarelli, P.; Ciusani, E.; Viganò, L.; Locatelli, A.; Sisto, F.; Doglia, S.M.; et al. Paclitaxel is incorporated by mesenchymal stromal cells and released in exosomes that inhibit in vitro tumor growth: A new approach for drug delivery. J. Control. Release 2014, 192, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Haney, M.J.; Zhao, Y.; Mahajan, V.; Deygen, I.; Klyachko, N.L.; Inskoe, E.; Piroyan, A.; Sokolsky, M.; Okolie, O.; et al. Development of exosome-encapsulated paclitaxel to overcome MDR in cancer cells. Nanomedicine 2016, 12, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.H.; Wan, Y.L.; Lin, Y.; Zhang, W.; Yang, M.; Li, G.L.; Lin, H.M.; Shang, C.Z.; Chen, Y.J.; Min, J. Anticancer drugs causerelease of exosomes with heat shock proteins from humanhepatocellular carcinoma cells that elicit effective naturalkiller cell antitumor responses in vitro. J. Biol. Chem. 2012, 287, 15874–15885. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Xiang, X.; Grizzle, W.; Sun, D.; Zhang, S.; Axtell, R.C.; Ju, S.; Mu, J.; Zhang, L.; Steinman, L.; et al. Treatment of brain inflammatory diseases by delivering exosome encapsulated anti-inflammatory drugs from the nasal region to the brain. Mol. Ther. 2011, 19, 1769–1779. [Google Scholar] [CrossRef] [PubMed]
- Haney, M.J.; Klyachko, N.L.; Zhaoa, Y.L.; Gupta, R.; Plotnikova, E.G.; He, Z.J.; Patel, T.; Piroyan, A.; Sokolsky, M.; Kabanov, A.V.; et al. Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J. Control. Release 2015, 207, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef] [PubMed]
- Nakase, I.; Futaki, S. Combined treatment with a pH-sensitive fusogenic peptide and cationic lipids achieves enhanced cytosolic delivery of exosomes. Sci. Rep. 2015, 5, 10112. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, G.; Serio, A.; Mazo, M.; Nair, R.; Stevens, M.M. Active loading into extracellular vesicles significantly improves the cellular uptake and photodynamic effect of porphyrins. J. Control. Release 2015, 205, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Wahlgren, J.; Karlson, T.D.L.; Brisslert, M.; Vaziri Sani, F.; Telemo, E.; Sunnerhagen, P.; Valadi, H. Plasma exosomes can deliver exogenous short interfering RNA to monocytes and lymphocytes. Nucleic Acids Res. 2012, 40, e130. [Google Scholar] [CrossRef] [PubMed]
- Lamichhane, T.N.; Jeyaram, A.; Patel, D.B.; Parajuli, B.; Livingston, N.K.; Arumugasaamy, N.; Schardt, J.S.; Jay, S.M. Oncogeneknockdown via active loading of small RNAs intoextracellular vesicles by sonication. Cell Mol. Bioeng. 2016, 9, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Shtam, T.A.; Kovalev, R.A.; Varfolomeeva, E.Y.; Makarov, E.M.; Kil, Y.V.; Filatov, M.V. Exosomes are natural carriers of exogenous siRNA to human cells in vitro. Cell Commun. Signal. 2013, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Didiot, M.C.; Hall, L.M.; Coles, A.H.; Haraszti, R.A.; Godinho, B.M.; Chase, K.; Sapp, E.; Ly, S.; Alterman, J.F.; Hassler, M.R.; et al. Exosome-mediated delivery of hydrophobically modified siRNA forHuntingtin mRNA silencing. Mol. Ther. 2016, 24, 1836–1847. [Google Scholar] [CrossRef] [PubMed]
- Stremersch, S.; Vandenbroucke, R.E.; Van Wonterghem, E.; Hendrix, A.; De Smedt, S.C.; Raemdonck, K. Comparing exosome-like vesicles with liposomes forthe functional cellular delivery of small RNAs. J. Control. Release 2016, 232, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Haney, M.J.; Zhao, Y.; Yuan, D.; Deygen, I.; Klyachko, N.L.; Kabanov, A.V.; Batrakova, E.V. Engineering for targeted paclitaxel delivery to pulmonary metastases: In vitro and in vivo evaluations. Nanomedicine 2018, 14, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Camussi, G.; Deregibus, M.C.; Bruno, S.; Grange, C.; Fonsato, V.; Tetta, C. Exosome/microvesicle-mediated epigenetic reprogramming of cells. Am. J. Cancer Res. 2011, 1, 98–110. [Google Scholar] [PubMed]
- Meckes, D.G., Jr.; Raab-Traub, N. Microvesicles and viral infection. J. Virol. 2011, 85, 12844–12854. [Google Scholar] [CrossRef] [PubMed]
- Kim, O.Y.; Lee, J.; Gho, Y.S. Extracellular vesicle mimetics: Novel alternatives to extracellular vesicle-based theranostics, drug delivery, and vaccines. Semin. Cell Dev. Biol. 2017, 67, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Zhao, X.; Xing, H.; Xun, Z.; Zhu, S.; Lang, L.; Yang, T.; Cai, C.; Wang, D.; Ding, P. Comparison of exosome-mimicking liposomes with conventional liposomes for intracellular delivery of siRNA. Int. J. Pharm. 2018, 550, 100–113. [Google Scholar] [CrossRef] [PubMed]
- De La Peña, H.; Madrigal, J.A.; Rusakiewicz, S.; Bencsik, M.; Cave, G.W.; Selman, A.; Rees, R.C.; Travers, P.J.; Dodi, I.A. Artificial exosomes as tools for basic and clinical immunology. J. Immunol. Methods 2009, 344, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lorenzo, M.J.; Anel, A.; Saez-Gutierrez, B.; Royo-Canas, M.; Bosque, A.; Alava, M.A.; Piñeiro, A.; Lasierra, P.; Asín-Ungría, J.; Larrad, L. Rheumatoid synovial fluid T cells are sensitive to APO2L/TRAIL. Clin. Immunol. 2007, 122, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lostao, L.; García-Alvarez, F.C.; Basáñez, G.; Alegre-Aguarón, E.; Desportes, P.; Larrad, L.; Naval, J.; Martínez-Lorenzo, M.J.; Anel, A. Liposome-bound APO2L/TRAIL is an effective treatment in a rabbit model of rheumatoid arthritis. Arthritis Rheum. 2010, 62, 2272–2282. [Google Scholar] [CrossRef] [PubMed]
- De Miguel, D.; Basáñez, G.; Sánchez, D.; Malo, P.G.; Marzo, I.; Larrad, L.; Naval, J.; Pardo, J.; Anel, A.; Martinez-Lostao, L. Liposomes Decorated with Apo2L/TRAIL Overcome Chemoresistance of Human Hematologic Tumor Cells. Mol. Pharm. 2013, 10, 893–904. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.B.; Zhuang, X.; Ju, S.; Xiang, X.; Mu, J.; Liu, Y.; Jiang, H.; Zhang, L.; Mobley, J.; McClain, C.; et al. Exosome-like nanoparticles from intestinal mucosal cells carry prostaglandin E2 and suppress activation of liver NKT cells. J. Immunol. 2013, 190, 3579–3589. [Google Scholar] [CrossRef] [PubMed]
- Goh, W.J.; Zhou, S.; Ong, W.Y.; Torta, F.; Alexandra, A.F.; Schiffelers, R.M.; Storm, G.; Wang, J.W.; Czarny, B.; Pastorin, G. Bioinspired Cell-Derived Nanovesicles versus Exosomes as Dryg Delivery Systems: A Cost-Effective Alternative. Sci. Rep. 2017, 7, 14322. [Google Scholar] [CrossRef] [PubMed]
- Goh, W.J.; Lee, C.K.; Zou, S.; Woon, E.C.; Czarny, B.; Pastorin, G. Doxorubicin-loaded cell-derived nanovesicles: An alternative targeted approach for anti-tumor therapy. Int. J. Nanomed. 2017, 12, 2759–2767. [Google Scholar] [CrossRef] [PubMed]
- Jo, W.; Kim, J.; Yoon, J.; Jeong, D.; Cho, S.; Jeong, H.; Yoon, Y.J.; Kim, S.C.; Gho, Y.S.; Park, J. Large-scale generation of cell-derived nanovesicles. Nanoscale 2014, 6, 12056–12064. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.Y.; Ji, A.L.; Wang, Z.X.; Qiang, G.H.; Qu, Z.; Wu, J.H.; Jiang, C.P. Exosome-Mimetic Nanovesicles from Hepatocytes promote hepatocyte proliferation in vitro and liver regeneration in vivo. Sci. Rep. 2018, 8, 2471. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.; Jo, W.; Jeong, D.; Kim, J.; Jeong, H.; Park, J. Generation of nanovesicles with sliced cellular membrane fragments for exogenous material delivery. Biomaterials 2015, 59, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.C.; Kim, O.Y.; Yoon, C.M.; Choi, D.S.; Roh, T.Y.; Park, J.; Nilsson, J.; Lötvall, J.; Kim, Y.K.; Gho, Y.S. Bioinspired exosome-mimetic nanovesicles for targeted delivery of chemotherapeutics to malignant tumors. ACS Nano 2013, 7, 7698–7710. [Google Scholar] [CrossRef] [PubMed]
- Lunavat, T.R.; Jang, S.C.; Nilsson, L.; Park, H.T.; Repiska, G.; Lässer, C.; Nilsson, J.A.; Gho, Y.S.; Lötvall, J. RNAi delivery by exosome-mimetic nanovesicles—Implications for targeting c-Myc in cancer. Biomaterials 2016, 102, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hensley, L.; McKnight, K.L.; Hu, F.; Madden, V.; Ping, L.; Jeong, S.H.; Walker, C.; Lanford, R.E.; Lemon, S.M. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature 2013, 496, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Zucker, D.; Marcus, D.; Barenholz, Y.; Goldblum, A. Liposome drugs’ loading efficiency: A working model based on loading conditions and drug’s physicochemical properties. J. Control. Release 2009, 139, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Hood, R.R.; Vreeland, W.N.; Devoe, D.L. Microfluidic remote loading for rapid single-step liposomal drug preparation. Lab. Chip 2014, 14, 3359–3367. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Angsantikul, P.; Ying, M.; Zhuang, J.; Zhang, Q.; Wei, X.; Jiang, Y.; Zhang, Y.; Dehaini, D.; Chen, M.; et al. Remote Loading of Small-Molecule Therapeutics into Cholesterol-Enriched Cell-Membrane-Derived Vesicles. Angew. Chem. Int. Ed. Engl. 2017, 56, 14075–14079. [Google Scholar] [CrossRef] [PubMed]
- Ying, M.; Zhuang, J.; Wei, X.; Zhang, X.; Zhang, X.; Jiang, Y.; Dehaini, D.; Chen, M.; Gu, S.; Gao, W.; et al. Remote-Loaded Platelet Vesicles for Disease-Targeted Delivery of Therapeutics. Adv. Funct. Mat. 2018, 28, 1801032. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, J.; Jeong, M.; Lee, H.; Goh, U.; Kim, H.; Kim, B.; Park, J.H. Liposome-based engineering of cells to package hydrophobic compounds in membrane vesicles for tumor penetration. Nano Lett. 2015, 15, 2938–2944. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Lee, H.; Goh, U.; Kim, J.; Jeong, M.; Lee, J.; Park, J.H. Cellular Engineering with Membrane Fusogenic Liposomes to Produce Functionalized Extracellular Vesicles. ACS Appl. Mater. Interfaces 2016, 8, 6790–6795. [Google Scholar] [CrossRef] [PubMed]
- Akao, Y.; Nakagawa, Y.; Hirata, I.; Iio, A.; Itoh, T.; Kojima, K.; Nakashima, R.; Kitade, Y.; Naoe, T. Role of anti-oncomirs miR-143 and -145 in human colorectal tumors. Cancer Gene Ther. 2010, 17, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S.; Takanashi, M.; Sudo, K.; Ueda, S.; Ishikawa, A.; Matsuyama, N.; Fujita, K.; Mizutani, T.; Ohgi, T.; Ochiya, T.; et al. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol. Ther. 2013, 21, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, D.; Chen, X.; Li, J.; Li, L.; Bian, Z.; Sun, F.; Lu, J.; Yin, Y.; Cai, X.; et al. Secreted monocytic miR-150 enhances targeted endothelial cell migration. Mol. Cell 2010, 39, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Xin, H.; Li, Y.; Buller, B.; Katakowski, M.; Zhang, Y.; Wang, X.; Shang, X.; Zhang, Z.G.; Chopp, M. Exosome-mediatedtransfer of miR-133b from multipotent mesenchymal stromal cells to neural cells contributes to neurite outgrowth. Stem Cells 2012, 30, 1556–1564. [Google Scholar] [CrossRef] [PubMed]
- Maguire, C.A.; Balaj, L.; Sivaraman, S.; Crommentuijn, M.H.; Ericsson, M.; Mincheva-Nilsson, L.; Baranov, V.; Gianni, D.; Tannous, B.A.; Sena-Esteves, M.; et al. Microvesicle-associated AAV vector as a novel gene delivery system. Mol. Ther. 2012, 20, 960–971. [Google Scholar] [CrossRef] [PubMed]
- Kooijmans, S.A.; Stremersch, S.; Braeckmans, K.; de Smedt, S.C.; Hendrix, A.; Wood, M.J.; Schiffelers, R.M.; Raemdonck, K.; Vader, P. Electroporation-induced siRNA precipitation obscures the efficiency of siRNA loading into extracellular vesicles. J. Control. Release 2013, 172, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, K.B.; Gudbergsson, J.M.; Skov, M.N.; Christiansen, G.; Gurevich, L.; Moos, T.; Duroux, M. Evaluation of electroporation-induced adverse effects on adipose-derived stem cell exosomes. Cytotechnology 2016, 68, 2125–2138. [Google Scholar] [CrossRef] [PubMed]
- Lamichhane, T.N.; Raiker, R.S.; Jay, S.M. Exogenous DNA loading into extracellular vesicles via electroporation is size-dependent and enables limited gene delivery. Mol. Pharm. 2015, 12, 3650–3657. [Google Scholar] [CrossRef] [PubMed]
- Podolak, I.; Galanty, A.; Sobolewska, D. Saponins as cytotoxic agents: A review. Phytochem. Rev. 2010, 9, 425–474. [Google Scholar] [CrossRef] [PubMed]
- Hood, J.L. Post isolation modification of exosomes for nanomedicine applications. Nanomedicine (Lond) 2016, 11, 1745–1756. [Google Scholar] [CrossRef] [PubMed]
- Mentkowski, K.I.; Snitzer, J.D.; Rusnak, S.; Lang, J.K. Therapeutic Potential of Engineered Extracellular Vesicles. AAPS J. 2018, 20, 50. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, D.; Liu, Z.; Zhou, Y.; Chu, D.; Li, X.; Jiang, X.; Hou, D.; Chen, X.; Chen, Y.; et al. Targeted exosome-mediated delivery of opioid receptor Mu siRNA for the treatment of morphine relapse. Sci. Rep. 2015, 5, 17543. [Google Scholar] [CrossRef] [PubMed]
- Oude Blenke, E.; Klaasse, G.; Merten, H.; Pluckthun, A.; Mastrobattista, E.; Martin, N.I. Liposome functionalization with copper-free “click chemistry”. J. Control. Release 2015, 202, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Altinoglu, S.; Takeda, Y.S.; Xu, Q. Integrating protein engineering and bioorthogonal click conjugation for extracellular vesicle modulation and intracellular delivery. PLoS ONE 2015, 10, e0141860. [Google Scholar] [CrossRef] [PubMed]
- Smyth, T.; Petrova, K.; Payton, N.M.; Persaud, I.; Redzic, J.S.; Graner, M.W.; Smith-Jones, P.; Anchordoquy, T.J. Surface functionalization of exosomes using click chemistry. Bioconjugate Chem. 2014, 25, 1777–1784. [Google Scholar] [CrossRef] [PubMed]
- Lakrishnan, G.; Danelon, C.; Izewska, P.; Prummer, M.; Bolinger, P.-Y.; Geissbühler, I.; Demurtas, D.; Dubochet, J.; Vogel, H. Multifunctional Lipid/Quantum Dot Hybrid Nanocontainers for Controlled Targeting of Live Cells. Angew. Chem. Int. Ed. 2006, 45, 5478–5483. [Google Scholar] [CrossRef] [PubMed]
- Nel, A.E.; Mädler, L.; Velegol, D.; Xia, T.; Hoek, E.M.; Somasundaran, P.; Klaessig, F.; Castranova, V.; Thompson, M. Understanding biophysicochemical interactions at the nano-bio interface. Nat. Mater. 2009, 8, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Liu, C.; Long, L.; Ren, Y.; Zhang, S.; Chang, X.; Qian, X.; Jia, H.; Zhao, J.; Sun, J.; et al. Blood exosomes endowed with magnetic and targeting properties for cancer therapy. ACS Nano 2016, 10, 3323–3333. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Wu, N.; Yang, J.M.; Gould, S.J. Protein targeting to exosomes/microvesicles by plasma membrane anchors. J. Biol. Chem. 2011, 286, 14383–14395. [Google Scholar] [CrossRef] [PubMed]
- Marcus, M.E.; Leonard, J.N. FedExosomes: Engineering Therapeutic Biological Nanoparticles that Truly Deliver. Pharmaceuticals (Basel) 2013, 6, 659–680. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Ran, N.; Dong, X.; Zuo, B.; Yang, R.; Zhuo, Q.; Moulton, H.M.; Seow, Y.; Yin, H. Anchor peptide captures, targets, and loads exosomes of diverse origins for diagnostics and therapy. Sci. Transl. Med. 2018, 10, eaat0195. [Google Scholar] [CrossRef] [PubMed]
- Ran, R.; Middelberg, A.P.J.; Zhao, C.X. Microfluidic synthesis of multifunctional liposomes for tumour targeting. Colloids Surf. B Biointerfaces 2016, 148, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Kibria, G.; Ramos, E.K.; Wan, Y.; Gius, D.R.; Liu, H. Exosomes as a Drug Delivery System in Cancer Therapy: Potential and Challenges. Mol. Pharm. 2018, 15, 3625–3633. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, A.; Silva, A.M.; Teixeira, J.H.; Gonçalves, R.M.; Almeida, M.I.; Barbosa, M.A.; Santos, S.G. Extracellular vesicles: Intelligent delivery strategies for therapeutic applications. J. Control. Release 2018, 289, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Namee, N.M.; O’Driscoll, L. Extracellular vesicles and anti-cancer drug resistance. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Bei, Y.; Das, S.; Rodosthenous, R.S.; Holvoet, P.; Vanhaverbeke, M.; Monteiro, M.C.; Monteiro, V.V.S.; Radosinska, J.; Bartekova, M.; Jansen, F.; et al. Extracellular vesicles in cardiovascular theranostics. Theranostics 2017, 7, 4168–4182. [Google Scholar] [CrossRef] [PubMed]
- Rackov, G.; Garcia-Romero, N.; Esteban-Rubio, S.; Carrión-Navarro, J.; Belda-Iniesta, C.; Ayuso-Sacido, A. Vesicle-mediated control of cell function: The role of extracellular matrix and microenvironment. Front. Physiol. 2018, 9, 651. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.; Prabhakar, S.; Balaj, L.; Lai, C.P.; Cerione, R.A.; Breakefield, X.O. Delivery of Therapeutic Proteins via Extracellular Vesicles: Review and Potential Treatments for Parkinson’s Disease, Glioma, and Schwannoma. Cell. Mol. Neurobiol. 2016, 36, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Ridolfi, B.; Abdel-Haq, H. Neurodegenerative disorders treatment: The microRNA role. Curr. Gene Ther. 2017, 17, 327–363. [Google Scholar] [CrossRef] [PubMed]
- Cunha, S.; Amaral, M.H.; Sousa Lobo, J.M.; Silva, A.C. Therapeutic strategies for Alzheimer’s and Parkinson’s diseases by means of drug delivery systems. Curr. Med. Chem. 2016, 23, 3618–3631. [Google Scholar] [CrossRef] [PubMed]
- Bellavia, D.; Raimondi, L.; Costa, V.; De Luca, A.; Carina, V.; Maglio, M.; Fini, M.; Alessandro, R.; Giavaresi, G. Engineered exosomes: A new promise for the management of musculoskeletal diseases. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 1893–1901. [Google Scholar] [CrossRef] [PubMed]
- Yao, K.; Ricardo, S.D. Mesenchymal stem cells as novel micro-ribonucleic acid delivery vehicles in kidney disease. Nephrology (Carlton) 2016, 21, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, J.P.K.; Stevens, M.M. Strategic design of extracellular vesicle drug delivery systems. Adv. Drug Deliv. Rev. 2018, 130, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.; Zhao, Y.; Banks, W.A.; Bullock, K.M.; Haney, M.; Batrakova, E.; Kabanov, A.V. Macrophage Exosomes as Natural Nanocarriers for Protein Delivery to Inflamed Brain. Biomaterials 2017, 142, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Xing, F.; Wu, S.Y.; Watabe, K. Extracellular vesicles as emerging targets in cancer: Recent development from bench to bedside. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 538–563. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, G.; Chandrawati, R.; Parmar, P.A.; Keane, T.J.; Maynard, S.A.; Bertazzo, S.; Stevens, M.M. Engineering Extracellular Vesicles with the Tools of Enzyme Prodrug Therapy. Adv. Mater. 2018, 30, e1706616. [Google Scholar] [CrossRef] [PubMed]
- Reátegui, E.; van der Vos, K.E.; Lai, C.P.; Zeinali, M.; Atai, N.A.; Aldikacti, B.; Floyd, F.P., Jr.; Khankhel, A.H.; Thapar, V.; Hochberg, F.H.; et al. Engineered nanointerfaces for microfluidic isolation and molecular profiling of tumor-specific extracellular vesicles. Nat. Commun. 2018, 9, 175. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Advantages | Disadvantages | Drug Loaded/Application |
|---|---|---|---|
| Treatment of parental cells with drug | Relatively simple Does not require addition of drug into the system | Low loading efficiency Drugs may be cytotoxic to cells | Paclitaxel (Ptx) [104] Hydrophobic sensitizers (model drug) [141] |
| Incubation with drug | Simplest method Do not require extra equipement | Low loading efficiency | Curcumin [92,107]; si RNAs [116]; Porphyrins [111]; Catalase [108]; PTX [105]; DOX [129] |
| Electroporation | Loading with large molecules possible | Disrupts EX integrity siRNA aggregation Low loading efficiency (hydrophobic drugs) | siRNA [112,114]; Porphyrins [111]; DOX [109]; Dextran macromolecules [110]; PTX [105] |
| Sonication | Increased loading efficiency (compared to other methods) Applicable for small RNAs | Potential deformation of membrane Not efficient for hydrophobic drugs | PTX [105]; Catalase [108]; siRNA, miRNA, ssDNA [114,149] |
| Extrusion | High drug loading efficiency | Potential deformation of membrane | Porphyrins [111]; Catalase [108] |
| Freeze/thaw method | Medium loading Fusion of membranes possible [54] | Exosomes may aggregate Low loading Efficiency | Catalase [108]; DOX [129] |
| Saponin-assisted loading | High drug loading, compared to the other methods used in early reports | Generates pores in EXs Haemolysis/Toxicity concerns [150] Saponin conc. Control & Washing required | Catalase [108]; Porphyrins [111]; DOX [129] |
| Status | Study Title | Conditions | Interventions | Phase | NCT Number |
|---|---|---|---|---|---|
| Not yet recruiting | Allogenic Mesenchymal Stem Cell Derived Exosome in Patients With Acute Ischemic Stroke | Cerebrovascular Disorders | Biological: exosome | Phase 1 Phase 2 | 03384433 |
| Enrolling by invitation | Effect of Plasma Derived Exosomes on Cutaneous Wound Healing | Ulcer | Other: plasma-derived exosomes | Early Phase 1 | 02565264 |
| Active, not recruiting | Study Investigating the Ability of Plant Exosomes to Deliver Curcumin to Normal and Colon Cancer Tissue | Colon Cancer | curcumin Curcumin conjugated with plant exosomes | Phase 1 | 01294072 |
| Recruiting | Dendritic Cells-Derived Exosomes in Human Sepsis | Sepsis | Drug: Antibiotics | 02957279 | |
| Not yet recruiting | Plant Exosomes and Patients Diagnosed With Polycystic Ovary Syndrome (PCOS) 17 | Polycystic Ovary Syndrome | Ginger exosomes Aloe exosomes Placebo | Not Applicable | 03493984 |
| Unknown | Effect of Microvesicles and Exosomes Therapy on β-cell Mass in Type I Diabetes Mellitus (T1DM) | Diabetes Mellitus Type 1 | Biological: MSC exosomes. | Phase 2 Phase 3 | 02138331 |
| Not yet recruiting | iExosomes in Treating Participants With Metastatic Pancreas Cancer With KrasG12D Mutation | KRAS NP_004976.2:p.G12D Metastatic Pancreatic Adenocarcinoma | Drug: Mesenchymal Stromal Cells-derived Exosomes with KRAS G12D siRNA | Phase 1 | 03608631 |
| Recruiting | MSC-Exos Promote Healing of MHs | Macular Holes | Biological: exosomes derived from mesenchymal stem cells (MSC-Exo) | Early Phase 1 | 03437759 |
| Active, not recruiting | Edible Plant Exosome Ability to Prevent Oral Mucositis Associated With Chemoradiation Treatment of Head and Neck Cancer | Head and Neck Cancer Oral Mucositis | Dietary Supplement: Grape extract Drug: Lortab, Fentanyl patch, mouthwash | Phase 1 | 01668849 |
| Completed | Trial of a Vaccination With Tumor Antigen-loaded Dendritic Cell-derived Exosomes | Non Small Cell Lung Cancer | Biological: Dex2 | Phase 2 | 01159288 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antimisiaris, S.G.; Mourtas, S.; Marazioti, A. Exosomes and Exosome-Inspired Vesicles for Targeted Drug Delivery. Pharmaceutics 2018, 10, 218. https://doi.org/10.3390/pharmaceutics10040218
Antimisiaris SG, Mourtas S, Marazioti A. Exosomes and Exosome-Inspired Vesicles for Targeted Drug Delivery. Pharmaceutics. 2018; 10(4):218. https://doi.org/10.3390/pharmaceutics10040218
Chicago/Turabian StyleAntimisiaris, Sophia G., Spyridon Mourtas, and Antonia Marazioti. 2018. "Exosomes and Exosome-Inspired Vesicles for Targeted Drug Delivery" Pharmaceutics 10, no. 4: 218. https://doi.org/10.3390/pharmaceutics10040218
APA StyleAntimisiaris, S. G., Mourtas, S., & Marazioti, A. (2018). Exosomes and Exosome-Inspired Vesicles for Targeted Drug Delivery. Pharmaceutics, 10(4), 218. https://doi.org/10.3390/pharmaceutics10040218