A Tailored Thermosensitive PLGA-PEG-PLGA/Emulsomes Composite for Enhanced Oxcarbazepine Brain Delivery via the Nasal Route

 ,
,  , ,
, , 
 and
and
Abstract
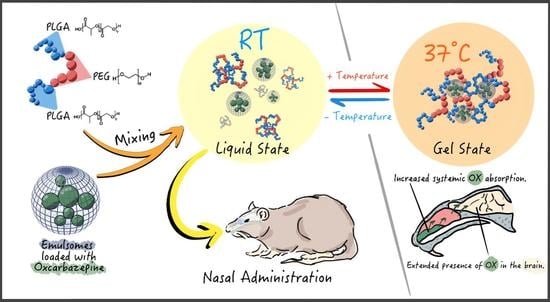
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of PLGA-PEG-PLGA Tri-Block Copolymers
2.3. Characterization of PLGA-PEG-PLGA Copolymers
2.3.1. Nuclear Magnetic Resonance (1H-NMR)
2.3.2. Gel Permeation Chromatography (GPC)
2.3.3. Differential Scanning Calorimetry (DSC)
2.3.4. Size Measurement of Copolymer Solution by Dynamic Light Scattering (DLS)
2.4. Preparation and Characterization of PLGA-PEG-PLGA Thermogel Solutions Loaded with Emulsomes
2.4.1. Sample Preparation
2.4.2. Macroscopic Phase Behaviour of Aqueous Copolymer Solutions and Determination of Gelation Temperature
2.4.3. Viscosity Measurement
2.5. In Vitro Drug Release
2.6. Mucoadhesion Studies
2.7. In Vivo Studies
2.7.1. Pharmacokinetic Study
2.7.2. Histopathological Study
2.8. Statistical Analysis
3. Results
3.1. Preparation and Characterization of PLGA-PEG-PLGA Triblock Copolymers
3.1.1. NMR Spectroscopy
3.1.2. GPC
3.1.3. DSC
3.1.4. DLS
3.2. Preparation and Characterization of Plain and Emulsome-Loaded PLGA-PEG-PLGA Thermogel Solutions
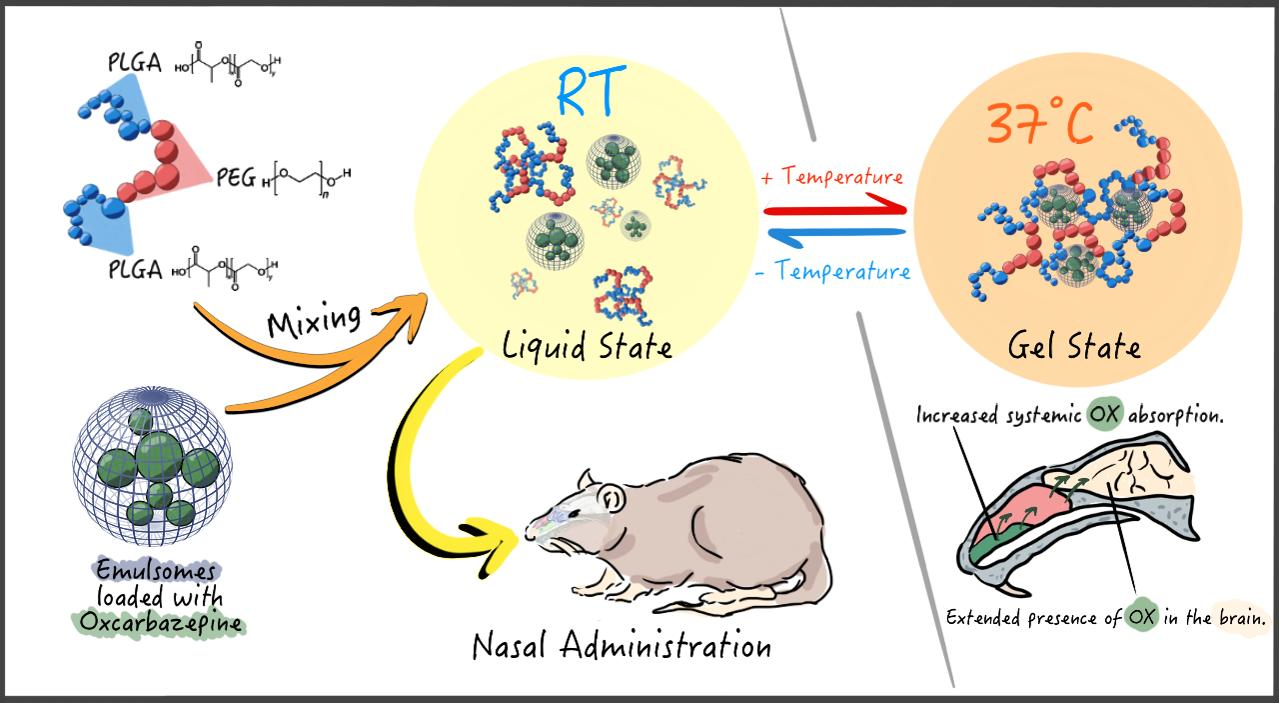
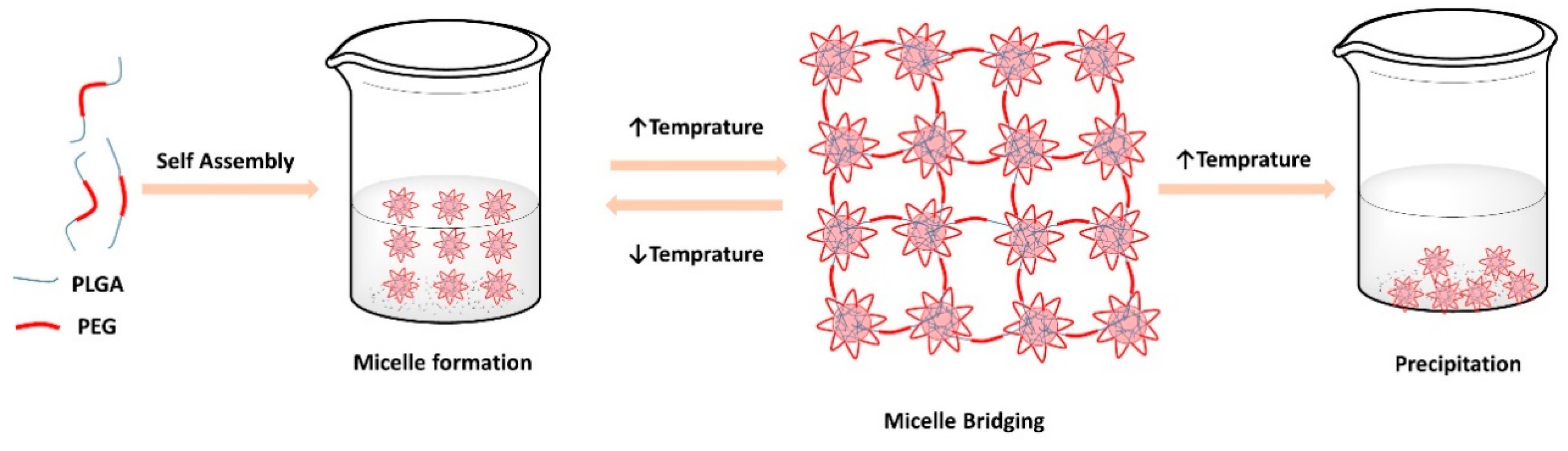
3.2.1. Thermogelation of the Copolymer Solutions
3.2.2. Viscosity Measurement
3.3. In Vitro Release Study
3.4. Mucoadhesion Study
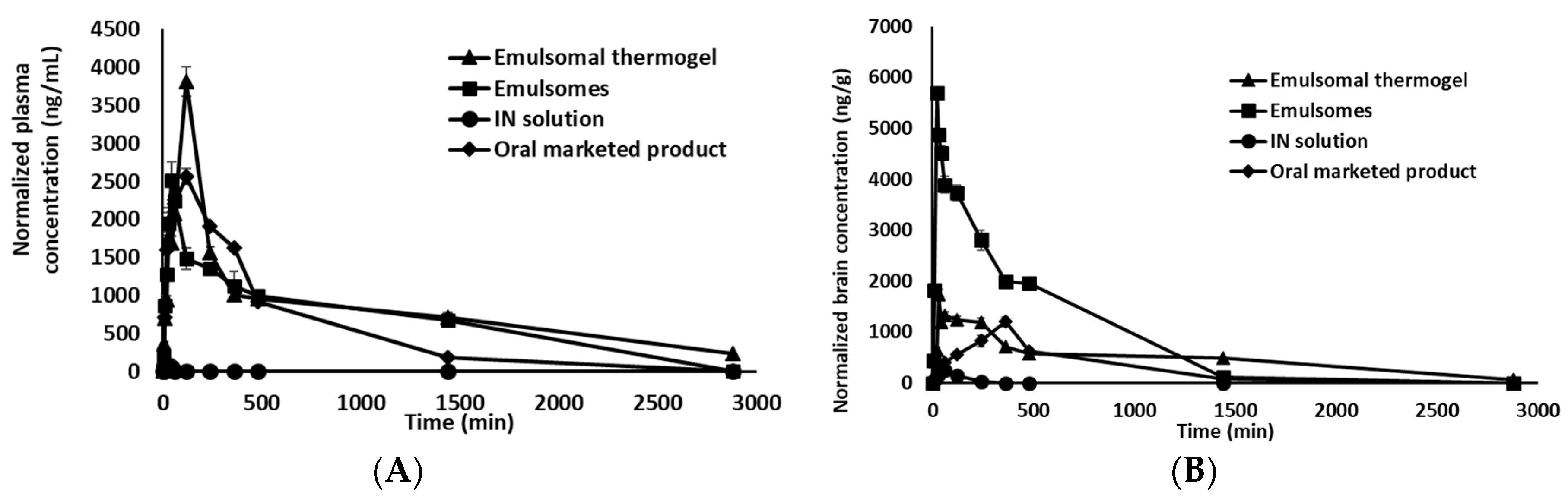
3.5. Pharmacokinetic Study
3.5.1. Plasma Pharmacokinetic Parameters
3.5.2. Brain Pharmacokinetic Parameters
3.6. Histopathological Examination
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yan, H.; Fujiwara, H.; Sasaki, K.; Tsujii, K. Rapid Swelling/Collapsing Behavior of Thermoresponsive Poly(N-isopropylacrylamide) Gel Containing Poly(2-(methacryloyloxy)decyl phosphate) Surfactant. Angew. Chem. 2005, 44, 1951–1954. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.J.; Suh, J.M.; Sohn, Y.S.; Bae, Y.H.; Kim, S.W.; Jeong, B. Thermogelling poly (caprolactone-b-ethylene glycol-b-caprolactone) aqueous solutions. Macromolecules 2005, 38, 5260–5265. [Google Scholar] [CrossRef]
- Ji, S.; Ding, J. A Macroscopic Helix Formation Induced by the Shrinking of a Cylindrical Polymeric Hydrogel. Polym. J. 2001, 33, 701–703. [Google Scholar] [CrossRef]
- Nayak, S.; Lyon, L.A. Photoinduced phase transitions in poly(N-isopropylacrylamide) microgels. Chem. Mater. 2004, 16, 2623–2627. [Google Scholar] [CrossRef]
- Kim, H.-J.; Lee, J.-H.; Lee, M. Stimuli-Responsive Gels from Reversible Coordination Polymers. Angew. Chem. 2005, 44, 5810–5814. [Google Scholar] [CrossRef] [PubMed]
- Suri, J.T.; Cordes, D.B.; Cappuccio, F.E.; Wessling, R.A.; Singaram, B. Continuous Glucose Sensing with a Fluorescent Thin-Film Hydrogel. Angew. Chem. 2003, 42, 5857–5859. [Google Scholar] [CrossRef] [PubMed]
- Zentner, G.M.; Rathi, R.; Shih, C.; McRea, J.C.; Seo, M.-H.; Oh, H.; Rhee, B.; Mestecky, J.; Moldoveanu, Z.; Morgan, M. Biodegradable block copolymers for delivery of proteins and water-insoluble drugs. J. Control. Release 2001, 72, 203–215. [Google Scholar] [CrossRef]
- Tiller, J.C. Increasing the Local Concentration of Drugs by Hydrogel Formation. Angew. Chem. 2003, 42, 3072–3075. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhu, W.; Wang, B.; Ding, J. A novel microgel and associated post-fabrication encapsulation technique of proteins. J. Control. Release 2005, 105, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Beebe, D.J.; Moore, J.S.; Bauer, J.M.; Yu, Q.; Liu, R.H.; Devadoss, C.; Jo, B.-H. Functional hydrogel structures for autonomous flow control inside microfluidic channels. Nature 2000, 404, 588–590. [Google Scholar] [CrossRef] [PubMed]
- Eeckman, F.; Moës, A.J.; Amighi, K. Synthesis and characterization of thermosensitive copolymers for oral controlled drug delivery. Eur. Polym. J. 2004, 40, 873–881. [Google Scholar] [CrossRef]
- Casey, D.J.; Jarrett, P.K.; Rosati, L. Diblock and Triblock Copolymers. Patents US4716203A, 29 December 1987. [Google Scholar]
- Cerrai, P.; Tricoli, M.; Andruzzi, F.; Paci, M.; Paci, M. Polyether-polyester block copolymers by non-catalysed polymerization of ɛ-caprolactone with poly (ethylene glycol). Polymer 1989, 30, 338–343. [Google Scholar] [CrossRef]
- Youxin, L.; Kissel, T. Synthesis and properties of biodegradable ABA triblock copolymers consisting of poly (l-lactic acid) or poly (l-lactic-co-glycolic acid) A-blocks attached to central poly (oxyethylene) B-blocks. J. Control. Release 1993, 27, 247–257. [Google Scholar] [CrossRef]
- Jeong, B.; Bae, Y.H.; Kim, S.W. Drug release from biodegradable injectable thermosensitive hydrogel of PEG–PLGA–PEG triblock copolymers. J. Control. Release 2000, 63, 155–163. [Google Scholar] [CrossRef]
- Jeong, B.; Lee, K.M.; Gutowska, A.; An, Y.H. % Thermogelling Biodegradable Copolymer Aqueous Solutions for Injectable Protein Delivery and Tissue Engineering. Biomacromolecules 2002, 3, 865–868. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-J.; Lee, J.-H.; Lee, M. Stimuli-responsive gels from reversible coordination polymers. Angew. Chem. 2005, 117, 5960–5964. [Google Scholar] [CrossRef]
- Ruel-Gariepy, E.; Leroux, J.-C. In situ-forming hydrogels—Review of temperature-sensitive systems. Eur. J. Pharm. Biopharm. 2004, 58, 409–426. [Google Scholar] [CrossRef] [PubMed]
- Katakam, M.; Ravis, W.R.; Banga, A.K. Controlled release of human growth hormone in rats following parenteral administration of poloxamer gels. J. Control. Release 1997, 49, 21–26. [Google Scholar] [CrossRef]
- Wenzel, J.G.; Balaji, K.S.; Koushik, K.; Navarre, C.; Duran, S.H.; Rahe, C.H.; Kompella, U.B. Pluronic® F127 gel formulations of Deslorelin and GnRH reduce drug degradation and sustain drug release and effect in cattle. J. Control. Release 2002, 85, 51–59. [Google Scholar] [CrossRef]
- Huh, K.M.; Cho, Y.W.; Park, K. PLGA-PEG block copolymers for drug formulations. Drug Dev. Deliv. 2003, 3, 1–11. [Google Scholar]
- Zhang, J. Switchable and Responsive Surfaces and Materials for Biomedical Applications; Elsevier: New York, NY, USA, 2014. [Google Scholar]
- Nguyen, M.K.; Lee, D.S. Injectable biodegradable hydrogels. Macromol. Biosci. 2010, 10, 563–579. [Google Scholar] [CrossRef] [PubMed]
- Shim, M.S.; Lee, H.T.; Shim, W.S.; Park, I.; Lee, H.; Chang, T.; Kim, S.W.; Lee, D.S. Poly (d,l-lactic acid-co-glycolic acid)-b-poly (ethylene glycol)-b-poly (d,l-lactic acid-co-glycolic acid) triblock copolymer and thermoreversible phase transition in water. J. Biomed. Mater. Res. A 2002, 61, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.L.; Venkataraman, S.; Fox, C.H.; Coady, D.J.; Frank, C.W.; Hedrick, J.L.; Yang, Y.Y. Modular composite hydrogels from cholesterol-functionalized polycarbonates for antimicrobial applications. J. Mater. Chem. B 2015, 3, 6953–6963. [Google Scholar] [CrossRef]
- Karavasili, C.; Fatouros, D.G. Smart materials: In situ gel-forming systems for nasal delivery. Drug Discov. Today 2016, 21, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Toledano, R.; Gil-Nagel, A. Adverse effects of antiepileptic drugs. Semin. Neurol. 2008, 28, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Minn, A.; Leclerc, S.; Heydel, J.-M.; Minn, A.-L.; Denizot, C.; Cattarelli, M.; Netter, P.; Gradinaru, D. Drug transport into the mammalian brain: The nasal pathway and its specific metabolic barrier. J. Drug Target. 2002, 10, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Chandrika, M.V.; Babu, M.K. Design and development of Candesartan cilexetil emulsomal drug delivery systems for the effective management of cardiovascular diseases. Indian J. Res. Pharm. Biotechnol. 2014, 2, 1283. [Google Scholar]
- Ucisik, M.H.; Küpcü, S.; Schuster, B.; Sleytr, U.B. Characterization of CurcuEmulsomes: Nanoformulation for enhanced solubility anddelivery of curcumin. J. Nanobiotechnol. 2013, 11, 37. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Sharma, D.; Singh, G.; Singh, M.; Rathore, M.S. Lipoidal soft hybrid biocarriers of supramolecular construction for drug delivery. ISRN Pharm. 2012, 2012, 474830. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-C.; Hsieh, W.-Y.; Lee, W.-F.; Zeng, D.-T. Effects of surface modification of PLGA-PEG-PLGA nanoparticles on loperamide delivery efficiency across the blood–brain barrier. J. Biomater. Appl. 2013, 27, 909–922. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Li, J.; Tan, H.; Fu, Q. Self-assembly of biodegradable polyurethanes for controlled delivery applications. Soft Matter 2012, 8, 5414–5428. [Google Scholar] [CrossRef]
- Duvvuri, S.; Janoria, K.G.; Mitra, A.K. Development of a novel formulation containing poly (d, l-lactide-co-glycolide) microspheres dispersed in PLGA–PEG–PLGA gel for sustained delivery of ganciclovir. J. Control. Release 2005, 108, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Senesi, A. Analysis of Rheological Properties of Thermogel Copolymers Based PLGA-PEG1500-PLGA: Effect of the Molecular Weight of the Side Chains; University of Camerino: Camerino, Italy, 2013. [Google Scholar]
- Ghahremankhani, A.A.; Dorkoosh, F.; Dinarvand, R. PLGA-PEG-PLGA tri-block copolymers as an in-situ gel forming system for calcitonin delivery. Polym. Bull. 2007, 59, 637–646. [Google Scholar] [CrossRef]
- Song, Z.; Feng, R.; Sun, M.; Guo, C.; Gao, Y.; Li, L.; Zhai, G. Curcumin-loaded PLGA-PEG-PLGA triblock copolymeric micelles: Preparation, pharmacokinetics and distribution in vivo. J. Colloid Interface Sci. 2011, 354, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Ren, F.; Ding, B.; Sun, N.; Liu, X.; Ding, X.; Gao, S. A thermo-sensitive PLGA-PEG-PLGA hydrogel for sustained release of docetaxel. J. Drug Target. 2011, 19, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Paliwal, R.; Paliwal, S.R.; Mishra, N.; Mehta, A.; Vyas, S.P. Engineered chylomicron mimicking carrier emulsome for lymph targeted oral delivery of methotrexate. Int. J. Pharm. 2009, 380, 181–188. [Google Scholar] [CrossRef] [PubMed]
- El-Zaafarany, G.M.; Soliman, M.E.; Mansour, S.; Awad, G.A.S. Identifying lipidic emulsomes for improved oxcarbazepine brain targeting: In vitro and rat in vivo studies. Int. J. Pharm. 2016, 503, 127–140. [Google Scholar] [CrossRef] [PubMed]
- Khodaverdi, E.; Hadizadeh, F.; Tekie, F.S.M.; Jalali, A.; Mohajeri, S.A.; Ganji, F. Preparation and analysis of a sustained drug delivery system by PLGA–PEG–PLGA triblock copolymers. Polym. Bull. 2012, 69, 429–438. [Google Scholar] [CrossRef]
- Basu, S.; Bandyopadhyay, A.K. Development and characterization of mucoadhesive in situ nasal gel of midazolam prepared with Ficus carica mucilage. AAPS PharmSciTech 2010, 11, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.M.; Prajapati, B.G.; Patel, M.M. Formulation, evaluation, and comparison of bilayered and multilayered mucoadhesive buccal devices of propranolol hydrochloride. AAPS PharmSciTech 2007, 8, E147–E154. [Google Scholar] [CrossRef] [PubMed]
- Gabal, Y.M.; Kamel, A.O.; Sammour, O.A.; Elshafeey, A.H. Effect of surface charge on the brain delivery of nanostructured lipid carriers in situ gels via the nasal route. Int. J. Pharm. 2014, 473, 442–457. [Google Scholar] [CrossRef] [PubMed]
- Qiao, M.; Chen, D.; Ma, X.; Liu, Y. Injectable biodegradable temperature-responsive PLGA–PEG–PLGA copolymers: Synthesis and effect of copolymer composition on the drug release from the copolymer-based hydrogels. Int. J. Pharm. 2005, 294, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Shit, S.C.; Maiti, S. Application of NMR spectroscopy in molecular weight determination of polymers. Eur. Polym. J. 1986, 22, 1001–1008. [Google Scholar] [CrossRef]
- Perinelli, D.; Bonacucina, G.; Cespi, M.; Naylor, A.; Whitaker, M.; Palmieri, G.; Giorgioni, G.; Casettari, L. Evaluation of P (L) LA-PEG-P (L) LA as processing aid for biodegradable particles from gas saturated solutions (PGSS) process. Int. J. Pharm. 2014, 468, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Kwon, G.S.; Cho, H. Thermogel Formulation for Combination Drug Delivery. Patents US20150025106, 22 July 2014. [Google Scholar]
- Chen, Y.; Li, Y.; Shen, W.; Li, K.; Yu, L.; Chen, Q.; Ding, J. Controlled release of liraglutide using thermogelling polymers in treatment of diabetes. Sci. Rep. 2016, 6, 31593. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Zhang, Z.; Ding, J. Influence of LA and GA sequence in the PLGA block on the properties of thermogelling PLGA-PEG-PLGA block copolymers. Biomacromolecules 2011, 12, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ci, T.; Yu, L.; Ding, J. Effects of molecular weight and its distribution of peg block on micellization and thermogellability of plga–peg–plga copolymer aqueous solutions. Macromolecules 2015, 48, 3662–3671. [Google Scholar] [CrossRef]
- Jeong, B.; Kim, S.W.; Bae, Y.H. Thermosensitive sol–gel reversible hydrogels. Adv. Drug Deliv. Rev. 2012, 64, 154–162. [Google Scholar] [CrossRef]
- Guo, X.; Cui, F.; Xing, Y.; Mei, Q.; Zhang, Z. Investigation of a new injectable thermosensitive hydrogel loading solid lipid nanoparticles. Die Pharmazie 2011, 66, 948–952. [Google Scholar] [PubMed]
- Yang, Y.; Wang, J.; Zhang, X.; Lu, W.; Zhang, Q. A novel mixed micelle gel with thermo-sensitive property for the local delivery of docetaxel. J. Control. Release 2009, 135, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Gwaltney, J.J.M.; Hendley, J.O.; Phillips, C.D.; Bass, C.R.; Mygind, N.; Winther, B. Nose blowing propels nasal fluid into the paranasal sinuses. Clin. Infect. Dis. 2000, 30, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Clarot, T.; Hensley, C. System for Delivering a Composition to the Nasal Membrane and Method of Using Same. U.S. Patent US20060275343A1, 7 December 2006. [Google Scholar]
- Hsu, Y.Y.; Gresser, J.D.; Stewart, R.R.; Trantolo, D.J.; Lyons, C.M.; Simons, G.A.; Gangadharam, P.R.; Wise, D.L. Mechanisms of isoniazid release from poly (d, I-lactide-co-glycolide) matrices prepared by dry-mixing and low density polymeric foam methods. J. Pharm. Sci. 1996, 85, 706–713. [Google Scholar] [CrossRef] [PubMed]
- Athanasiou, K.A.; Niederauer, G.G.; Agrawal, C.M. Sterilization, toxicity, biocompatibility and clinical applications of polylactic acid/polyglycolic acid copolymers. Biomaterials 1996, 17, 93–102. [Google Scholar] [CrossRef]
- Sawant, D.; Dandagi, P.M.; Gadad, A.P. Formulation and evaluation of sparfloxacin emulsomes-loaded thermosensitive in situ gel for ophthalmic delivery. J. Sol-Gel Sci. Technol. 2016, 77, 654–665. [Google Scholar] [CrossRef]
- Khodaverdi, E.; Tekie, F.S.M.; Mohajeri, S.A.; Ganji, F.; Zohuri, G.; Hadizadeh, F. Preparation and investigation of sustained drug delivery systems using an injectable, thermosensitive, in situ forming hydrogel composed of PLGA–PEG–PLGA. AAPS PharmSciTech 2012, 13, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.S.; Woolfson, A.D.; Djokic, J.; Coulter, W. Development and mechanical characterization of bioadhesive semi-solid, polymeric systems containing tetracycline for the treatment of periodontal diseases. Pharm. Res. 1996, 13, 1734–1738. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.S.; Woolfson, A.D.; Brown, A.F.; Coulter, W.A.; McClelland, C.; Irwin, C.R. Design, characterisation and preliminary clinical evaluation of a novel mucoadhesive topical formulation containing tetracycline for the treatment of periodontal disease. J. Control. Release 2000, 67, 357–368. [Google Scholar] [CrossRef]
- Eouani, C.; Piccerelle, P.; Prinderre, P.; Bourret, E.; Joachim, J. In-vitro comparative study of buccal mucoadhesive performance of different polymeric films. Eur. J. Pharm. Biopharm. 2001, 52, 45–55. [Google Scholar] [CrossRef]
- Yu, T.; Wang, Y.-Y.; Yang, M.; Schneider, C.; Zhong, W.; Pulicare, S.; Choi, W.-J.; Mert, O.; Fu, J.; Lai, S.K. Biodegradable mucus-penetrating nanoparticles composed of diblock copolymers of polyethylene glycol and poly (lactic-co-glycolic acid). Drug Deliv. Transl. Res 2012, 2, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, F.C.; Bruschi, M.L.; Evangelista, R.C.; Gremião, M.P.D. Mucoadhesive drug delivery systems. Braz. J. Pharm. Sci. 2010, 46, 1–17. [Google Scholar] [CrossRef]
- Soane, R.; Frier, M.; Perkins, A.; Jones, N.; Davis, S.; Illum, L. Evaluation of the clearance characteristics of bioadhesive systems in humans. Int. J. Pharm. 1999, 178, 55–65. [Google Scholar] [CrossRef]
- Gao, K.; Jiang, X. Influence of particle size on transport of methotrexate across blood brain barrier by polysorbate 80-coated polybutylcyanoacrylate nanoparticles. Int. J. Pharm. 2006, 310, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Chen, Z. Preparation and performance evaluation of emulsomes as a drug delivery system for silybin. Arch. Pharm. Res. 2015, 38, 2193–2200. [Google Scholar] [CrossRef] [PubMed]
- Shorvon, S. Oxcarbazepine: A review. Seizure 2000, 9, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Hellewell, J.S. Oxcarbazepine (Trileptal) in the treatment of bipolar disorders: A review of efficacy and tolerability. J. Affect. Disord. 2002, 72, S23–S34. [Google Scholar] [CrossRef]
- Martinez, W.; Ingenito, A.; Blakeslee, M.; Barkley, G.L.; McCague, K.; D’Souza, J. Efficacy, safety, and tolerability of oxcarbazepine monotherapy. Epilepsy Behav. 2006, 9, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Kaur, I.P.; Bhandari, R.; Bhandari, S.; Kakkar, V. Potential of solid lipid nanoparticles in brain targeting. J. Control. Release 2008, 127, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Xie, L.; Childs, J.; Sun, Y.; Mao, X.O.; Logvinova, A.; Greenberg, D.A. Cerebral neurogenesis is induced by intranasal administration of growth factors. Ann. Neurol. 2003, 53, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Misra, A.; Mishra, A.; Mishra, P.; Pathak, K. Mucoadhesive nanoemulsion-based intranasal drug delivery system of olanzapine for brain targeting. J. Drug Target. 2008, 16, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Akhter, S.; Jain, G.K.; Khan, Z.I.; Khar, R.K.; Ahmad, F.J. Antiepileptic intranasal Amiloride loaded mucoadhesive nanoemulsion: Development and safety assessment. J. Biomed. Nanotechnol. 2011, 7, 142–143. [Google Scholar] [CrossRef] [PubMed]
- Perucca, E. Extended-release formulations of antiepileptic drugs: Rationale and comparative value. Epilepsy Curr. 2009, 9, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Steinhoff, B.; Stefan, H.; Schulze-Bonhage, A.; Hueber, R.; Paulus, W.; Wangemann, M.; Elger, C. Retardiertes vs. schnell freisetzendes Oxcarbazepin bei therapierefraktärer fokaler Epilepsie. Der Nervenarzt 2012, 83, 1292–1299. [Google Scholar] [CrossRef] [PubMed]
- Moffatt, S.; Cristiano, R.J. Uptake characteristics of NGR-coupled stealth PEI/pDNA nanoparticles loaded with PLGA-PEG-PLGA tri-block copolymer for targeted delivery to human monocyte-derived dendritic cells. Int. J. Pharm. 2006, 321, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-W.; Choi, D.; Kim, W.J.; Yockman, J.W.; Christensen, L.V.; Kim, Y.-H.; Kim, S.W. Non-ionic amphiphilic biodegradable PEG–PLGA–PEG copolymer enhances gene delivery efficiency in rat skeletal muscle. J. Control. Release 2007, 118, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Chang, G.; Li, C.; Lu, W.; Ding, J. N-Boc-Histidine-Capped PLGA-PEG-PLGA as a Smart Polymer for Drug Delivery Sensitive to Tumor Extracellular pH. Macromol. Biosci. 2010, 10, 1248–1256. [Google Scholar] [CrossRef] [PubMed]
- Castrén, K.; Pienimäki, P.; Arvela, P.; Vähäkangas, K. Metabolites and DNA-binding of carbamazepine and oxcarbazepine in vitro by rat liver microsomes. Hum. Exp. Toxicol. 1996, 15, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Aktaş, Z.; Cansu, A.; Erdoğan, D.; Take, G.; Goktas, G.; Ozdek, S.; Serdaroglu, A. Retinal ganglion cell toxicity due to oxcarbazepine and valproic acid treatment in rat. Seizure 2009, 18, 396–399. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Emulsome Concentration % v/v | Gelation Temperature (°C ± SD) | |||
|---|---|---|---|---|
| PLGA-PEG-PLGA Copolymer (% w/w) | ||||
| 5% | 10% | 20% | 30% | |
| 0% | G1 No gelation | G2 Opaque solution | G3 30.2 ± 0.05 °C | G4 28.5 ± 0.1 °C |
| 10% | G5 No gelation | G6 Opaque solution | G7 31.5 ± 0.1 °C * | G8 29.0 ± 0.06 °C * |
| 25% | G9 No gelation | G10 Opaque solution | G11 32.0 ± 0.06 °C * | G12 30.1 ± 0.05 °C * |
| 50% | G13 No gelation | G14 Opaque solution | G15 33.4 ± 0.05 °C * | G16 31.0 ± 0.0 °C * |
| Parameter | Plain G4 Thermogel (Mean ± SD) | G16 Emulsomal Thermogel (Mean ± SD) | Carbopol 980 Gel (Mean ± SD) |
|---|---|---|---|
| Peak detachment force (N) | 21.9 ± 1.7 * | 18.7 ± 2.3 * | 10.52 ± 0.1 |
| Deformation peak (mm) | 10.5 ± 2 * | 8.2 ± 1.3 | 5.4 ± 0.8 |
| Work of adhesion (mJ) | 788.3 ± 23.7 * | 746.6 ± 52.2 * | 304.8 ± 35 |
| Final load (N) | 20.2 ± 1.7 | 17.84 ± 2.9 | 15.41 ± 1.9 |
| Parameter | Plasma | Brain | ||||||
|---|---|---|---|---|---|---|---|---|
| IN Emulsomes * | IN Emulsomal Thermogel | IN OX Solutions | Trileptal® Suspension * | IN Emulsomes * | IN Emulsomal Thermogel | IN OX Solutions | Trileptal® Suspension * | |
| Cmax (ng/mL)/(ng/g) | 2514.4 | 3818.8 | 80.9 | 2567.6 | 5699.9 | 1733.2 | 249.1 | 1198.9 |
| Tmax (min) | 45 | 120 | 20 | 120 | 20 | 30 | 60 | 360 |
| AUC0–2880min (µg/mL·min)/(µg/g·min) | 1943.4 | 2757.7 | 111.2 | 1524.6 | 2445.1 | 1440.5 | 57.7 | 742.8 |
| T1/2 (min) | 1581.8 | 1919.6 | 302.1 | 364 | 250.9 | 734 | 197.4 | 285.6 |
| MRT (min) | 2024.3 | 2638.1 | 34.5 | 465.4 | 378 | 1007.8 | 95.4 | 504.5 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Zaafarany, G.M.; Soliman, M.E.; Mansour, S.; Cespi, M.; Palmieri, G.F.; Illum, L.; Casettari, L.; Awad, G.A.S. A Tailored Thermosensitive PLGA-PEG-PLGA/Emulsomes Composite for Enhanced Oxcarbazepine Brain Delivery via the Nasal Route. Pharmaceutics 2018, 10, 217. https://doi.org/10.3390/pharmaceutics10040217
El-Zaafarany GM, Soliman ME, Mansour S, Cespi M, Palmieri GF, Illum L, Casettari L, Awad GAS. A Tailored Thermosensitive PLGA-PEG-PLGA/Emulsomes Composite for Enhanced Oxcarbazepine Brain Delivery via the Nasal Route. Pharmaceutics. 2018; 10(4):217. https://doi.org/10.3390/pharmaceutics10040217
Chicago/Turabian StyleEl-Zaafarany, Ghada M., Mahmoud E. Soliman, Samar Mansour, Marco Cespi, Giovanni Filippo Palmieri, Lisbeth Illum, Luca Casettari, and Gehanne A. S. Awad. 2018. "A Tailored Thermosensitive PLGA-PEG-PLGA/Emulsomes Composite for Enhanced Oxcarbazepine Brain Delivery via the Nasal Route" Pharmaceutics 10, no. 4: 217. https://doi.org/10.3390/pharmaceutics10040217
APA StyleEl-Zaafarany, G. M., Soliman, M. E., Mansour, S., Cespi, M., Palmieri, G. F., Illum, L., Casettari, L., & Awad, G. A. S. (2018). A Tailored Thermosensitive PLGA-PEG-PLGA/Emulsomes Composite for Enhanced Oxcarbazepine Brain Delivery via the Nasal Route. Pharmaceutics, 10(4), 217. https://doi.org/10.3390/pharmaceutics10040217