An Evaluation of the Potential of NMR Spectroscopy and Computational Modelling Methods to Inform Biopharmaceutical Formulations



Abstract
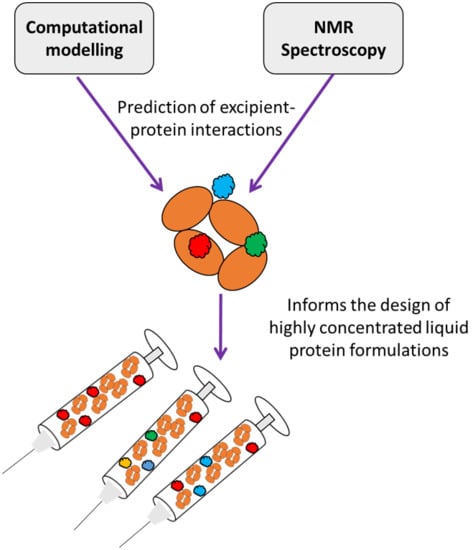
1. Introduction
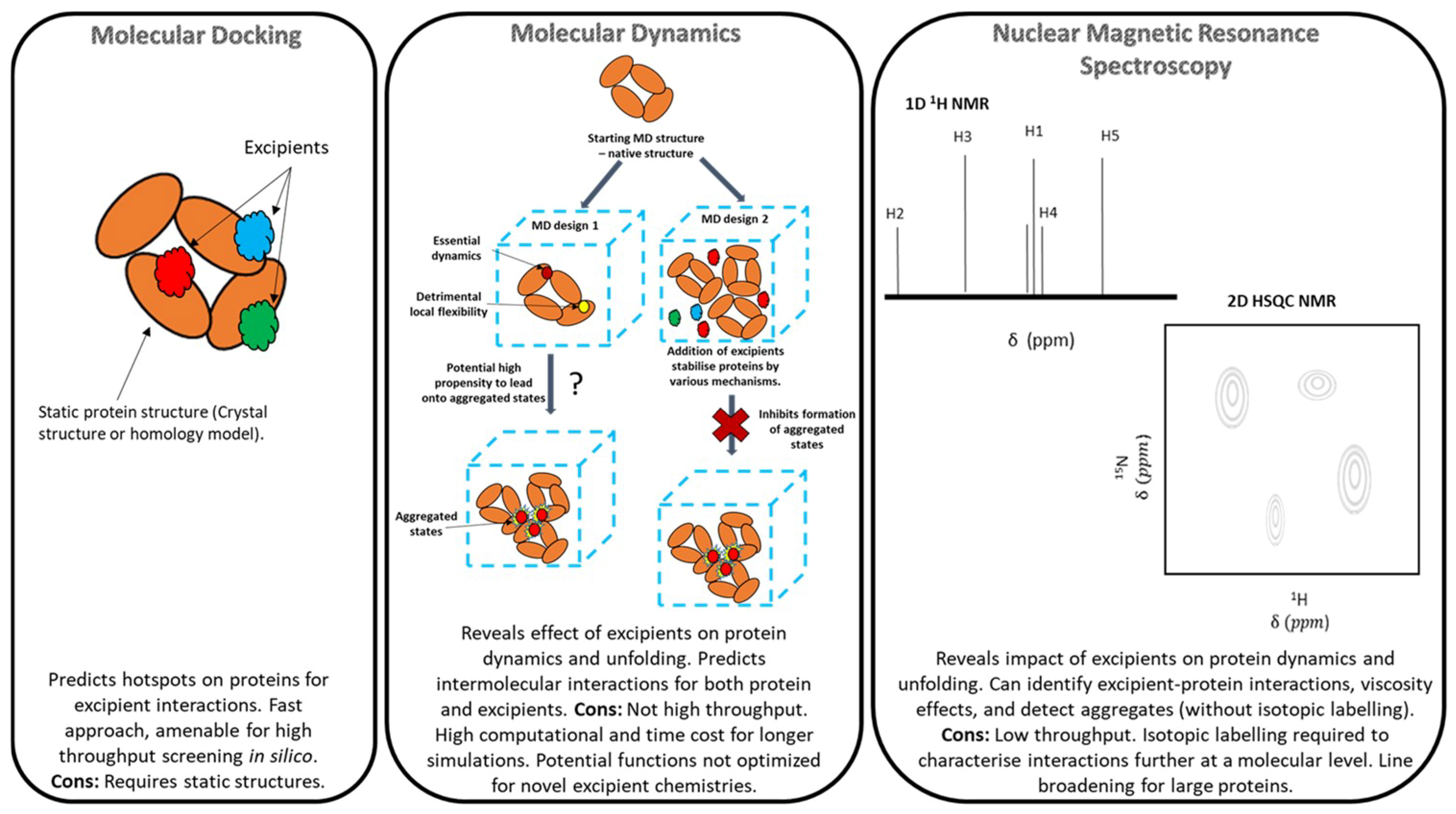
2. Overview of Molecular Modelling, Methodologies and Limitations
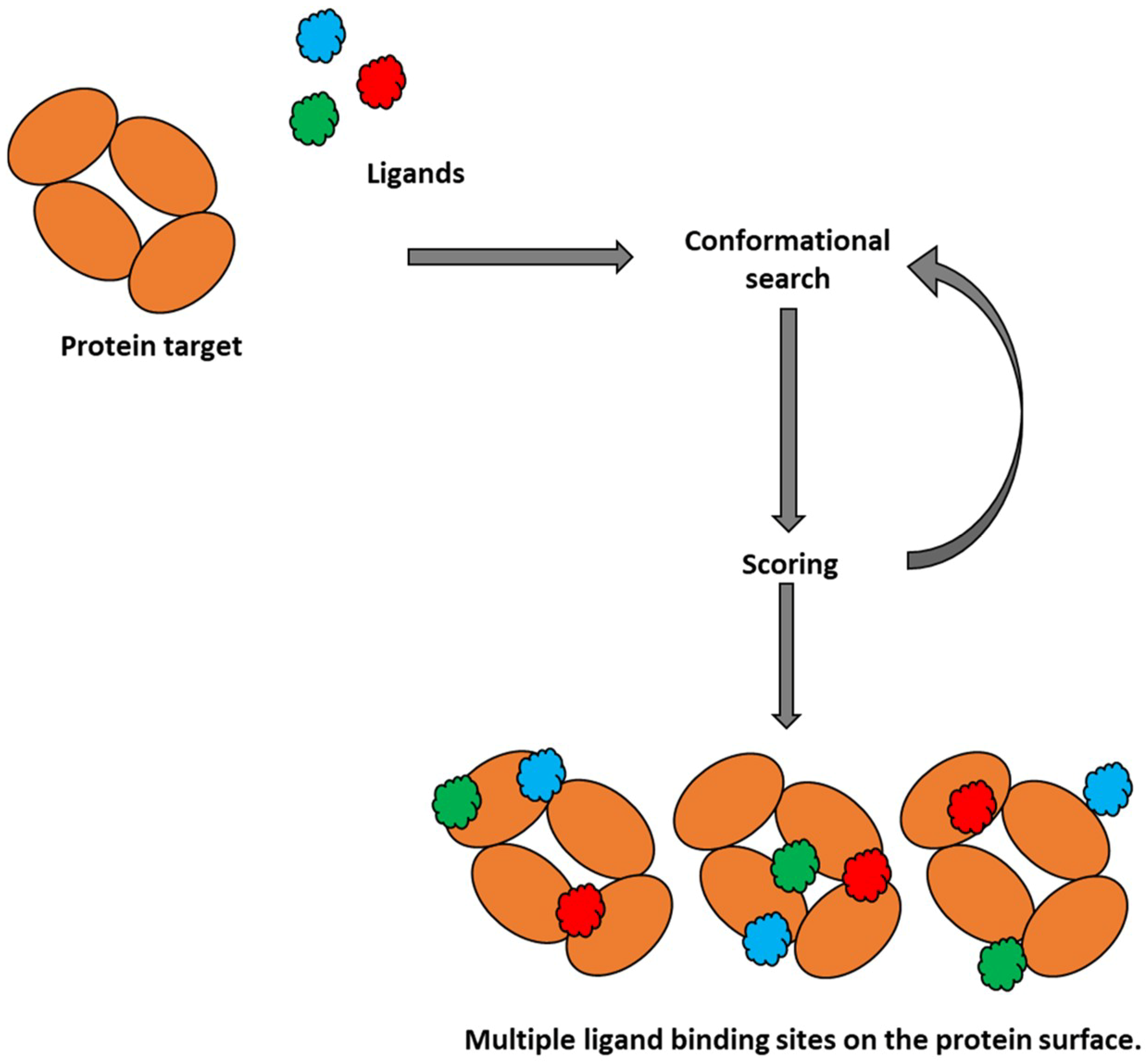
2.1. Molecular Docking
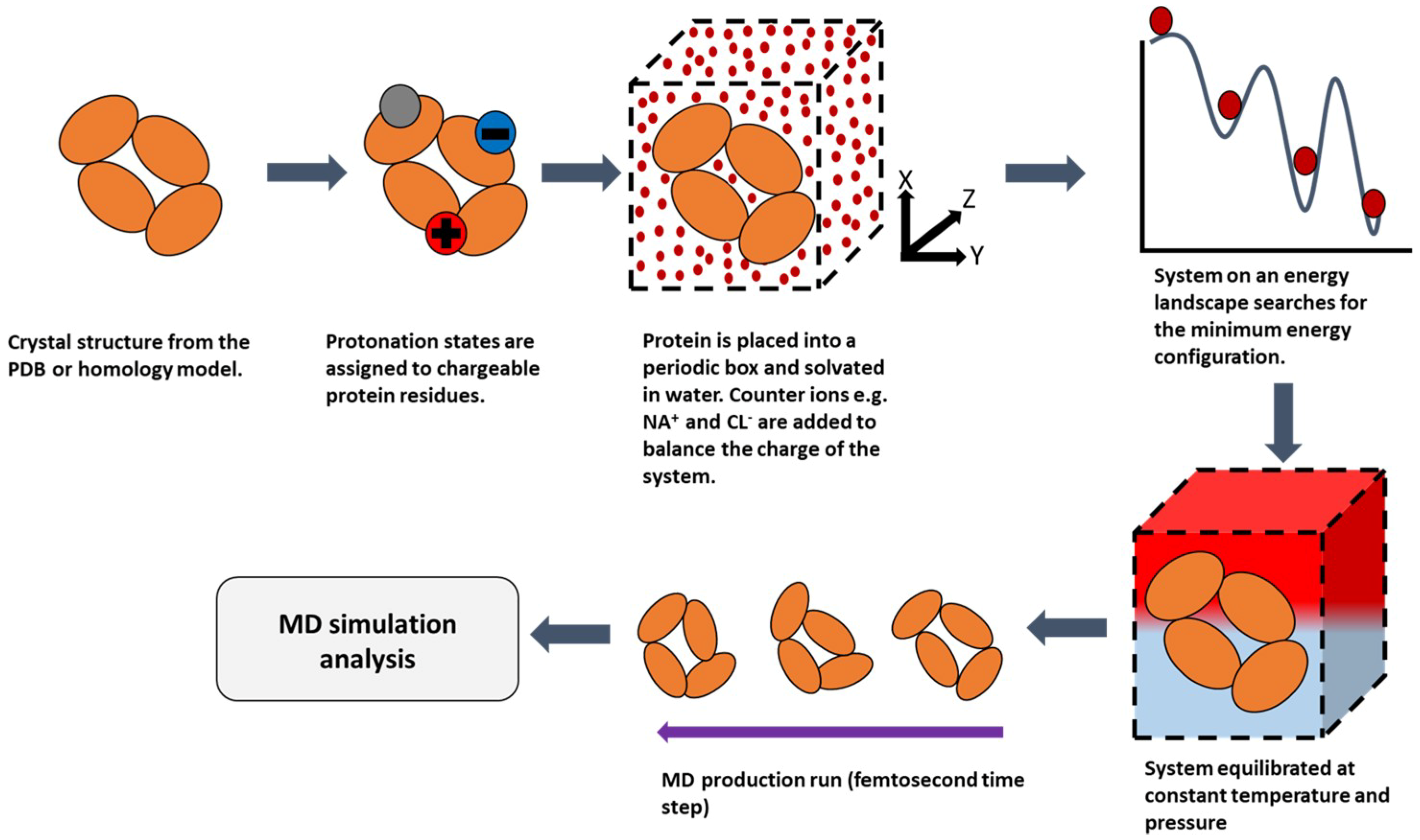
2.2. Molecular Dynamics (MD)
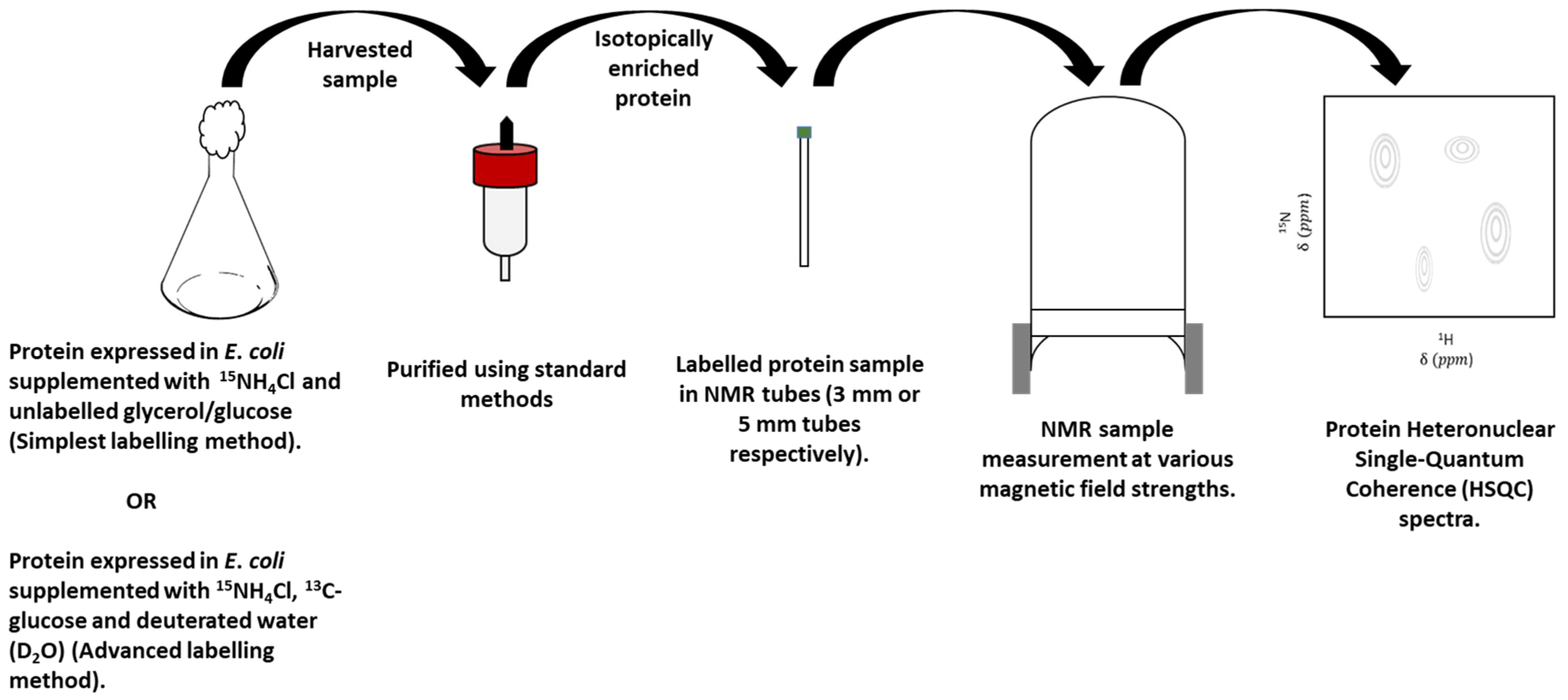
3. Overview of Nuclear Magnetic Resonance (NMR) Spectroscopy
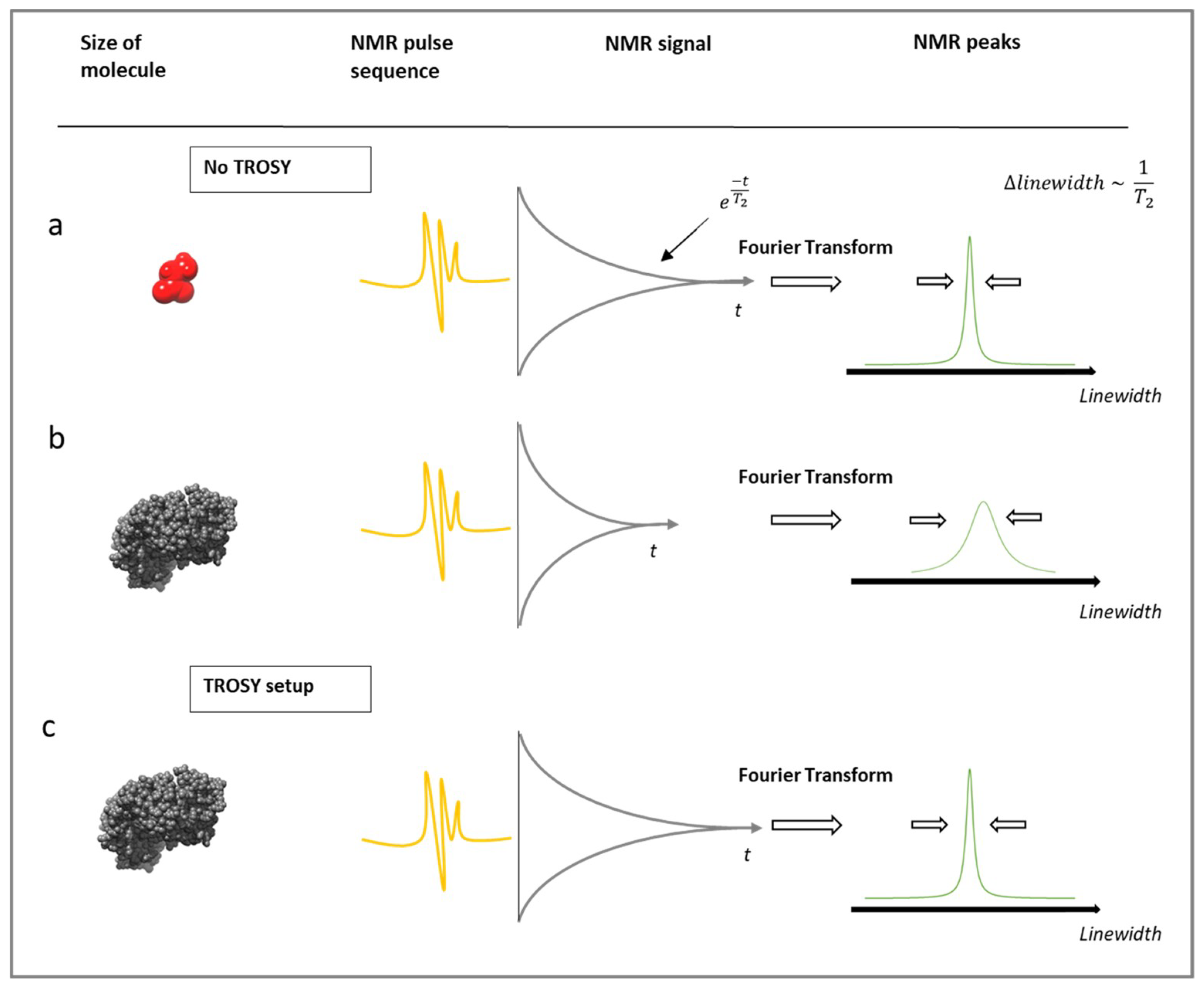
3.1. Limitations
3.2. Protein-Observe Methods
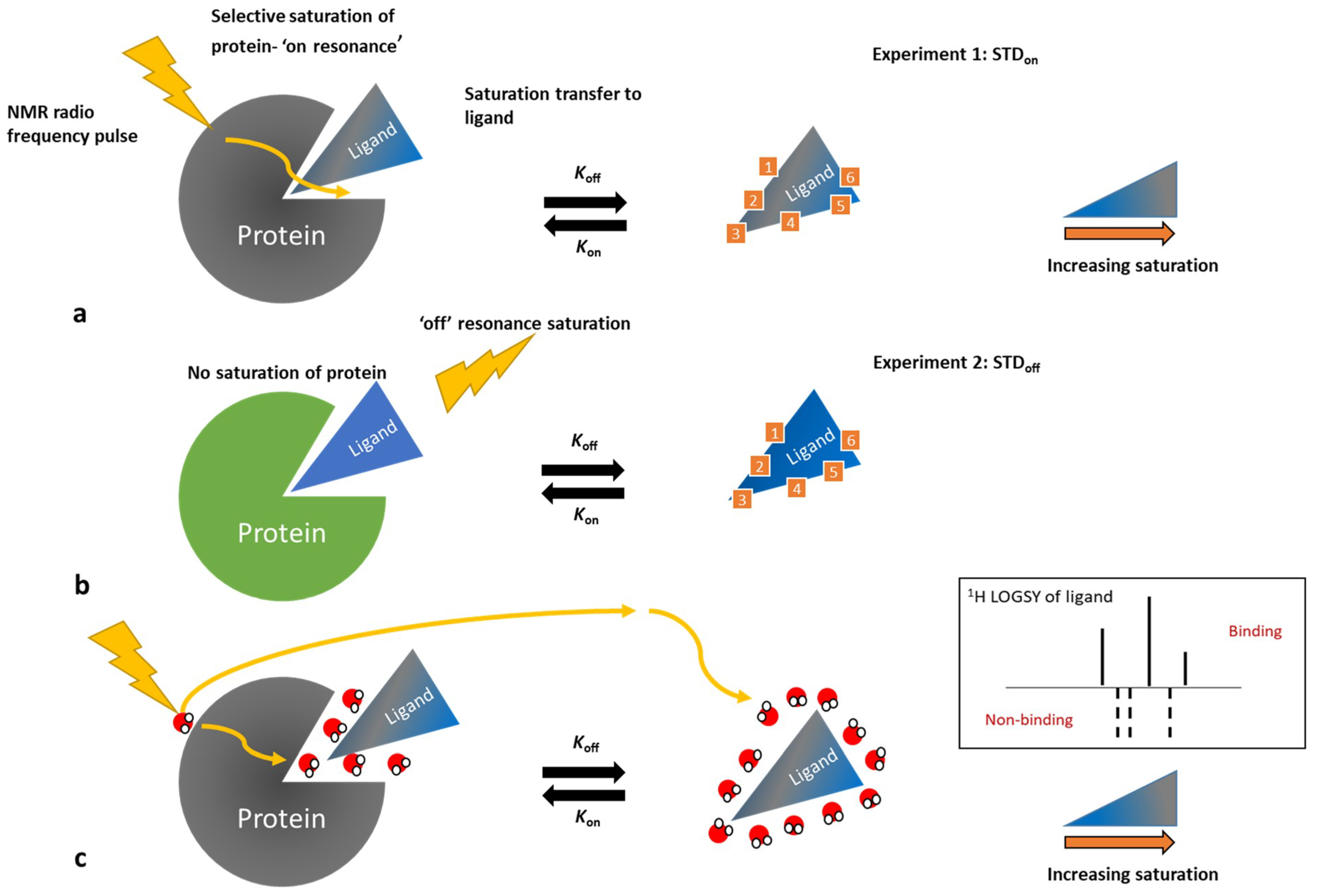
3.3. Ligand-Observe Methods
4. Nuclear Magnetic Resonance (NMR) Spectroscopy Applications in Aggregation and Formulation
5. Molecular Modelling Applications in Aggregation and Formulation Design
6. Future Perspectives
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wang, W.; Singh, S.; Zeng, D.L.; King, K.; Nema, S. Antibody structure, instability, and formulation. J. Pharm. Sci. 2007, 96, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Garidel, P.; Kuhn, A.B.; Schäfer, L.V.; Karow-Zwick, A.R.; Blech, M. High-concentration protein formulations: How high is high? Eur. J. Pharm. Biopharm. 2017, 119, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.J. Protein aggregation and its impact on product quality. Curr. Opin. Biotechnol. 2014, 30, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Smart, A.L.; Gaisford, S.; Basit, A.W. Oral peptide and protein delivery: Intestinal obstacles and commercial prospects. Expert Opin. Drug Deliv. 2014, 11, 1323–1335. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yu, M.; Fan, W.; Gan, Y.; Hovgaard, L.; Yang, M. Orally active-targeted drug delivery systems for proteins and peptides. Expert Opin. Drug Deliv. 2014, 11, 1435–1447. [Google Scholar] [CrossRef] [PubMed]
- Bye, J.W.; Platts, L.; Falconer, R.J. Biopharmaceutical liquid formulation: A review of the science of protein stability and solubility in aqueous environments. Biotechnol. Lett. 2014, 36, 869–875. [Google Scholar] [CrossRef] [PubMed]
- Wang, W. Advanced protein formulations. Protein Sci. 2015, 24, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Cromwell, M.E.M.; Hilario, E.; Jacobson, F. Protein aggregation and bioprocessing. AAPS J. 2006, 8, E572–E579. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Rey, M.; Lang, D.A. Aggregates in monoclonal antibody manufacturing processes. Biotechnol. Bioeng. 2011, 108, 1494–1508. [Google Scholar] [CrossRef] [PubMed]
- Philo, J.S.; Arakawa, T. Mechanisms of protein aggregation. Curr. Pharm. Biotechnol. 2009, 10, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Nema, S.; Teagarden, D. Protein aggregation-Pathways and influencing factors. Int. J. Pharm. 2010, 390, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Menzen, T.; Friess, W. Temperature-ramped studies on the aggregation, unfolding, and interaction of a therapeutic monoclonal antibody. J. Pharm. Sci. 2014, 103, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Maddux, N.R.; Iyer, V.; Cheng, W.; Youssef, A.M.K.; Joshi, S.B.; Volkin, D.B.; Ralston, J.P.; Winter, G.; Russell Middaugh, C. High throughput prediction of the long-term stability of pharmaceutical macromolecules from short-term multi-instrument spectroscopic data. J. Pharm. Sci. 2014, 103, 828–839. [Google Scholar] [CrossRef] [PubMed]
- Brader, M.L.; Estey, T.; Bai, S.; Alston, R.W.; Lucas, K.K.; Lantz, S.; Landsman, P.; Maloney, K.M. Examination of thermal unfolding and aggregation profiles of a series of developable therapeutic monoclonal antibodies. Mol. Pharm. 2015, 12, 1005–1017. [Google Scholar] [CrossRef] [PubMed]
- Chakroun, N.; Hilton, D.; Ahmad, S.S.; Platt, G.W.; Dalby, P.A. Mapping the Aggregation Kinetics of a Therapeutic Antibody Fragment. Mol. Pharm. 2016, 13, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.J.; Matejtschuk, P.; Bristow, A.F.; Dalby, P.A. Tm-Values and Unfolded Fraction Can Predict Aggregation Rates for Granulocyte Colony Stimulating Factor Variant Formulations but Not under Predominantly Native Conditions. Mol. Pharm. 2018, 15, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Samad, M.; Yu, H.; Chakroun, N.; Hilton, D.; Dalby, P.A. Computational-design to reduce conformational flexibility and aggregation rates of an antibody Fab fragment. Mol. Pharm. 2018, 15, 3079–3092. [Google Scholar] [CrossRef] [PubMed]
- Kamerzell, T.J.; Esfandiary, R.; Joshi, S.B.; Middaugh, C.R.; Volkin, D.B. Protein-excipient interactions: Mechanisms and biophysical characterization applied to protein formulation development. Adv. Drug Deliv. Rev. 2011, 63, 1118–1159. [Google Scholar] [CrossRef] [PubMed]
- Timasheff, S.N. Protein-solvent preferential interactions, protein hydration, and the modulation of biochemical reactions by solvent components. Proc. Natl. Acad. Sci. USA 2002, 99, 9721–9726. [Google Scholar] [CrossRef] [PubMed]
- Shukla, D.; Schneider, C.P.; Trout, B.L. Molecular level insight into intra-solvent interaction effects on protein stability and aggregation. Adv. Drug Deliv. Rev. 2011, 63, 1074–1085. [Google Scholar] [CrossRef] [PubMed]
- Fox, S.; Farr-Jones, S.; Sopchak, L.; Boggs, A.; Nicely, H.W.; Khoury, R.; Biros, M. High-throughput screening: Update on practices and success. J. Biomol. Screen. 2006, 11, 864–869. [Google Scholar] [CrossRef] [PubMed]
- Macarron, R.; Banks, M.N.; Bojanic, D.; Burns, D.J.; Cirovic, D.A.; Garyantes, T.; Green, D.V.S.; Hertzberg, R.P.; Janzen, W.P.; Paslay, J.W.; et al. Impact of high-throughput screening. Nature 2011, 10, 188–195. [Google Scholar] [CrossRef]
- Mayr, L.M.; Bojanic, D. Novel trends in high-throughput screening. Curr. Opin. Pharmacol. 2009, 9, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Bhambhani, A.; Kissmann, J.M.; Joshi, S.B.; Volkin, D.B.; Kashi, R.S.; Middaugh, C.R. Formulation design and high-throughput excipient selection based on structural integrity and conformational stability of dilute and highly concentrated IgG1 monoclonal antibody solutions. J. Pharm. Sci. 2012, 101, 1120–1135. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.Q. Raman spectroscopy of protein pharmaceuticals. J. Pharm. Sci. 2007, 96, 2861–2878. [Google Scholar] [CrossRef] [PubMed]
- Woody, R.W. On the analysis of membrane protein circular dichroism spectra. Protein Sci. 2004, 13, 100–112. [Google Scholar] [CrossRef]
- Joshi, S.B.; Kamerzell, T.J.; McNown, C.; Middaugh, C.R. The interaction of heparin/polyanions with bovine, porcine, and human growth hormone. J. Pharm. Sci. 2008, 97, 1368–1385. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Hogan, S.; Latypov, R.F.; Narhi, L.O.; Razinkov, V.I. High throughput thermostability screening of monoclonal antibody formulations. J. Pharm. Sci. 2010, 99, 1707–1720. [Google Scholar] [CrossRef] [PubMed]
- Kamerzell, T.J.; Ramsey, J.D.; Middaugh, C.R. Immunoglobulin dynamics, conformational fluctuations, and nonlinear elasticity and their effects on stability. J. Phys. Chem. B 2008, 112, 3240–3250. [Google Scholar] [CrossRef] [PubMed]
- Harn, N.; Allan, C.; Oliver, C.; Middaugh, C.R. Highly concentrated monoclonal antibody solutions: Direct analysis of physical structure and thermal stability. J. Pharm. Sci. 2007, 96, 532–546. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, D.S.; Bishop, S.M.; Shah, A.U.; Sathish, H.A. Formulation development of therapeutic monoclonal antibodies using high-throughput fluorescence and static light scattering techniques: Role of conformational and colloidal stability. J. Pharm. Sci. 2011, 100, 1306–1315. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Phan, D.H.; Hogan, S.; Bailey, R.; Becker, G.W.; Narhi, L.O.; Razinkov, V.I. Detection of IgG Aggregation by a High Throughput Method Based on Extrinsic Fluorescence. J. Pharm. Sci. 2010, 99, 2598–2608. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Mach, H.; Blue, J.T. High Throughput Formulation Screening for Global Aggregation Behaviors of Three Monoclonal Antibodies. J. Pharm. Sci. 2011, 100, 2120–2135. [Google Scholar] [CrossRef] [PubMed]
- Espargaró, A.; Castillo, V.; de Groot, N.S.; Ventura, S. The in Vivo and in Vitro Aggregation Properties of Globular Proteins Correlate with Their Conformational Stability: The SH3 Case. J. Mol. Biol. 2008, 378, 1116–1131. [Google Scholar] [CrossRef] [PubMed]
- Chi, E.; Krishnan, S.; Kendrick, B. Roles of conformational stability and colloidal stability in the aggregation of recombinant human granulocyte colony-stimulating factor. Protein Sci. 2003, 12, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Amyloid formation by globular proteins under native conditions. Nat. Chem. Biol. 2009, 5, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, J.A.; O’Brien, C.J.; Tiller, K.; Tamargo, E.; Robinson, A.S.; Roberts, C.J.; Fernandez, E.J. Conformational stability as a design target to control protein aggregation. Protein Eng. Des. Sel. 2014, 27, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Neudecker, P.; Robustelli, P.; Cavalli, A.; Walsh, P.; Lundström, P.; Zarrine-Afsar, A.; Sharpe, S.; Vendruscolo, M.; Kay, L.E. Structure of an Intermediate State in Protein Folding and Aggregation. Science 2012, 336, 362. [Google Scholar] [CrossRef] [PubMed]
- Kontoyianni, M. Docking and Virtual Screening in Drug Discovery. In Proteomics for Drug Discovery: Methods and Protocols; Lazar, I.M., Kontoyianni, M., Lazar, A.C., Eds.; Springer: New York, NY, USA, 2017; pp. 255–266. ISBN 978-1-4939-7201-2. [Google Scholar]
- Novikov, F.N.; Chilov, G.G. Molecular docking: Theoretical background, practical applications and perspectives. Mendeleev Commun. 2009, 19, 237–242. [Google Scholar] [CrossRef]
- Huang, N.; Kalyanaraman, C.; Irwin, J.J.; Jacobson, M.P. Physics-based scoring of protein—Ligand complexes: Enrichment of known inhibitors in large-scale virtual screening. J. Chem. Inf. Model. 2006, 46, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Lu, Y.; Wang, S. Comparative evaluation of 11 scoring functions for molecular docking. J. Med. Chem. 2003, 46, 2287–2303. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-Y.; Grinter, S.Z.; Zou, X. Scoring functions and their evaluation methods for protein–ligand docking: Recent advances and future directions. Phys. Chem. Chem. Phys. 2010, 12, 12899. [Google Scholar] [CrossRef] [PubMed]
- Sousa, S.F.; Ribeiro, A.J.M.; Coimbra, J.T.S.; Neves, R.P.P.; Martins, S.A.; Moorthy, N.S.H.N.; Fernandes, P.A.; Ramos, M.J. Protein-Ligand Docking in the New Millennium—A Retrospective of 10 Years in the Field. Curr. Med. Chem. 2013, 20, 2296–2314. [Google Scholar] [CrossRef] [PubMed]
- Macindoe, G.; Mavridis, L.; Venkatraman, V.; Devignes, M.D.; Ritchie, D.W. HexServer: An FFT-based protein docking server powered by graphics processors. Nucleic Acids Res. 2010, 38, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Tovchigrechko, A.; Vakser, I.A. GRAMM-X public web server for protein-protein docking. Nucleic Acids Res. 2006, 34, 310–314. [Google Scholar] [CrossRef] [PubMed]
- Lyskov, S.; Gray, J.J. The RosettaDock server for local protein-protein docking. Nucleic Acids Res. 2008, 36, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.; Huey, R. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C. Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J. Mol. Biol. 1995, 245, 43–53. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.M.; Chen, C.C. GEMDOCK: A Generic Evolutionary Method for Molecular Docking. Proteins Struct. Funct. Genet. 2004, 55, 288–304. [Google Scholar] [CrossRef] [PubMed]
- B-Rao, C.; Subramanian, J.; Sharma, S.D. Managing protein flexibility in docking and its applications. Drug Discov. Today 2009, 14, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A. NIH Public Access. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A Fast Flexible Docking Method using an Incremental Construction Algorithm. J. Mol. Biol. 1996, 261, 470–489. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Linares, I.; Pérez-Sánchez, H.; Cecilia, J.M.; García, J.M. High-Throughput parallel blind Virtual Screening using BINDSURF. BMC Bioinform. 2012, 13. [Google Scholar] [CrossRef] [PubMed]
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef] [PubMed]
- De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem. 2016, 59, 4035–4061. [Google Scholar] [CrossRef] [PubMed]
- González, M.A. Force fields and molecular dynamics simulations. Collect. SFN 2011, 12, 169–200. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef] [PubMed]
- MacKerell, A.D.; Banavali, N.; Foloppe, N. Development and current status of the CHARMM force field for nucleic acids. Biopolymers 2000, 56, 257–265. [Google Scholar] [CrossRef]
- Weiner, P.K.; Kollman, P.A. AMBER: Assisted model building with energy refinement. A general program for modeling molecules and their interactions. J. Comput. Chem. 1981, 2, 287–303. [Google Scholar] [CrossRef]
- Scott, W.R.P.; Hünenberger, P.H.; Tironi, I.G.; Mark, A.E.; Billeter, S.R.; Fennen, J.; Torda, A.E.; Huber, T.; Krüger, P.; Van Gunsteren, W.F. The GROMOS biomolecular simulation program package. J. Phys. Chem. A 1999, 103, 3596–3607. [Google Scholar] [CrossRef]
- Lindahl, E.; Hess, B. GROMACS 3.0: A package for molecular simulation and trajectory analysis. Mol. Model. Annu. 2001, 306–317. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.; Brooks, C.; MacKerell, A.; Nilsson, L.; Petrella, R.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The Biomolecular Simulation Program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Klepeis, J.L.; Lindorff-Larsen, K.; Dror, R.O.; Shaw, D.E. Long-timescale molecular dynamics simulations of protein structure and function. Curr. Opin. Struct. Biol. 2009, 19, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Lane, T.J.; Shukla, D.; Beauchamp, K.A.; Pande, V.S. To milliseconds and beyond: Challenges in the simulation of protein folding. Curr. Opin. Struct. Biol. 2013, 23, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Dror, R.O.; Jensen, M.Ø.; Borhani, D.W.; Shaw, D.E. Exploring atomic resolution physiology on a femtosecond to millisecond timescale using molecular dynamics simulations. J. Gen. Physiol. 2010, 135, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S. Characterization of biopharmaceuticals by NMR spectroscopy. TrAC Trends Anal. Chem. 2013, 48, 96–111. [Google Scholar] [CrossRef]
- Osawa, M.; Takeuchi, K.; Ueda, T.; Nishida, N.; Shimada, I. Functional dynamics of proteins revealed by solution NMR. Curr. Opin. Struct. Biol. 2012, 22, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Tang, C.; Liu, M. Protein dynamics elucidated by NMR technique. Protein Cell 2013, 4, 726–730. [Google Scholar] [CrossRef] [PubMed]
- Saitô, H.; Ando, I.; Ramamoorthy, A. Chemical shift tensor—The heart of NMR: Insights into biological aspects of proteins. Prog. Nucl. Magn. Reson. Spectrosc. 2010, 57, 181–228. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S. Interpreting protein chemical shift data. Prog. Nucl. Magn. Reson. Spectrosc. 2011, 58, 62–87. [Google Scholar] [CrossRef] [PubMed]
- Neudecker, P.; Lundström, P.; Kay, L.E. Relaxation dispersion NMR spectroscopy as a tool for detailed studies of protein folding. Biophys. J. 2009, 96, 2045–2054. [Google Scholar] [CrossRef] [PubMed]
- Farber, P.J.; Mittermaier, A. Relaxation dispersion NMR spectroscopy for the study of protein allostery. Biophys. Rev. 2015, 7, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Ban, D.; Smith, C.A.; de Groot, B.L.; Griesinger, C.; Lee, D. Recent advances in measuring the kinetics of biomolecules by NMR relaxation dispersion spectroscopy. Arch. Biochem. Biophys. 2017, 628, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Sapienza, P.; Lee, A. Using NMR to study fast dynamics in proteins: Methods and applications. Curr. Opin. Pharmacol. 2010, 10, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Charlier, C.; Cousin, S.F.; Ferrage, F. Protein dynamics from nuclear magnetic relaxation. Chem. Soc. Rev. 2016, 45, 2410–2422. [Google Scholar] [CrossRef] [PubMed]
- Gossert, A.D.; Jahnke, W. NMR in drug discovery: A practical guide to identification and validation of ligands interacting with biological macromolecules. Prog. Nucl. Magn. Reson. Spectrosc. 2016, 97, 82–125. [Google Scholar] [CrossRef] [PubMed]
- Becker, W.; Bhattiprolu, K.C.; Gubensäk, N.; Zangger, K. Investigating protein-ligand interactions by solution NMR spectroscopy. ChemPhysChem 2018, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Lee, D.H.; Jeong, K.J. Enhanced production of human full-length immunoglobulin G1 in the periplasm of Escherichia coli. Appl. Microbiol. Biotechnol. 2014, 98, 1237–1246. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, L.P.; Dahlquist, F.W. Biosynthetic Incorporation of 15N and 13C for Assignment and Interpretation of Nuclear Magnetic Resonance Spectra of Proteins. Q. Rev. Biophys. 1990, 23, 1–38. [Google Scholar] [CrossRef] [PubMed]
- Williamson, M.P. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 73, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Lemaster, D.M. Uniform and selective deuteration in two-dimensional NMR of proteins. Annu. Rev. Biophys. Biophys. Chem. 1990, 19, 243–266. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, R.; Kay, L.E. Bringing Dynamic Molecular Machines into Focus by Methyl-TROSY NMR. Annu. Rev. Biochem. 2014, 83, 291–315. [Google Scholar] [CrossRef] [PubMed]
- Tugarinov, V.; Kay, L.E. Methyl groups as probes of structure and dynamics in NMR studies of high-molecular-weight proteins. ChemBioChem 2005, 6, 1567–1577. [Google Scholar] [CrossRef] [PubMed]
- Tugarinov, V.; Kanelis, V.; Kay, L.E. Isotope labeling strategies for the study of high- molecular-weight proteins by solution NMR spectroscopy. Nat. Protoc. 2006, 1, 749–754. [Google Scholar] [CrossRef] [PubMed]
- Ayala, I.; Sounier, R.; Usé, N.; Gans, P.; Boisbouvier, J. An efficient protocol for the complete incorporation of methyl-protonated alanine in perdeuterated protein. J. Biomol. NMR 2009, 43, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Ruschak, A.M.; Velyvis, A.; Kay, L.E. A simple strategy for 13C,1H labeling at the Ile-γ2 methyl position in highly deuterated proteins. J. Biomol. NMR 2010, 48, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Ayala, I.; Hamelin, O.; Amero, C.; Pessey, O.; Plevin, M.J. An optimized isotopic labelling strategy of isoleucine-γ2 methyl groups for solution NMR studies of high molecular weight proteins. Chem. Commun. 2012, 1434–1436. [Google Scholar] [CrossRef] [PubMed]
- Lichtenecker, R.J.; Coudevylle, N.; Konrat, R.; Schmid, W. Selective Isotope Labelling of Leucine Residues by Using α-Ketoacid Precursor Compounds. ChemBioChem 2013, 14, 818–821. [Google Scholar] [CrossRef] [PubMed]
- Pervushin, K.; Riek, R.; Wider, G.; Wuthrich, K. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc. Natl. Acad. Sci. USA 1997, 94, 12366–12371. [Google Scholar] [CrossRef] [PubMed]
- Pervushin, K.V.; Wider, G.; Riek, R.; Wüthrich, K. The 3D NOESY-[(1)H,(15)N,(1)H]-ZQ-TROSY NMR experiment with diagonal peak suppression. Proc. Natl. Acad. Sci. USA 1999, 96, 9607–9612. [Google Scholar] [CrossRef] [PubMed]
- Fernández, C.; Wider, G. TROSY in NMR studies of the structure and function of large biological macromolecules. Curr. Opin. Struct. Biol. 2003, 13, 570–580. [Google Scholar] [CrossRef] [PubMed]
- Arbogast, L.W.; Brinson, R.G.; Marino, J.P. Mapping Monoclonal Antibody Structure by 2D 13C NMR at Natural Abundance. Anal. Chem. 2015, 87, 3556–3561. [Google Scholar] [CrossRef] [PubMed]
- Amezcua, C.A.; Szabo, C.M. Assessment of higher order structure comparability in therapeutic proteins using nuclear magnetic resonance spectroscopy. J. Pharm. Sci. 2013, 102, 1724–1733. [Google Scholar] [CrossRef] [PubMed]
- Fávero-Retto, M.P.; Palmieri, L.C.; Souza, T.A.C.B.; Almeida, F.C.L.; Lima, L.M.T.R. Structural meta-analysis of regular human insulin in pharmaceutical formulations. Eur. J. Pharm. Biopharm. 2013, 85, 1112–1121. [Google Scholar] [CrossRef] [PubMed]
- Quinternet, M.; Starck, J.P.; Delsuc, M.A.; Kieffer, B. Heteronuclear NMR provides an accurate assessment of therapeutic insulin’s quality. J. Pharm. Biomed. Anal. 2013, 78–79, 252–254. [Google Scholar] [CrossRef] [PubMed]
- Pellecchia, M.; Sem, D.S.; Wüthrich, K. NMR in Drug Discovery. Nat. Rev. Drug Discov. 2002, 1, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Wagstaff, J.L.; Taylor, S.L.; Howard, M.J. Recent developments and applications of saturation transfer difference nuclear magnetic resonance (STD NMR) spectroscopy. Mol. BioSyst. 2013, 9, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Dalvit, C.; Pevarello, P.; Tato, M.; Veronesi, M.; Vulpetti, A.; Sundström, M. Identification of compounds with binding affinity to proteins via magnetization transfer from bulk water. J. Biomol. NMR 2000, 18, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Dalvit, C.; Fogliatto, G.P.; Stewart, A.; Veronesi, M.; Stockman, B. WaterLOGSY as a method for primary NMR screening: Practical aspects and range of applicability. J. Biomol. NMR 2001, 21, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Pagès, G.; Gilard, V.; Martino, R.; Malet-Martino, M. Pulsed-field gradient nuclear magnetic resonance measurements (PFG NMR) for diffusion ordered spectroscopy (DOSY) mapping. Analyst 2017, 142, 3771–3796. [Google Scholar] [CrossRef] [PubMed]
- Bernadó, P.; Åkerud, T.; García de la Torre, J.; Akke, M.; Pons, M. Combined use of NMR relaxation measurements and hydrodynamic calculations to study protein association. Evidence for tetramers of low molecular weight protein tyrosine phosphatase in solution. J. Am. Chem. Soc. 2003, 125, 916–923. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.-P.; Baum, J. Detection of transient interchain interactions in the intrinsically disordered protein alpha-synuclein by NMR paramagnetic relaxation enhancement. J. Am. Chem. Soc. 2010, 132, 5546–5547. [Google Scholar] [CrossRef] [PubMed]
- Dedmon, M.M.; Lindorff-Larsen, K.; Christodoulou, J.; Vendruscolo, M.; Dobson, C.M. Mapping long-range interactions in α-synuclein using spin-label NMR and ensemble molecular dynamics simulations. J. Am. Chem. Soc. 2005, 127, 476–477. [Google Scholar] [CrossRef] [PubMed]
- Fawzi, N.L.; Ying, J.; Ghirlando, R.; Torchia, D.A.; Clore, G.M. Atomic-resolution dynamics on the surface of amyloid-β protofibrils probed by solution NMR. Nature 2011, 480, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Carr, H.Y.; Purcell, E.M. Effects of diffusion on free precession in nuclear magnetic resonance experiments. Phys. Rev. 1954, 94, 630–638. [Google Scholar] [CrossRef]
- Meiboom, S.; Gill, D. Modified spin-echo method for measuring nuclear relaxation times. Rev. Sci. Instrum. 1958, 29, 688–691. [Google Scholar] [CrossRef]
- Mukherjee, S.; Pondaven, S.P.; Jaroniec, C.P. Conformational flexibility of a human immunoglobulin light chain variable domain by relaxation dispersion nuclear magnetic resonance spectroscopy: Implications for protein misfolding and amyloid assembly. Biochemistry 2011, 50, 5845–5857. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Taraban, M.B.; Yu, Y.B. Water proton NMR—A sensitive probe for solute association. Chem. Commun. 2015, 51, 6804–6807. [Google Scholar] [CrossRef] [PubMed]
- Taraban, M.B.; Truong, H.C.; Feng, Y.; Jouravleva, E.V.; Anisimov, M.A.; Yu, Y.B. Water Proton NMR for in Situ Detection of Insulin Aggregates. J. Pharm. Sci. 2015, 104, 4132–4141. [Google Scholar] [CrossRef] [PubMed]
- Taraban, M.B.; Depaz, R.A.; Lobo, B.; Yu, Y.B. Water Proton NMR: A Tool for Protein Aggregation Characterization. Anal. Chem. 2017, 89, 5494–5502. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.M.; Keire, D.A.; Chen, K. Comparison of NMR and Dynamic Light Scattering for Measuring Diffusion Coefficients of Formulated Insulin: Implications for Particle Size Distribution Measurements in Drug Products. AAPS J. 2017, 19, 1760–1766. [Google Scholar] [CrossRef] [PubMed]
- Rossini, A.J.; Widdifield, C.M.; Zagdoun, A.; Lelli, M.; Schwarzwälder, M.; Copéret, C.; Lesage, A.; Emsley, L. Dynamic nuclear polarization enhanced NMR spectroscopy for pharmaceutical formulations. J. Am. Chem. Soc. 2014, 136, 2324–2334. [Google Scholar] [CrossRef] [PubMed]
- Pisklak, D.M.; Zielińska-Pisklak, M.; Szeleszczuk, Ł.; Wawer, I. 13C cross-polarization magic-angle spinning nuclear magnetic resonance analysis of the solid drug forms with low concentration of an active ingredient-propranolol case. J. Pharm. Biomed. Anal. 2014, 93, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Maciej, D.; Zieli, M.A.; Szeleszczuk, Ł. 13C solid-state NMR analysis of the most common pharmaceutical excipients used in solid drug formulations, Part I: Chemical shifts assignment. J. Pharm. Biomed. Anal. 2016, 122, 81–89. [Google Scholar] [CrossRef]
- Pisklak, D.M.; Zielińska-Pisklak, M.; Szeleszczuk, Ł.; Wawer, I. 13C solid-state NMR analysis of the most common pharmaceutical excipients used in solid drug formulations Part II: CP kinetics and relaxation analysis. J. Pharm. Biomed. Anal. 2016, 122, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Panjwani, N.; Hodgson, D.J.; Sauvé, S.; Aubin, Y. Assessment of the effects of pH, formulation and deformulation on the conformation of interferon alpha-2 by NMR. J. Pharm. Sci. 2010, 99, 3334–3342. [Google Scholar] [CrossRef] [PubMed]
- Aubin, Y.; Gingras, G.; Sauvé, S. Assessment of the three-dimensional structure of recombinant protein therapeutics by NMR fingerprinting: Demonstration on recombinant human granulocyte macrophage-colony stimulation factor. Anal. Chem. 2008, 80, 2623–2627. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.M.; Bandi, S.; Jones, D.N.M.; Mallela, K.M.G. Effect of Polysorbate 20 and Polysorbate 80 on the Higher-Order Structure of a Monoclonal Antibody and Its Fab and Fc Fragments Probed Using 2D Nuclear Magnetic Resonance Spectroscopy. J. Pharm. Sci. 2017, 106, 3486–3498. [Google Scholar] [CrossRef] [PubMed]
- Kheddo, P.; Cliff, M.J.; Uddin, S.; Van Der Walle, C.F.; Golovanov, A.P.; Kheddo, P.; Cliff, M.J.; Uddin, S.; Van Der, C.F.; Golovanov, A.P.; et al. Characterizing monoclonal antibody formulations in arginine glutamate solutions using 1H NMR spectroscopy. Mabs 2016, 8, 1245–1258. [Google Scholar] [CrossRef] [PubMed]
- Poppe, L.; Jordan, J.B.; Lawson, K.; Jerums, M.; Apostol, I.; Schnier, P.D. Profiling Formulated Monoclonal Antibodies by 1 H NMR Spectroscopy. Anal. Chem. 2013, 85, 9623–9629. [Google Scholar] [CrossRef] [PubMed]
- Poppe, L.; Jordan, J.B.; Rogers, G.; Schnier, P.D. On the analytical superiority of 1D NMR for fingerprinting the higher order structure of protein therapeutics compared to multidimensional NMR methods. Anal. Chem. 2015, 87, 5539–5545. [Google Scholar] [CrossRef] [PubMed]
- Bednarek, E.; Sitkowski, J.; Bocian, W.; Borowicz, P.; Płucienniczak, G.; Stadnik, D.; Surmacz-Chwedoruk, W.; Jaworska, B.; Kozerski, L. Structure and pharmaceutical formulation development of a new long-acting recombinant human insulin analog studied by NMR and MS. J. Pharm. Biomed. Anal. 2017, 135, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Morin, A.; Meiler, J.; Mizoue, L.S. Computational design of protein-ligand interfaces: Potential in therapeutic development. Trends Biotechnol. 2011, 29, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Li, L.; Hsu, W.-L.; Pilcher, M.N.; Uversky, V.; Zhou, Y.; Dunker, A.K.; Meroueh, S.O. Exploring the Molecular Design of Protein Interaction Sites with Molecular Dynamics Simulations and Free Energy Calculations. Biochemistry 2009, 48, 399–414. [Google Scholar] [CrossRef] [PubMed]
- Childers, M.C.; Daggett, V. Insights from molecular dynamics simulations for computational protein design. Mol. Syst. Des. Eng. 2017. [Google Scholar] [CrossRef] [PubMed]
- Daggett, V.; Levitt, M. A model of the molten globule state from molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 1992, 89, 5142–5146. [Google Scholar] [CrossRef] [PubMed]
- Alonso, D.O.V.; DeArmond, S.J.; Cohen, F.E.; Daggett, V. Mapping the early steps in the pH-induced conformational conversion of the prion protein. Proc. Natl. Acad. Sci. USA 2001, 98, 2985–2989. [Google Scholar] [CrossRef] [PubMed]
- Alonso, D.O.V.; An, C.; Daggett, V. Simulations of biomolecules: Characterization of the early steps in the pH-induced conformational conversion of the hamster, bovine and human forms of the prion protein. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2002, 360, 1165–1178. [Google Scholar] [CrossRef] [PubMed]
- Daggett, V. α-sheet: The toxic conformer in amyloid diseases? Acc. Chem. Res. 2006, 39, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Kellock, J.; Hopping, G.; Caughey, B.; Daggett, V. Peptides Composed of Alternating L- and D-Amino Acids Inhibit Amyloidogenesis in Three Distinct Amyloid Systems Independent of Sequence. J. Mol. Biol. 2016, 428, 2317–2328. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.J.; Kumar, S.; Wang, X.; Helk, B.; Singh, S.K.; Trout, B.L. Aggregation in Protein-Based Biotherapeutics: Computational Studies and Tools to Identify Aggregation-Prone Regions. J. Pharm. Sci. 2011, 100, 5081–5095. [Google Scholar] [CrossRef] [PubMed]
- Conchillo-Solé, O.; de Groot, N.S.; Avilés, F.X.; Vendrell, J.; Daura, X.; Ventura, S. AGGRESCAN: A server for the prediction and evaluation of “hot spots” of aggregation in polypeptides. BMC Bioinform. 2007, 8. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Escamilla, A.M.; Rousseau, F.; Schymkowitz, J.; Serrano, L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat. Biotechnol. 2004, 22, 1302–1306. [Google Scholar] [CrossRef] [PubMed]
- Trovato, A.; Seno, F.; Tosatto, S.C.E. The PASTA server for protein aggregation prediction. Protein Eng. Des. Sel. 2007, 20, 521–523. [Google Scholar] [CrossRef] [PubMed]
- Chennamsetty, N.; Voynov, V.; Kayser, V.; Helk, B.; Trout, B. Design of therapeutic proteins with enhanced stability. Proc. Natl. Acad. Sci. USA 2009, 106, 11937–11942. [Google Scholar] [CrossRef] [PubMed]
- Chennamsetty, N.; Voynov, V.; Kayser, V.; Helk, B.; Trout, B.L. Prediction of aggregation prone regions of therapeutic proteins. J. Phys. Chem. B 2010, 114, 6614–6624. [Google Scholar] [CrossRef] [PubMed]
- Chennamsetty, N.; Helk, B.; Voynov, V.; Kayser, V.; Trout, B. Aggregation-Prone Motifs in Human Immunoglobulin G. J. Mol. Biol. 2009, 391, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, J.X.; Le, Y.; Chen, J.F. Studies of bicalutamide-excipients interaction by combination of molecular docking and molecular dynamics simulation. Mol. Pharm. 2013, 10, 2362–2369. [Google Scholar] [CrossRef] [PubMed]
- Jha, P.K.; Larson, R.G. Assessing the efficiency of polymeric excipients by atomistic molecular dynamics simulations. Mol. Pharm. 2014, 11, 1676–1686. [Google Scholar] [CrossRef] [PubMed]
- Shukla, D.; Trout, B. Interaction of Arginine with Proteins and the Mechanism by Which It Inhibits Aggregation. J. Phys. Chem. B 2010, 114, 13426–13438. [Google Scholar] [CrossRef] [PubMed]
- Kheddo, P.; Tracka, M.; Armer, J.; Dearman, R.J.; Uddin, S.; Van Der Walle, C.F.; Golovanov, A.P. The effect of arginine glutamate on the stability of monoclonal antibodies in solution. Int. J. Pharm. 2014, 473, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Kheddo, P.; Golovanov, A.P.; Mellody, K.T.; Uddin, S.; Van Der Walle, C.F.; Dearman, R.J. The effects of arginine glutamate, a promising excipient for protein formulation, on cell viability: Comparisons with NaCl. Toxicol. In Vitro 2016, 33, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Shukla, D.; Trout, B. Understanding the Synergistic Effect of Arginine and Glutamic Acid Mixtures on Protein Solubility. J. Phys. Chem. B 2011, 115, 11831–11839. [Google Scholar] [CrossRef] [PubMed]
- Shukla, D.; Zamolo, L.; Cavallotti, C.; Trout, B.L. Understanding the role of arginine as an eluent in affinity chromatography via molecular computations. J. Phys. Chem. B 2011, 115, 2645–2654. [Google Scholar] [CrossRef] [PubMed]
- Kurinomaru, T.; Kuwada, K.; Tomita, S.; Kameda, T.; Shiraki, K. Noncovalent PEGylation through Protein-Polyelectrolyte Interaction: Kinetic Experiment and Molecular Dynamics Simulation. J. Phys. Chem. B 2017, 121, 6785–6791. [Google Scholar] [CrossRef] [PubMed]
- Rawat, S.; Gupta, P.; Kumar, A.; Garg, P.; Suri, C.; Sahoo, D. Molecular Mechanism of Poly(vinyl alcohol) Mediated Prevention of Aggregation and Stabilization of Insulin in Nanoparticles. Mol. Pharm. 2015, 12, 1018–1030. [Google Scholar] [CrossRef] [PubMed]
- Kale, S.S.; Akamanchi, K.G. Trehalose Monooleate: A Potential Antiaggregation Agent for Stabilization of Proteins. Mol. Pharm. 2016, 13, 4082–4093. [Google Scholar] [CrossRef] [PubMed]
- Messina, M.S.; Ko, J.H.; Yang, Z.; Strouse, M.J.; Houk, K.N.; Maynard, H.D. Effect of trehalose polymer regioisomers on protein stabilization. Polym. Chem. 2017, 8, 4781–4788. [Google Scholar] [CrossRef]
- Kale, S.; Akamanchi, K. Rational approach for design and evaluation of anti-aggregation agents for protein stabilization: A case study of trehalose phenylalaninate. Int. J. Pharm. 2017, 524, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Arsiccio, A.; Pisano, R. Stability of Proteins in Carbohydrates and Other Additives during Freezing: The Human Growth Hormone as a Case Study. J. Phys. Chem. B 2017, 121, 8652–8660. [Google Scholar] [CrossRef] [PubMed]
- Arsiccio, A.; Pisano, R. Clarifying the role of cryo-and lyo-protectants in the biopreservation of proteins. Phys. Chem. Chem. Phys. 2018, 20, 8267–8277. [Google Scholar] [CrossRef] [PubMed]
- Barata, T.; Zhang, C.; Dalby, P.; Brocchini, S.; Zloh, M. Identification of Protein–Excipient Interaction Hotspots Using Computational Approaches. Int. J. Mol. Sci. 2016, 17, 853. [Google Scholar] [CrossRef] [PubMed]
- Westermaier, Y.; Veurink, M.; Riis-Johannessen, T.; Guinchard, S.; Gurny, R.; Scapozza, L. Identification of aggregation breakers for bevacizumab (Avastin®) self-association through similarity searching and interaction studies. Eur. J. Pharm. Biopharm. 2013, 85, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Veurink, M.; Westermaier, Y.; Gurny, R.; Scapozza, L. Breaking the aggregation of the monoclonal antibody bevacizumab (Avastin®) by dexamethasone phosphate: Insights from molecular modelling and asymmetrical flow field-flow fractionation. Pharm. Res. 2013, 30, 1176–1187. [Google Scholar] [CrossRef] [PubMed]
- Robustelli, P.; Kohlhoff, K.; Cavalli, A.; Vendruscolo, M. Using NMR chemical shifts as structural restraints in molecular dynamics simulations of proteins. Structure 2010, 18, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Robustelli, P.; Stafford, K.A.; Palmer, A.G. Interpreting protein structural dynamics from NMR chemical shifts. J. Am. Chem. Soc. 2012, 134, 6365–6374. [Google Scholar] [CrossRef] [PubMed]
- Stark, J.; Powers, R. Rapid protein-ligand costructures using chemical shift perturbations. J. Am. Chem. Soc. 2008, 130, 535–545. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biophysical Method | Details | Limitations | Application References |
|---|---|---|---|
| Raman spectroscopy | Measures shifts in energy (wavelength) of photons re-emitted after interaction with molecular vibrational modes. Provides an empirical signature of protein structure, that can be used to monitor changes in intramolecular dynamics and intermolecular interactions. | Low sensitivity. Out of the millions of incoming photons interacting with molecules, there is only one scattered Raman photon. | [18,25] |
| Circular dichroism | Measures the difference in adsorption of circularly polarised light. Far-UV CD can determine the absolute and relative contributions of secondary structure types in proteins. Near UV CD can probe tertiary structure content. Can probe changes in protein structure in response to formulation. | A reference protein with known secondary structure is required to fit the experimental data. The quality of the fit also depends on the wavelengths used. | [18,26] |
| Isothermal titration calorimetry (ITC) | Measures the heat emitted or absorbed during the titration of a protein with a ligand. The amount of heat indicates the proportion of excipient that binds the protein and its associated enthalpy. | ITC can be used to determine the excipient mechanism directly and indirectly. However, no structural information of the protein is given. | [18,27] |
| Differential scanning calorimetry (DSC) | Routinely used in high-throughput screening of excipients for formulations. Determines the impact of excipients on the thermal stability of the protein, measured as the melting temperature and enthalpy of unfolding. | Useful for identifying excipients that preferentially interact with proteins, or that stabilise through crowding effects. Cannot be used to detect other mechanisms of action. Unable to characterise changes specific to the secondary or tertiary structure of proteins. | [16,28,29,30] |
| Differential scanning fluorimetry (DSF) | Uses a PCR thermocycler to scan the fluorescence of extrinsic dye-binding to proteins as a function of temperature in microtitre plates, and determine their melting temperatures. | The excitation source of the PCR equipment can potential limit the type extrinsic fluorescence dyes used. Unable to characterise excipient mechanisms of action and can only detect tertiary structure changes. | [16,31,32,33] |
| Protein Dynamics Event | MD Simulation Time Range |
|---|---|
| Vibrational motions | Femtoseconds (10−15) to picoseconds (10−12) |
| Rotational motions | Picoseconds (10−12) to nanoseconds (10−9) |
| Loop dynamics | Picoseconds (10−12) to milliseconds (10−3) |
| Ligand binding/unbinding | Nanoseconds (10−9) to seconds |
| Protein folding/unfolding | Microseconds (10−6) to seconds |
| Aggregation | Seconds and beyond |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pandya, A.; Howard, M.J.; Zloh, M.; Dalby, P.A. An Evaluation of the Potential of NMR Spectroscopy and Computational Modelling Methods to Inform Biopharmaceutical Formulations. Pharmaceutics 2018, 10, 165. https://doi.org/10.3390/pharmaceutics10040165
Pandya A, Howard MJ, Zloh M, Dalby PA. An Evaluation of the Potential of NMR Spectroscopy and Computational Modelling Methods to Inform Biopharmaceutical Formulations. Pharmaceutics. 2018; 10(4):165. https://doi.org/10.3390/pharmaceutics10040165
Chicago/Turabian StylePandya, Akash, Mark J. Howard, Mire Zloh, and Paul A. Dalby. 2018. "An Evaluation of the Potential of NMR Spectroscopy and Computational Modelling Methods to Inform Biopharmaceutical Formulations" Pharmaceutics 10, no. 4: 165. https://doi.org/10.3390/pharmaceutics10040165
APA StylePandya, A., Howard, M. J., Zloh, M., & Dalby, P. A. (2018). An Evaluation of the Potential of NMR Spectroscopy and Computational Modelling Methods to Inform Biopharmaceutical Formulations. Pharmaceutics, 10(4), 165. https://doi.org/10.3390/pharmaceutics10040165