Drying Technologies for the Stability and Bioavailability of Biopharmaceuticals



Abstract
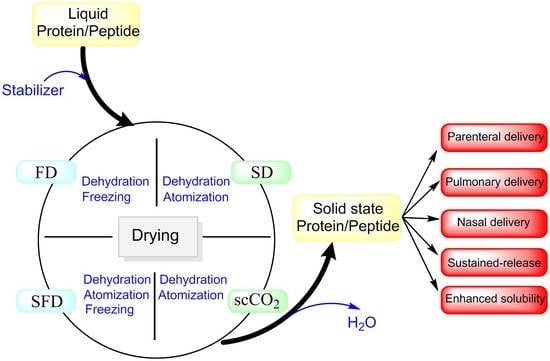
1. Introduction
2. Drying Techniques
2.1. Freeze Drying (FD)
2.2. Spray Drying (SD)
2.3. Spray Freeze Drying (SFD)
2.4. Supercritical Fluid Drying (SCFD)
2.5. Comparison of the Physical Characteristics of Dried Powders
3. Stabilizers for Dried-Powder Protein Formulations
3.1. The Water Replacement Hypothesis
3.2. The Glassy Matrix Hypothesis
3.3. Reducing Surface Adsorption
4. Delivery and Pharmacokinetics of Dried-Powder Proteins
4.1. Pulmonary Delivery
4.1.1. Local Delivery
4.1.2. Systemic Delivery
4.2. Nasal Delivery
4.3. Sustained-Release Delivery
4.4. Enhancing Solubility and Bioavailability of Cyclosporine A
4.5. Pharmacokinetics of Inhaled Insulins
5. Future Perspectives and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ajmera, A.; Scherliess, R. Stabilisation of proteins via mixtures of amino acids during spray drying. Int. J. Pharm. 2014, 463, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Ramezani, V.; Vatanara, A.; Najafabadi, A.R.; Shokrgozar, M.A.; Khabiri, A.; Seyedabadi, M. A comparative study on the physicochemical and biological stability of IgG 1 and monoclonal antibodies during spray drying process. DARU J. Pharm. Sci. 2014, 22, 31. [Google Scholar] [CrossRef] [PubMed]
- Cicerone, M.T.; Pikal, M.J.; Qian, K.K. Stabilization of proteins in solid form. Adv. Drug Deliv. Rev. 2015, 93, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, A.L.; Mrsny, R.J. Formulation and delivery issues for monoclonal antibody therapeutics. Adv. Drug Deliv. Rev. 2006, 58, 686–706. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Sharma, V.K.; Kalonia, D.S. In situ precipitation and vacuum drying of interferon alpha-2a: Development of a single-step process for obtaining dry, stable protein formulation. Int. J. Pharm. 2009, 366, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Na, D.H.; Youn, Y.S.; Lee, I.B.; Park, E.J.; Park, C.J.; Lee, K.C. Effect of molecular size of pegylated recombinant human epidermal growth factor on the biological activity and stability in rat wound tissue. Pharm. Dev. Technol. 2006, 11, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Park, E.J.; Kwak, S.; Lee, K.C.; Na, D.H.; Bae, J.-S. Trimeric peg-conjugated exendin-4 for the treatment of sepsis. Biomacromolecules 2016, 17, 1160–1169. [Google Scholar] [CrossRef] [PubMed]
- Park, E.J.; Tak, T.H.; Na, D.H.; Lee, K.C. Effect of PEGylation on stability of peptide in poly(lactide-co-glycolide) microispheres. Arch. Pharm. Res. 2010, 33, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- Na, D.H.; DeLuca, P.P. PEGylation of octreotide: I. Separation of positional isomers and stability against acylation by poly(d,l-lactide-co-gycolide). Pharm. Res. 2005, 22, 736–742. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Park, E.J.; Na, D.H. Recent progress in dendrimer-based nanomedicine. Arch. Pharm. Res. 2018, 41, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Szlachcic, A.; Zakrzewska, M.; Otlewski, J. Longer action means better drug: Tuning up protein therapeutics. Biotechnol. Adv. 2011, 29, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Mohtashamian, S.; Boddohi, S. Nanostructured polysaccharide-based carriers for antimicrobial peptide delivery. J. Pharm. Investig. 2017, 47, 85–94. [Google Scholar] [CrossRef]
- Kim, C.H.; Lee, S.G.; Kang, M.J.; Lee, S.; Choi, Y.W. Surface modification of lipid-based nanocarriers for cancer cell-specific drug targeting. J. Pharm. Investig. 2017, 47, 203–227. [Google Scholar] [CrossRef]
- Depreter, F.; Pilcer, G.; Amighi, K. Inhaled proteins: Challenges and perspectives. Int. J. Pharm. 2013, 447, 251–280. [Google Scholar] [CrossRef] [PubMed]
- Maltesen, M.J.; Van De Weert, M. Drying methods for protein pharmaceuticals. Drug Discov. Today Technol. 2008, 5, e81–e88. [Google Scholar] [CrossRef] [PubMed]
- Smales, C.M.; James, D.C. Therapeutic Proteins: Methods and Protocols; Springer: Berlin, Germany, 2005; Volume 308. [Google Scholar]
- Liao, Y.H.; Brown, M.B.; Martin, G.P. Investigation of the stabilisation of freeze-dried lysozyme and the physical properties of the formulations. Eur. J. Pharm. Biopharm. 2004, 58, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Sane, S.; Rytting, J.H. Calorimetric investigation of protein/amino acid interactions in the solid state. Int. J. Pharm. 2006, 310, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.H.; Kirwan, S.M.; Abraham, S.N.; Staats, H.F.; Hickey, A.J. Stable dry powder formulation for nasal delivery of anthrax vaccine. J. Pharm. Sci. 2012, 101, 31–47. [Google Scholar] [CrossRef] [PubMed]
- Mensink, M.A.; Frijlink, H.W.; van der Voort Maarschalk, K.; Hinrichs, W.L. How sugars protect proteins in the solid state and during drying (review): Mechanisms of stabilization in relation to stress conditions. Eur. J. Pharm. Biopharm. 2017, 114, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Maa, Y.-F.; Nguyen, P.-A.; Sweeney, T.; Shire, S.J.; Hsu, C.C. Protein inhalation powders: Spray drying vs. spray freeze drying. Pharm. Res. 1999, 16, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Nuchuchua, O.; Every, H.A.; Hofland, G.W.; Jiskoot, W. Scalable organic solvent free supercritical fluid spray drying process for producing dry protein formulations. Eur. J. Pharm. Biopharm. 2014, 88, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Walters, R.H.; Bhatnagar, B.; Tchessalov, S.; Izutsu, K.; Tsumoto, K.; Ohtake, S. Next generation drying technologies for pharmaceutical applications. J. Pharm. Sci. 2014, 103, 2673–2695. [Google Scholar] [CrossRef] [PubMed]
- Weers, J.G.; Miller, D.P. Formulation design of dry powders for inhalation. J. Pharm. Sci. 2015, 104, 3259–3288. [Google Scholar] [CrossRef] [PubMed]
- Langford, A.; Bhatnagar, B.; Walters, R.; Tchessalov, S.; Ohtake, S. Drying of biopharmaceuticals: Recent developments, new technologies and future direction. Jpn. J. Food Eng. 2018, 19, 15–24. [Google Scholar]
- Langford, A.; Bhatnagar, B.; Walters, R.; Tchessalov, S.; Ohtake, S. Drying technologies for biopharmaceutical applications: Recent developments and future direction. Dry. Technol. 2018, 36, 677–684. [Google Scholar] [CrossRef]
- Abdul-Fattah, A.M.; Kalonia, D.S.; Pikal, M.J. The challenge of drying method selection for protein pharmaceuticals: Product quality implications. J. Pharm. Sci. 2007, 96, 1886–1916. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, M.; Albet, C.; Palomer, A.; Guglietta, A. Drying in pharmaceutical and biotechnological industries. Food Sci. Technol. Int. 2003, 9, 237–243. [Google Scholar] [CrossRef]
- Shukla, S. Freeze drying process: A review. Int. J. Pharm. Sci. Res. 2011, 2, 3061. [Google Scholar]
- Faghihi, H.; Khalili, F.; Amini, M.; Vatanara, A. The effect of freeze-dried antibody concentrations on its stability in the presence of trehalose and hydroxypropyl-β-cyclodextrin: A box–behnken statistical design. Pharm. Dev. Technol. 2017, 22, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Roy, I.; Gupta, M.N. Freeze-drying of proteins: Some emerging concerns. Biotechnol. Appl. Biochem. 2004, 39, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Pikal, M.J. Design of freeze-drying processes for pharmaceuticals: Practical advice. Pharm. Res. 2004, 21, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.Y.; Kim, N.A.; Lim, D.G.; Kim, K.H.; Choi, D.H.; Jeong, S.H. Process cycle development of freeze drying for therapeutic proteins with stability evaluation. J. Pharm. Investig. 2016, 46, 519–536. [Google Scholar] [CrossRef]
- Pieters, S.; De Beer, T.; Kasper, J.C.; Boulpaep, D.; Waszkiewicz, O.; Goodarzi, M.; Tistaert, C.; Friess, W.; Remon, J.P.; Vervaet, C.; et al. Near-infrared spectroscopy for in-line monitoring of protein unfolding and its interactions with lyoprotectants during freeze-drying. Anal. Chem. 2012, 84, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Oetjen, G.W. Freeze-Drying; Wiley: Hoboken, NJ, USA, 2004. [Google Scholar]
- Costantino, H.R.; Firouzabadian, L.; Wu, C.; Carrasquillo, K.G.; Griebenow, K.; Zale, S.E.; Tracy, M.A. Protein spray freeze drying. 2. Effect of formulation variables on particle size and stability. J. Pharm. Sci. 2002, 91, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Okuda, T.; Lu, X.-Y.; Chan, H.-K. Amorphous powders for inhalation drug delivery. Adv. Drug Deliv. Rev. 2016, 100, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Tonnis, W.; Mensink, M.; De Jager, A.; Van Der Voort Maarschalk, K.; Frijlink, H.; Hinrichs, W. Size and molecular flexibility of sugars determine the storage stability of freeze-dried proteins. Mol. Pharm. 2015, 12, 684–694. [Google Scholar] [CrossRef] [PubMed]
- Pilcer, G.; Amighi, K. Formulation strategy and use of excipients in pulmonary drug delivery. Int. J. Pharm. 2010, 392, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Sosnik, A.; Seremeta, K.P. Advantages and challenges of the spray-drying technology for the production of pure drug particles and drug-loaded polymeric carriers. Adv. Colloid Interface Sci. 2015, 223, 40–54. [Google Scholar] [CrossRef] [PubMed]
- Ameri, M.; Maa, Y.-F. Spray drying of biopharmaceuticals: Stability and process considerations. Dry. Technol. 2006, 24, 763–768. [Google Scholar] [CrossRef]
- Faghihi, H.; Vatanara, A.; Najafabadi, A.R.; Ramezani, V.; Gilani, K. The use of amino acids to prepare physically and conformationally stable spray-dried IgG with enhanced aerosol performance. Int. J. Pharm. 2014, 466, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Maa, Y.-F.; Sellers, S.P. Solid-state protein formulation. In Therapeutic Proteins Methods Protocols; Humana Press: New York, NY, USA, 2005; pp. 265–285. [Google Scholar]
- Poursina, N.; Vatanara, A.; Rouini, M.R.; Gilani, K.; Rouholamini Najafabadi, A. Systemic delivery of parathyroid hormone (1–34) using spray freeze-dried inhalable particles. Pharm. Dev. Technol. 2017, 22, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Ramezani, V.; Vatanara, A.; Najafabadi, A.R.; Gilani, K.; Nabi-Meybodi, M. Screening and evaluation of variables in the formation of antibody particles by spray drying. Powder Technol. 2013, 233, 341–346. [Google Scholar] [CrossRef]
- Vehring, R. Pharmaceutical particle engineering via spray drying. Pharm. Res. 2008, 25, 999–1022. [Google Scholar] [CrossRef] [PubMed]
- Schüle, S.; Frieß, W.; Bechtold-Peters, K.; Garidel, P. Conformational analysis of protein secondary structure during spray-drying of antibody/mannitol formulations. Eur. J. Pharm. Biopharm. 2007, 65, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Li, H.Y.; Song, X.; Seville, P.C. The use of sodium carboxymethylcellulose in the preparation of spray-dried proteins for pulmonary drug delivery. Eur. J. Pharm. Sci. 2010, 40, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Wanning, S.; Suverkrup, R.; Lamprecht, A. Jet-vortex spray freeze drying for the production of inhalable lyophilisate powders. Eur. J. Pharm. Sci. 2017, 96, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Wanning, S.; Süverkrüp, R.; Lamprecht, A. Pharmaceutical spray freeze drying. Int. J. Pharm. 2015, 488, 136–153. [Google Scholar] [CrossRef] [PubMed]
- Rogers, T.L.; Johnston, K.P.; Williams, R.O., 3rd. Solution-based particle formation of pharmaceutical powders by supercritical or compressed fluid CO2 and cryogenic spray-freezing technologies. Drug Dev. Ind. Pharm. 2001, 27, 1003–1015. [Google Scholar] [CrossRef] [PubMed]
- Emami, F.; Vatanara, A.; Najafabadi, A.R.; Kim, Y.; Park, E.J.; Sardari, S.; Na, D.H. Effect of amino acids on the stability of spray freeze-dried immunoglobulin G in sugar-based matrices. Eur. J. Pharm. Sci. 2018, 119, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Poursina, N.; Vatanara, A.; Rouini, M.R.; Gilani, K.; Najafabadi, A.R. The effect of excipients on the stability and aerosol performance of salmon calcitonin dry powder inhalers prepared via spray freeze drying process. Acta Pharm. 2016, 66, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Rahmati, M.R.; Vatanara, A.; Parsian, A.R.; Gilani, K.; Khosravi, K.M.; Darabi, M.; Najafabadi, A.R. Effect of formulation ingredients on the physical characteristics of salmeterol xinafoate microparticles tailored by spray freeze drying. Adv. Powder Technol. 2013, 24, 36–42. [Google Scholar] [CrossRef]
- Parsian, A.R.; Vatanara, A.; Rahmati, M.R.; Gilani, K.; Khosravi, K.M.; Najafabadi, A.R. Inhalable budesonide porous microparticles tailored by spray freeze drying technique. Powder Technol. 2014, 260, 36–41. [Google Scholar] [CrossRef]
- Rogers, T.L.; Nelsen, A.C.; Hu, J.; Brown, J.N.; Sarkari, M.; Young, T.J.; Johnston, K.P.; Williams, R.O. A novel particle engineering technology to enhance dissolution of poorly water soluble drugs: Spray-freezing into liquid. Eur. J. Pharm. Biopharm. 2002, 54, 271–280. [Google Scholar] [CrossRef]
- Rogers, T.L.; Hu, J.; Yu, Z.; Johnston, K.P.; Williams, R.O. A novel particle engineering technology: Spray-freezing into liquid. Int. J. Pharm. 2002, 242, 93–100. [Google Scholar] [CrossRef]
- Maa, Y.F.; Ameri, M.; Shu, C.; Payne, L.G.; Chen, D. Influenza vaccine powder formulation development: Spray-freeze-drying and stability evaluation. J. Pharm. Sci. 2004, 93, 1912–1923. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, N.; Bouchard, A.; Sutter, M.; Van Speybroeck, M.; Hofland, G.W.; Witkamp, G.J.; Crommelin, D.J.; Jiskoot, W. Stable sugar-based protein formulations by supercritical fluid drying. Int. J. Pharm. 2008, 346, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Jovanović, N.; Bouchard, A.; Hofland, G.W.; Witkamp, G.-J.; Crommelin, D.J.; Jiskoot, W. Stabilization of proteins in dry powder formulations using supercritical fluid technology. Pharm. Res. 2004, 21, 1955–1969. [Google Scholar] [CrossRef] [PubMed]
- Garidel, P.; Pevestorf, B.; Bahrenburg, S. Stability of buffer-free freeze-dried formulations: A feasibility study of a monoclonal antibody at high protein concentrations. Eur. J. Pharm. Biopharm. 2015, 97, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Mohammad Zadeh, A.H.; Rouholamini Najafabadi, A.; Vatanara, A.; Faghihi, H.; Gilani, K. Effect of molecular weight and ratio of poly ethylene glycols’ derivatives in combination with trehalose on stability of freeze-dried IgG. Drug Dev. Ind. Pharm. 2017, 43, 1945–1951. [Google Scholar] [CrossRef] [PubMed]
- Imamura, K.; Ogawa, T.; Sakiyama, T.; Nakanishi, K. Effects of types of sugar on the stabilization of protein in the dried state. J. Pharm. Sci. 2003, 92, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Saluja, V.; Amorij, J.P.; Kapteyn, J.C.; de Boer, A.H.; Frijlink, H.W.; Hinrichs, W.L. A comparison between spray drying and spray freeze drying to produce an influenza subunit vaccine powder for inhalation. J. Control. Release 2010, 144, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Bittner, B.; Morlock, M.; Koll, H.; Winter, G.; Kissel, T. Recombinant human erythropoietin (rhEPO) loaded poly (lactide-co-glycolide) microspheres: Influence of the encapsulation technique and polymer purity on microsphere characteristics. Eur. J. Pharm. Biopharm. 1998, 45, 295–305. [Google Scholar] [CrossRef]
- Jovanovic, N.; Bouchard, A.; Hofland, G.W.; Witkamp, G.J.; Crommelin, D.J.; Jiskoot, W. Stabilization of IgG by supercritical fluid drying: Optimization of formulation and process parameters. Eur. J. Pharm. Biopharm. 2008, 68, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, N.; Bouchard, A.; Hofland, G.W.; Witkamp, G.J.; Crommelin, D.J.; Jiskoot, W. Distinct effects of sucrose and trehalose on protein stability during supercritical fluid drying and freeze-drying. Eur. J. Pharm. Sci. 2006, 27, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Amidi, M.; Pellikaan, H.C.; de Boer, A.H.; Crommelin, D.J.; Hennink, W.E.; Jiskoot, W. Preparation and physicochemical characterization of supercritically dried insulin-loaded microparticles for pulmonary delivery. Eur. J. Pharm. Biopharm. 2008, 68, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.E.; Lamprecht, A. Spray freeze drying for dry powder inhalation of nanoparticles. Eur. J. Pharm. Biopharm. 2014, 87, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Mueannoom, W.; Srisongphan, A.; Taylor, K.M.; Hauschild, S.; Gaisford, S. Thermal ink-jet spray freeze-drying for preparation of excipient-free salbutamol sulphate for inhalation. Eur. J. Pharm. Biopharm. 2012, 80, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Filipe, V.; Hawe, A.; Carpenter, J.F.; Jiskoot, W. Analytical approaches to assess the degradation of therapeutic proteins. TrAC Trends Anal. Chem. 2013, 49, 118–125. [Google Scholar] [CrossRef]
- Song, J.G.; Lee, S.H.; Han, H.-K. The stabilization of biopharmaceuticals: Current understanding and future perspectives. J. Pharm. Investig. 2017, 47, 475–496. [Google Scholar] [CrossRef]
- Wang, W.; Singh, S.; Zeng, D.L.; King, K.; Nema, S. Antibody structure, instability, and formulation. J. Pharm. Sci. 2007, 96, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.L.; Pikal, M.J. Mechanisms of protein stabilization in the solid state. J. Pharm. Sci. 2009, 98, 2886–2908. [Google Scholar] [CrossRef] [PubMed]
- Hertel, S.P.; Winter, G.; Friess, W. Protein stability in pulmonary drug delivery via nebulization. Adv. Drug Deliv. Rev. 2015, 93, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Grenha, A.; Remuñán-López, C.; Carvalho, E.L.; Seijo, B. Microspheres containing lipid/chitosan nanoparticles complexes for pulmonary delivery of therapeutic proteins. Eur. J. Pharm. Biopharm. 2008, 69, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.A.; Hanes, J.; Caponetti, G.; Hrkach, J.; Ben-Jebria, A.; Eskew, M.L.; Mintzes, J.; Deaver, D.; Lotan, N.; Langer, R. Large porous particles for pulmonary drug delivery. Science 1997, 276, 1868–1872. [Google Scholar] [CrossRef] [PubMed]
- Tsapis, N.; Bennett, D.; Jackson, B.; Weitz, D.A.; Edwards, D. Trojan particles: Large porous carriers of nanoparticles for drug delivery. Proc. Natl. Acad. Sci. USA 2002, 99, 12001–12005. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.; Arnold, J.J.; Khan, M.A.; Ahsan, F. Absorption enhancers in pulmonary protein delivery. J. Control. Release 2004, 94, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Siekmeier, R.; Scheuch, G. Treatment of systemic diseases by inhalation of biomolecule aerosols. J. Physiol. Pharmacol. 2009, 60, 15–26. [Google Scholar] [PubMed]
- Okumura, K.; Iwakawa, S.; Yoshida, T.; Seki, T.; Komada, F. Intratracheal delivery of insulin absorption from solution and aerosol by rat lung. Int. J. Pharm. 1992, 88, 63–73. [Google Scholar] [CrossRef]
- Todo, H.; Okamoto, H.; Iida, K.; Danjo, K. Improvement of stability and absorbability of dry insulin powder for inhalation by powder-combination technique. Int. J. Pharm. 2004, 271, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Yang, F.F.; Liao, Y.H. Pharmacokinetic Considerations of inhaled pharmaceuticals for systemic delivery. Curr. Pharm. Des. 2016, 22, 2532–2548. [Google Scholar] [CrossRef] [PubMed]
- Garmise, R.J.; Staats, H.F.; Hickey, A.J. Novel dry powder preparations of whole inactivated influenza virus for nasal vaccination. AAPS PharmSciTech 2007, 8, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.; Kim, M.-S.; Jung, M.-S.; Park, J.; Cha, K.-H.; Kim, J.-S.; Park, H.J.; Alhalaweh, A.; Velaga, S.P.; Hwang, S.-J. Design of salmon calcitonin particles for nasal delivery using spray-drying and novel supercritical fluid-assisted spray-drying processes. Int. J. Pharm. 2015, 478, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Mok, H.; Park, T.G. Water-free microencapsulation of proteins within PLGA microparticles by spray drying using PEG-assisted protein solubilization technique in organic solvent. Eur. J. Pharm. Biopharm. 2008, 70, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Cleland, J.L.; Duenas, E.T.; Park, A.; Daugherty, A.; Kahn, J.; Kowalski, J.; Cuthbertson, A. Development of poly-(d,l-lactide-coglycolide) microsphere formulations containing recombinant human vascular endothelial growth factor to promote local angiogenesis. J. Control. Release 2001, 72, 13–24. [Google Scholar] [CrossRef]
- Al-Qadi, S.; Grenha, A.; Carrión-Recio, D.; Seijo, B.; Remuñán-López, C. Microencapsulated chitosan nanoparticles for pulmonary protein delivery: In vivo evaluation of insulin-loaded formulations. J. Control. Release 2012, 157, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Chiou, H.; Chan, H.-K.; Heng, D.; Prud’homme, R.K.; Raper, J.A. A novel production method for inhalable cyclosporine a powders by confined liquid impinging jet precipitation. J. Aerosol Sci. 2008, 39, 500–509. [Google Scholar] [CrossRef]
- Yamasaki, K.; Kwok, P.C.L.; Fukushige, K.; Prud’homme, R.K.; Chan, H.-K. Enhanced dissolution of inhalable cyclosporine nano-matrix particles with mannitol as matrix former. Int. J. Pharm. 2011, 420, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Cavaiola, T.S.; Edelman, S. Inhaled insulin: A breath of fresh air? A review of inhaled insulin. Clin. Ther. 2014, 36, 1275–1289. [Google Scholar] [CrossRef] [PubMed]
- Rave, K.; Bott, S.; Heinemann, L.; Sha, S.; Becker, R.H.; Willavize, S.A.; Heise, T. Time-action profile of inhaled insulin in comparison with subcutaneously injected insulin lispro and regular human insulin. Diabetes Care 2005, 28, 1077–1082. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.J.; Barnett, A.H. Inhaled insulin: New formulation, new trial. Lancet 2010, 375, 2199–2201. [Google Scholar] [CrossRef]
- Quattrin, T.; Bélanger, A.; Bohannon, N.J.; Schwartz, S.L. Efficacy and safety of inhaled insulin (Exubera) compared with subcutaneous insulin therapy in patients with type 1 diabetes: Results of a 6-month, randomized, comparative trial. Diabetes Care 2004, 27, 2622–2627. [Google Scholar] [CrossRef] [PubMed]
- Skyler, J.S.; Weinstock, R.S.; Raskin, P.; Yale, J.-F.; Barrett, E.; Gerich, J.E.; Gerstein, H.C. Use of inhaled insulin in a basal/bolus insulin regimen in type 1 diabetic subjects: A 6-month, randomized, comparative trial. Diabetes Care 2005, 28, 1630–1635. [Google Scholar] [CrossRef] [PubMed]
- Pfützner, A.; Forst, T. Pulmonary insulin delivery by means of the technosphere™ drug carrier mechanism. Expert Opin. Drug Deliv. 2005, 2, 1097–1106. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, J.P.; Amin, N.; Marino, M.; Gotfried, M.; Meyer, T.; Sommerer, K.; Baughman, R.A. Insulin lung deposition and clearance following technosphere® insulin inhalation powder administration. Pharm. Res. 2011, 28, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Angelo, R.; Rousseau, K.; Grant, M.; Leone-Bay, A.; Richardson, P. Technosphere® insulin: Defining the role of technosphere particles at the cellular level. J. Diabetes Sci. Technol. 2009, 3, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Swierczewska, M.; Lee, K.C.; Lee, S. What is the future of PEGylated therapies? Expert Opin. Emerg. Drugs 2015, 20, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Park, E.J.; Lim, S.M.; Lee, K.C.; Na, D.H. Exendins and exendin analogs for diabetic therapy: A patent review (2012–2015). Expert Opin. Ther. Pat. 2016, 26, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Park, E.J.; Na, D.H. Antibody-drug conjugates for targeted anticancer drug delivery. J. Pharm. Investig. 2016, 46, 341–349. [Google Scholar] [CrossRef]
- Li, X.; Huang, Y.; Huang, Z.; Ma, X.; Dong, N.; Chen, W.; Pan, X.; Wu, C. Enhancing stability of exenatide-containing pressurized metered-dose inhaler via reverse microemulsion system. AAPS PharmSciTech 2018, 19, 2499–2508. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Drying Procedure | Process Parameters | Stress | Advantages | Limitations | Typical Powder Characteristics |
|---|---|---|---|---|---|
| Freeze drying |
|
|
|
|
|
| Spray drying |
|
|
|
|
|
| Spray freeze drying |
|
|
|
|
|
| Supercritical fluid drying |
|
|
|
|
|
| Process | Proteins/Peptides | Stabilizers | Mechanism of Stabilization | Applications | References | |
|---|---|---|---|---|---|---|
| Stability Improvement | Drug Delivery | |||||
| Freeze drying |
|
|
| ✓ ✓ ✓ ✓ | _ _ _ _ | [61,62] [17] [63] [18] |
| Spray drying |
|
|
| ✓ ✓ _ ✓ _ ✓ _ |
| [42,45,47] [2] [21] [1] [64] [48] [65] |
| Spray freeze drying |
|
|
- | ✓ ✓ _ ✓ ✓ _ ✓ ✓ ✓ | _
| [52] [36] [21] [44] [53] [64] [58] [57] [19] |
| Supercritical fluid drying |
|
| -
| ✓ ✓ ✓ ✓ | _ _ _
| [66] [22,59] [67] [68] |
| Stabilizers | Stabilization | References | ||
|---|---|---|---|---|
| Mechanism | Process | |||
| Proteins | Human or Bovine serum albumin | Water replacement (Hydrogen bonding) | ||
| Amino acids | Glycine, Alanine, Histidine, Leucine Phenylalanine, Arginine, Aspartic acid | Water replacement Bulking agent Buffering agent Prevent protein-protein interactions | Freezing Dehydration Thermal stress | [1,18,52,74] |
| Polyols | Polyethylene glycol, Mannitol, Sorbitol | Water replacement Glassy state Increase matrix density | Freezing Dehydration | [74] |
| Carbohydrate (reducing and non-reducing sugar) | Fructose, Glucose, Lactose, Maltose, Maltodextrin Trehalose, Sucrose, Inulin, Dextran | Water replacement Glassy state (reduce global protein mobility) Reduce local protein mobility Protein-sugar interactions | Freezing Dehydration Thermal stress | [20,52,74] |
| Buffer and Salt | HEPES buffer, Citrate buffer, Phosphate buffer saline, Ammonium sulfate | Buffering agent | Freezing | [64] |
| Surfactant | Polysorbate 20, 80, Oleic acid, Sodium glycolate | Prevent surface adsorption (Reduce interfacial stress) Prevent protein-protein interactions (Prevent intermolecular interactions) Slow dissolution rate | Shear stress Interfacial stress Freezing Reconstitution | [20,74] |
| Polymers and Polysaccharides | Cyclodextrin, Dextran, PLGA, Hydroxy propyl β-cyclodextrin, Na-Carboxy methylcellulose | Glassy state | Freezing | [20] |
| Metals | Zinc | Reduce surface area | Freezing Interfacial stress | [36] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Emami, F.; Vatanara, A.; Park, E.J.; Na, D.H. Drying Technologies for the Stability and Bioavailability of Biopharmaceuticals. Pharmaceutics 2018, 10, 131. https://doi.org/10.3390/pharmaceutics10030131
Emami F, Vatanara A, Park EJ, Na DH. Drying Technologies for the Stability and Bioavailability of Biopharmaceuticals. Pharmaceutics. 2018; 10(3):131. https://doi.org/10.3390/pharmaceutics10030131
Chicago/Turabian StyleEmami, Fakhrossadat, Alireza Vatanara, Eun Ji Park, and Dong Hee Na. 2018. "Drying Technologies for the Stability and Bioavailability of Biopharmaceuticals" Pharmaceutics 10, no. 3: 131. https://doi.org/10.3390/pharmaceutics10030131
APA StyleEmami, F., Vatanara, A., Park, E. J., & Na, D. H. (2018). Drying Technologies for the Stability and Bioavailability of Biopharmaceuticals. Pharmaceutics, 10(3), 131. https://doi.org/10.3390/pharmaceutics10030131