The Effects of Synthetically Modified Natural Compounds on ABC Transporters

 ,
,  ,
,
Abstract
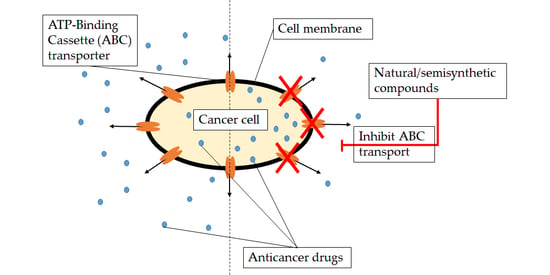
1. Introduction
2. ABCB1/P-gp
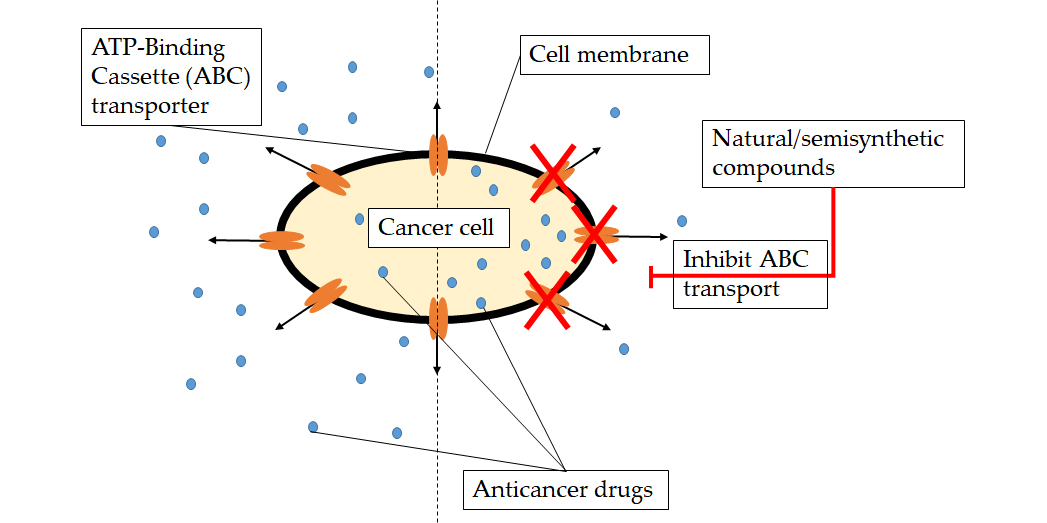
2.1. Ningalin B
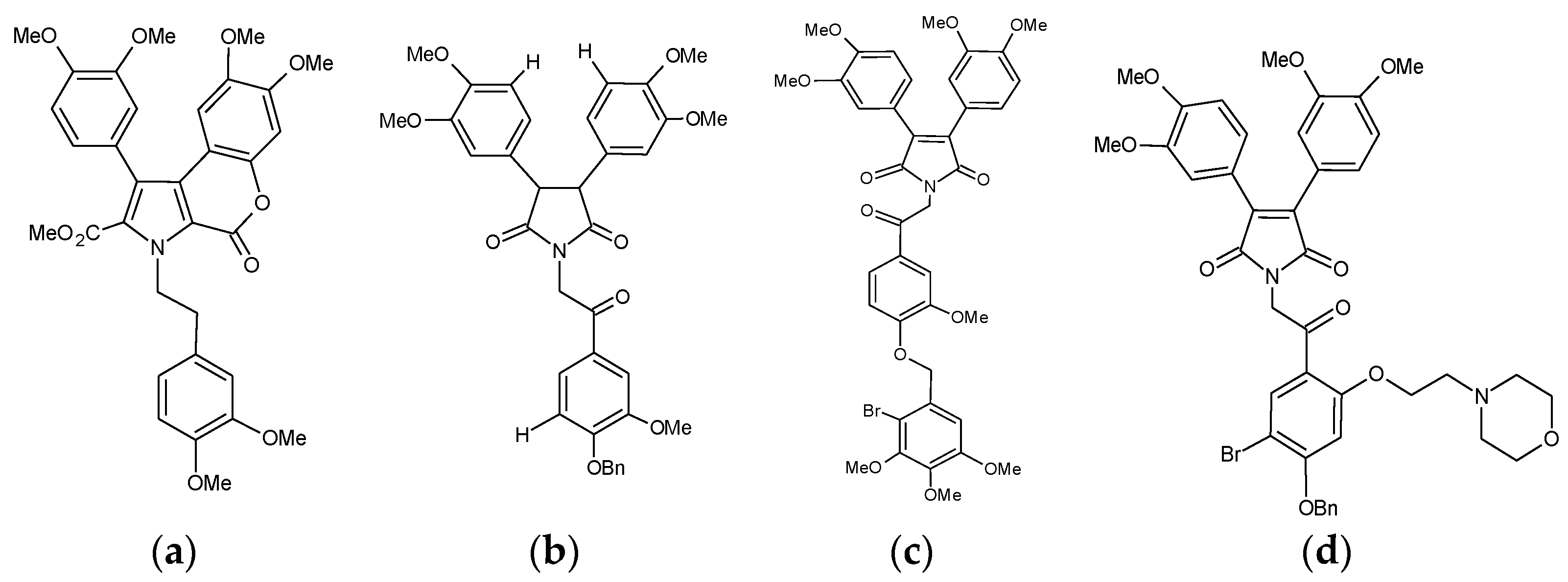
2.2. Tetrandrine
2.3. Terpenes
2.4. Other Notable Synthetic P-gp Inhibitors
3. ABCG2/BCRP
4. ABCC1/MRP1
5. Multi-Specific ABC Transporter Inhibitors
6. Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pastan, I.; Gottesman, M. Multiple-drug resistance in human cancer. N. Engl. J. Med. 1987, 316, 1388–1393. [Google Scholar] [PubMed]
- Gerlach, J.H.; Kartner, N.; Bell, D.R.; Ling, V. Multidrug resistance. Cancer Surv. 1986, 5, 25–46. [Google Scholar] [PubMed]
- Dean, M.; Hamon, Y.; Chimini, G. The human ATP-binding cassette (ABC) transporter superfamily. J. Lipid Res. 2001, 42, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Falasca, M.; Schlessinger, J.; Ferguson, K. Regulatory recruitment of signalling molecules to the cell membrane by pleckstrinhomology domains. Trends Cell Biol. 1997, 7, 237–242. [Google Scholar]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, L.J. Clinical reversal of drug resistance. Curr. Probl. Cancer 1995, 19, 65–124. [Google Scholar] [CrossRef]
- Joshi, P.; Vishwakarma, R.A.; Bharate, S.B. Natural alkaloids as P-gp inhibitors for multidrug resistance reversal in cancer. Eur. J. Med. Chem. 2017, 138, 273–292. [Google Scholar] [CrossRef] [PubMed]
- Fojo, T.; Bates, S. Strategies for reversing drug resistance. Oncogene 2003, 22, 7512–7523. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.C.; Zhang, Y.K.; Wang, Y.J.; Vispute, S.G.; Jain, S.; Chen, Y.M.; Li, J.; Youssef, D.T.A.; El Sayed, K.A.; Chen, Z.S. Esters of the marine-derived triterpene sipholenol a reverse P-gp-mediated drug resistance. Mar. Drugs 2015, 13, 2267–2286. [Google Scholar] [CrossRef] [PubMed]
- Keogh, J.P. Membrane transporters in drug development. Adv. Pharmacol. 2012, 63, 1–42. [Google Scholar] [PubMed]
- Prachayasittikul, V.; Prachayasittikul, V. P-glycoprotein transporter in drug development. EXCLI J. 2016, 15, 113–118. [Google Scholar] [PubMed]
- Dean, M.; Rzhetsky, A.; Allikmets, R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2002, 11, 1156–1166. [Google Scholar] [CrossRef]
- Basili, S.; Moro, S. Novel camptothecin derivatives as topoisomerase I inhibitors. Expert Opin. Ther. Pat. 2009, 19, 555–574. [Google Scholar] [CrossRef] [PubMed]
- Brangi, M.; Litman, T.; Ciotti, M.; Nishiyama, K.; Kohlhagen, G.; Takimoto, C.; Robey, R.; Pommier, Y.; Fojo, T.; Bates, S.E. Camptothecin resistance: Role of the ATP-binding cassette (ABC), mitoxantrone-resistance half-transporter (MXR), and potential for glucuronidation in MXR-expressing cells. Cancer Res. 1999, 59, 5938–5946. [Google Scholar] [PubMed]
- Lalloo, A.K.; Luo, F.R.; Guo, A.; Paranjpe, P.V.; Lee, S.H.; Vyas, V.; Rubin, E.; Sinko, P.J. Membrane transport of camptothecin: Facilitation by human P-glycoprotein (ABCB1) and multidrug resistance protein 2 (ABCC2). BMC Med. 2004, 2, 16. [Google Scholar] [CrossRef] [PubMed]
- Van de Wetering, K.; Burkon, A.; Feddema, W.; Bot, A.; de Jonge, H.; Somoza, V.; Borst, P. Intestinal breast cancer resistance protein (BCRP)/Bcrp1 and multidrug resistance protein 3 (MRP3)/Mrp3 are involved in the pharmacokinetics of resveratrol. Mol. Pharmacol. 2009, 75, 876–885. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Shukla, S.; Gupta, S. Apigenin and cancer chemoprevention: Progress, potential and promise (review). Int. J. Oncol. 2007, 30, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Gupta, S. Molecular targets for apigenin-induced cell cycle arrest and apoptosis in prostate cancer cell xenograft. Mol. Cancer Ther. 2006, 5, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Budhraja, A.; Gao, N.; Zhang, Z.; Son, Y.O.; Cheng, S.; Wang, X.; Ding, S.; Hitron, A.; Chen, G.; Luo, J.; et al. Apigenin induces apoptosis in human leukemia cells and exhibits anti-leukemic activity in vivo. Mol. Cancer Ther. 2012, 11, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Chuang, Y.J.; Yu, C.C.; Yang, J.S.; Lu, C.C.; Chiang, J.H.; Lin, J.P.; Tang, N.Y.; Huang, A.C.; Chung, J.G. Apigenin induces apoptosis through mitochondrial dysfunction in U-2 OS human osteosarcoma cells and inhibits osteosarcoma xenograft tumor growth in vivo. J. Agric. Food Chem. 2012, 60, 11395–11402. [Google Scholar] [CrossRef] [PubMed]
- Saeed, M.; Kadioglu, O.; Khalid, H.; Sugimoto, Y.; Efferth, T. Activity of the dietary flavonoid, apigenin, against multidrug-resistant tumor cells as determined by pharmacogenomics and molecular docking. J. Nutr. Biochem. 2015, 26, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Rajesh, E.; Sankari, L.S.; Malathi, L.; Krupaa, J.R. Naturally occurring products in cancer therapy. J. Pharm. Bioallied Sci. 2015, 7, S181–S183. [Google Scholar] [CrossRef] [PubMed]
- Rigalli, J.P.; Ciriaci, N.; Arias, A.; Ceballos, M.P.; Villanueva, S.S.; Luquita, M.G.; Mottino, A.D.; Ghanem, C.I.; Catania, V.A.; Ruiz, M.L. Regulation of multidrug resistance proteins by genistein in a hepatocarcinoma cell line: Impact on sorafenib cytotoxicity. PLoS ONE 2015, 10, e0119502. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Xu, H.; Wang, S.W.; Hu, M. Breast cancer resistance protein (BCRP) and sulfotransferases contribute significantly to the disposition of genistein in mouse intestine. AAPS J. 2010, 12, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Falasca, M.; Linton, K.J. Investigational ABC transporter inhibitors. Expert Opin. Investig. Drugs 2012, 21, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Revalde, J.L.; Li, Y.; Hawkins, B.C.; Rosengren, R.J.; Paxton, J.W. Heterocyclic cyclohexanone monocarbonyl analogs of curcumin can inhibit the activity of ATP-binding cassette transporters in cancer multidrug resistance. Biochem. Pharmacol. 2015, 93, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.A.; Gescher, A.J.; Steward, W.P. Curcumin: The story so far. Eur. J. Cancer 2005, 41, 1955–1968. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Ohnuma, S.; Fukuda, M.; Chufan, E.E.; Kudoh, K.; Kanehara, K.; Sugisawa, N.; Ishida, M.; Naitoh, T.; Shibata, H.; et al. Synthetic analogs of curcumin modulate the function of multidrug resistance-linked ATP-binding cassette transporter ABCG2. Drug Metab. Dispos. 2017, 45, 1166–1177. [Google Scholar] [CrossRef] [PubMed]
- Hilgendorf, C.; Ahlin, G.; Seithel, A.; Artursson, P.; Ungell, A.L.; Karlsson, J. Expression of thirty-six drug transporter genes in human intestine, liver, kidney, and organotypic cell lines. Drug Metab. Dispos. 2007, 35, 1333–1340. [Google Scholar] [CrossRef] [PubMed]
- Dano, K. Active outward transport of daunomycin in resistant ehrlich ascites tumor cells. Biochim. Biophys. Acta 1973, 323, 466–483. [Google Scholar] [CrossRef]
- Juliano, R.L.; Ling, V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim. Biophys. Acta 1976, 455, 152–162. [Google Scholar] [CrossRef]
- Ueda, K.; Cornwell, M.M.; Gottesman, M.M.; Pastan, I.; Roninson, I.B.; Ling, V.; Riordan, J.R. The mdr1 gene, responsible for multidrug-resistance, codes for P-glycoprotein. Biochem. Biophys. Res. Commun. 1986, 141, 956–962. [Google Scholar] [CrossRef]
- Li, Y.; Revalde, J.; Paxton, J.W. The effects of dietary and herbal phytochemicals on drug transporters. Adv. Drug Deliv. Rev. 2017, 116, 45–62. [Google Scholar] [CrossRef] [PubMed]
- Thiebaut, F.; Tsuruo, T.; Hamada, H.; Gottesman, M.M.; Pastan, I.; Willingham, M.C. Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc. Natl. Acad. Sci. USA 1987, 84, 7735–7738. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, I.; Kataoka, I.; Morishita, Y.; Hamada, H.; Tsuruo, T.; Itoyama, S.; Mori, S. Tissue distribution of P-glycoprotein encoded by a multidrug-resistant gene as revealed by a monoclonal antibody, MRK 16. Cancer Res. 1988, 48, 1926–1929. [Google Scholar] [PubMed]
- Fromm, M.F. P-glycoprotein: A defense mechanism limiting oral bioavailability and CNS accumulation of drugs. Int. J. Clin. Pharmacol. Ther. 2000, 38, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Pearce, H.L.; Safa, A.R.; Bach, N.J.; Winter, M.A.; Cirtain, M.C.; Beck, W.T. Essential features of the P-glycoprotein pharmacophore as defined by a series of reserpine analogs that modulate multidrug resistance. Proc. Natl. Acad. Sci. USA 1989, 86, 5128–5132. [Google Scholar] [CrossRef] [PubMed]
- Fojo, A.T.; Ueda, K.; Slamon, D.J.; Poplack, D.G.; Gottesman, M.M.; Pastan, I. Expression of a multidrug-resistance gene in human tumors and tissues. Proc. Natl. Acad. Sci. USA 1987, 84, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.L.; Goldstein, L.J.; Gottesman, M.M.; Pastan, I.; Tsai, C.M.; Johnson, B.E.; Mulshine, J.L.; Ihde, D.C.; Kayser, K.; Gazdar, A.F. MDR1 gene expression in lung cancer. J. Natl. Cancer Inst. 1989, 81, 1144–1150. [Google Scholar] [CrossRef] [PubMed]
- Kosztyu, P.; Dolezel, P.; Mlejnek, P. Can P-glycoprotein mediate resistance to nilotinib in human leukaemia cells? Pharmacol. Res. 2013, 67, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Ruzickova, E.; Janska, R.; Dolezel, P.; Mlejnek, P. Clinically relevant interactions of anti-apoptotic Bcl-2 protein inhibitors with ABC transporters. Pharmazie 2017, 72, 751–758. [Google Scholar] [PubMed]
- Long, S.; Sousa, E.; Kijjoa, A.; Pinto, M.M.M. Marine natural products as models to circumvent multidrug resistance. Molecules 2016, 21, 892. [Google Scholar] [CrossRef] [PubMed]
- Boger, D.L.; Soenen, D.R.; Boyce, C.W.; Hedrick, M.P.; Jin, Q. Total synthesis of ningalin B utilizing a heterocyclic azadiene Diels-alder reaction and discovery of a new class of potent multidrug resistant (MDR) reversal agents. J. Org. Chem. 2000, 65, 2479–2483. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wong, I.L.K.; Peng, K.; Liu, Z.; Wang, P.; Jiang, T.F.; Jiang, T.; Chow, L.M.C.; Wan, S.B. Extending the structure-activity relationship study of marine natural ningalin b analogues as P-glycoprotein inhibitors. Eur. J. Med. Chem. 2017, 125, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Fenical, W. Ningalins A–D: Novel aromatic alkaloids from a western australian ascidian of the genus didemnum. J. Org. Chem. 1997, 62, 3254–3262. [Google Scholar] [CrossRef] [PubMed]
- Soenen, D.R.; Hwang, I.; Hedrick, M.P.; Boger, D.L. Multidrug resistance reversal activity of key ningalin analogues. Bioorg. Med. Chem. Lett. 2003, 13, 1777–1781. [Google Scholar] [CrossRef]
- Tao, H.C.; Hwang, I.K.; Boger, D.L. Multidrug resistance reversal activity of permethyl ningalin B amide derivatives. Bioorg. Med. Chem. Lett. 2004, 14, 5979–5981. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C.; Guan, Y.; Soenen, D.R.; Danishefsky, S.J.; Boger, D.L. Potent reversal of multidrug resistance by ningalins and its use in drug combinations against human colon carcinoma xenograft in nude mice. Cancer Chemother. Pharmacol. 2005, 56, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Bin, J.W.; Wong, I.L.; Hu, X.; Yu, Z.X.; Xing, L.F.; Jiang, T.; Chow, L.M.; Biao, W.S. Structure-activity relationship study of permethyl ningalin B analogues as P-glycoprotein chemosensitizers. J. Med. Chem. 2013, 56, 9057–9070. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.Y.; Wong, I.L.; Yan, C.S.; Zhang, X.Y.; Jiang, T.; Chow, L.M.; Wan, S.B. Design and syntheses of permethyl ningalin B analogues: Potent multidrug resistance (MDR) reversal agents of cancer cells. J. Med. Chem. 2010, 53, 5108–5120. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wong, I.L.; Li, F.X.; Yang, C.; Liu, Z.; Jiang, T.; Jiang, T.F.; Chow, L.M.; Wan, S.B. Optimization of permethyl ningalin B analogs as P-glycoprotein inhibitors. Bioorg. Med. Chem. 2015, 23, 5566–5573. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.M.; Liang, Y.J.; Zhang, X.; Su, X.D.; Dai, C.L.; Wang, F.P.; Yan, Y.Y.; Tao, L.Y.; Fu, L.W. Reversal of P-gp-mediated multidrug resistance by bromotetrandrine in vivo is associated with enhanced accumulation of chemotherapeutical drug in tumor tissue. Anticancer Res. 2009, 29, 4597–4604. [Google Scholar] [PubMed]
- Sun, H.; Liu, X.D.; Liu, Q.; Wang, F.P.; Bao, X.Q.; Zhang, D. Reversal of P-glycoprotein-mediated multidrug resistance by the novel tetrandrine derivative w6. J. Asian Nat. Prod. Res. 2015, 17, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.Y.; Wang, R.M.; Lou, L.L.; Li, W.; Tang, G.H.; Bu, X.Z.; Yin, S. Jatrophane diterpenoids as modulators of P-glycoprotein-dependent multidrug resistance (MDR): Advances of structure-activity relationships and discovery of promising MDR reversal agents. J. Med. Chem. 2016, 59, 6353–6369. [Google Scholar] [CrossRef] [PubMed]
- Wong, I.L.K.; Wang, B.C.; Yuan, J.; Duan, L.X.; Liu, Z.; Liu, T.; Li, X.M.; Hu, X.S.; Zhang, X.Y.; Jiang, T.; et al. Potent and nontoxic chemosensitizer of P-glycoprotein-mediated multidrug resistance in cancer: Synthesis and evaluation of methylated epigallocatechin, gallocatechin, and dihydromyricetin derivatives. J. Med. Chem. 2015, 58, 4529–4549. [Google Scholar] [CrossRef] [PubMed]
- Wong, I.L.K.; Chan, K.F.; Tsang, K.H.; Lam, C.Y.; Zhao, Y.; Chan, T.H.; Chow, L.M.C. Modulation of multidrug resistance protein 1 (MRP1/ABCC1)-mediated multidrug resistance by bivalent apigenin homodimers and their derivatives. J. Med. Chem. 2009, 52, 5311–5322. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.W.; Zhang, Y.M.; Liang, Y.J.; Yang, X.P.; Pan, Q.C. The multidrug resistance of tumour cells was reversed by tetrandrine in vitro and in xenografts derived from human breast adenocarcinoma MCF-7/adr cells. Eur. J. Cancer 2002, 38, 418–426. [Google Scholar] [CrossRef]
- Wei, N.; Sun, H.; Wang, F.; Liu, G. H1, a novel derivative of tetrandrine reverse P-glycoprotein-mediated multidrug resistance by inhibiting transport function and expression of P-glycoprotein. Cancer Chemother. Pharmacol. 2011, 67, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Martinez, F.; Lu, P.; Cortes-Selva, F.; Perez-Victoria, J.M.; Jimenez, I.A.; Ravelo, A.G.; Sharom, F.J.; Gamarro, F.; Castanys, S. Celastraceae sesquiterpenes as a new class of modulators that bind specifically to human P-glycoprotein and reverse cellular multidrug resistance. Cancer Res. 2004, 64, 7130–7138. [Google Scholar] [CrossRef] [PubMed]
- Spivey, A.C.; Weston, M.; Woodhead, S. Celastraceae sesquiterpenoids: Biological activity and synthesis. Chem. Soc. Rev. 2002, 31, 43–59. [Google Scholar] [CrossRef] [PubMed]
- Szakacs, G.; Hall, M.D.; Gottesman, M.M.; Boumendjel, A.; Kachadourian, R.; Day, B.J.; Baubichon-Cortay, H.; Di Pietro, A. Targeting the achilles heel of multidrug-resistant cancer by exploiting the fitness cost of resistance. Chem. Rev. 2014, 114, 5753–5774. [Google Scholar] [CrossRef] [PubMed]
- Callies, O.; Sanchez-Canete, M.P.; Gamarro, F.; Jimenez, I.A.; Castanys, S.; Bazzocchi, I.L. Optimization by molecular fine tuning of dihydro-beta-agarofuran sesquiterpenoids as reversers of P-glycoprotein-mediated multidrug resistance. J. Med. Chem. 2016, 59, 1880–1890. [Google Scholar] [CrossRef] [PubMed]
- Barile, E.; Borriello, M.; Di Pietro, A.; Doreau, A.; Fattorusso, C.; Fattorusso, E.; Lanzotti, V. Discovery of a new series of jatrophane and lathyrane diterpenes as potent and specific P-glycoprotein modulators. Org. Biomol. Chem. 2008, 6, 1756–1762. [Google Scholar] [CrossRef] [PubMed]
- Reis, M.; Ferreira, R.J.; Santos, M.M.M.; dos Santos, D.J.V.A.; Molnar, J.; Ferreira, M.J.U. Enhancing macrocyclic diterpenes as multidrug-resistance reversers: Structure-activity studies on jolkinol D derivatives. J. Med. Chem. 2013, 56, 748–760. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, X.; Li, T.; Xu, J.; Ma, Y. A myrsinol diterpene isolated from a traditional herbal medicine, langdu reverses multidrug resistance in breast cancer cells. J. Ethnopharmacol. 2016, 194, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Teodori, E.; Dei, S.; Bartolucci, G.; Perrone, M.G.; Manetti, D.; Romanelli, M.N.; Contino, M.; Colabufo, N.A. Structure-activity relationship studies on 6,7-Dimethoxy-2-phenethyl-1,2,3,4-tetrahydroisoquinoline derivatives as multidrug resistance reversers. ChemMedChem 2017, 12, 1369–1379. [Google Scholar] [CrossRef] [PubMed]
- Bisi, A.; Cappadone, C.; Rampa, A.; Farruggia, G.; Sargenti, A.; Belluti, F.; Di Martino, R.M.C.; Malucelli, E.; Meluzzi, A.; Iotti, S.; et al. Coumarin derivatives as potential antitumor agents: Growth inhibition, apoptosis induction and multidrug resistance reverting activity. Eur. J. Med. Chem. 2017, 127, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Baggetto, L.G.; Dong, M.Q.; Bernaud, J.; Espinosa, L.; Rigal, D.; Bonvallet, R.; Marthinet, E. In vitro and in vivo reversal of cancer cell multidrug resistance by the semi-synthetic antibiotic tiamulin. Biochem. Pharmacol. 1998, 56, 1219–1228. [Google Scholar] [CrossRef]
- Vaclavikova, R.; Bourmendjel, A.; Ehrlichova, M.; Kovar, J.; Gut, I. Modulation of paclitaxel transport by flavonoid derivatives in human breast cancer cells. Is there a correlation between binding affinity to NBD of P-gp and modulation of transport? Bioorg. Med. Chem. 2006, 14, 4519–4525. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Rhee, Y.H.; Jeong, S.J.; Lee, H.J.; Lee, H.J.; Jung, M.H.; Kim, S.H.; Lee, E.O.; Ahn, K.S.; Ahn, K.S.; et al. Hydrocinchonine, cinchonine, and quinidine potentiate paclitaxel-induced cytotoxicity and apoptosis via multidrug resistance reversal in MES-SA/DX5 uterine sarcoma cells. Environ. Toxicol. 2011, 26, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Wong, I.L.K.; Jiang, T.; Wang, S.W.; Liu, T.; Wen, B.J.; Chow, L.M.C.; Sheng, B.W. Synthesis of methylated quercetin derivatives and their reversal activities on P-gp- and BCRP-mediated multidrug resistance tumour cells. Eur. J. Med. Chem. 2012, 54, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.; Toth, N.; Vanyolos, A.; Beni, Z.; Zupko, I.; Molnar, J.; Bathori, M.; Hunyadi, A. Significant activity of ecdysteroids on the resistance to doxorubicin in mammalian cancer cells expressing the human ABCB1 transporter. J. Med. Chem. 2012, 55, 5034–5043. [Google Scholar] [CrossRef] [PubMed]
- Han, H.K.; Van Anh, L.T. Modulation of P-glycoprotein expression by honokiol, magnolol and 4-O-methylhonokiol, the bioactive components of Magnolia officinalis. Anticancer Res. 2012, 32, 4445–4452. [Google Scholar] [PubMed]
- Podszun, M.C.; Jakobi, M.; Birringer, M.; Weiss, J.; Frank, J. The long chain alpha-tocopherol metabolite alpha-13’-cooh and gamma-tocotrienol induce P-glycoprotein expression and activity by activation of the pregnane X receptor in the intestinal cell line LS 180. Mol. Nutr. Food Res. 2017, 61, 1600605. [Google Scholar] [CrossRef] [PubMed]
- Henrich, C.J.; Bokesch, H.R.; Dean, M.; Bates, S.E.; Robey, R.W.; Goncharova, E.I.; Wilson, J.A.; McMahon, J.B. A high-throughput cell-based assay for inhibitors of ABCG2 activity. J. Biomol. Screen. 2006, 11, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.C.; Xiao, X.; Zhang, Y.K.; Talele, T.T.; Salim, A.A.; Chen, Z.S.; Capon, R.J. Lamellarin O, a pyrrole alkaloid from an australian marine sponge, ianthella sp., reverses BCRP mediated drug resistance in cancer cells. Mar. Drugs 2014, 12, 3818–3837. [Google Scholar] [CrossRef] [PubMed]
- Krapf, M.K.; Gallus, J.; Wiese, M. Synthesis and biological investigation of 2,4-substituted quinazolines as highly potent inhibitors of breast cancer resistance protein (ABCG2). Eur. J. Med. Chem. 2017, 139, 587–611. [Google Scholar] [CrossRef] [PubMed]
- Doyle, L.; Ross, D.D. Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2). Oncogene 2003, 22, 7340–7358. [Google Scholar] [CrossRef] [PubMed]
- Mao, Q.C.; Unadkat, J.D. Role of the breast cancer resistance protein (BCRP/ABCG2) in drug transport—An update. AAPS J. 2015, 17, 65–82. [Google Scholar] [CrossRef] [PubMed]
- Zander, S.A.; Sol, W.; Greenberger, L.; Zhang, Y.; van Tellingen, O.; Jonkers, J.; Borst, P.; Rottenberg, S. Ezn-2208 (PEG-SN38) overcomes ABCG2-mediated topotecan resistance in BRCA1-deficient mouse mammary tumors. PLoS ONE 2012, 7, e45248. [Google Scholar] [CrossRef] [PubMed]
- Nicolle, E.; Boccard, J.; Guilet, D.; Dijoux-Franca, M.G.; Zelefac, F.; Macalou, S.; Grosselin, J.; Schmidt, J.; Carrupt, P.A.; Di Pietro, A.; et al. Breast cancer resistance protein (BCRP/ABCG2): New inhibitors and QSAR studies by a 3D linear solvation energy approach. Eur. J. Pharm. Sci. 2009, 38, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Sim, H.M.; Wu, C.P.; Ambudkar, S.V.; Go, M.L. In vitro and in vivo modulation of ABCG2 by functionalized aurones and structurally related analogs. Biochem. Pharmacol. 2011, 82, 1562–1571. [Google Scholar] [CrossRef] [PubMed]
- Kraege, S.; Stefan, K.; Juvale, K.; Ross, T.; Willmes, T.; Wiese, M. The combination of quinazoline and chalcone moieties leads to novel potent heterodimeric modulators of breast cancer resistance protein (BCRP/ABCG2). Eur. J. Med. Chem. 2016, 117, 212–229. [Google Scholar] [CrossRef] [PubMed]
- Danko, B.; Toth, S.; Martins, A.; Vagvolgyi, M.; Kusz, N.; Molnar, J.; Chang, F.R.; Wu, Y.C.; Szakacs, G.; Hunyadi, A. Synthesis and sar study of anticancer protoflavone derivatives: Investigation of cytotoxicity and interaction with ABCB1 and ABCG2 multidrug efflux transporters. ChemMedChem 2017, 12, 850–859. [Google Scholar] [CrossRef] [PubMed]
- Talukdar, S.; Emdad, L.; Das, S.K.; Sarkar, D.; Fisher, P.B. Evolving strategies for therapeutically targeting cancer stem cells. Adv. Cancer Res. 2016, 131, 159–191. [Google Scholar] [PubMed]
- Dhillon, N.; Aggarwal, B.B.; Newman, R.A.; Wolff, R.A.; Kunnumakkara, A.B.; Abbruzzese, J.L.; Ng, C.S.; Badmaev, V.; Kurzrock, R. Phase II trial of curcumin in patients with advanced pancreatic cancer. Clin. Cancer Res. 2008, 14, 4491–4499. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.L.; Huang, T.S.; Lin, J.K. Curcumin, an antioxidant and anti-tumor promoter, induces apoptosis in human leukemia cells. Biochim. Biophys. Acta 1996, 1317, 95–100. [Google Scholar] [CrossRef]
- Chearwae, W.; Anuchapreeda, S.; Nandigama, K.; Ambudkar, S.V.; Limtrakul, P. Biochemical mechanism of modulation of human P-glycoprotein (ABCB1) by curcumin I, II, and III purified from turmeric powder. Biochem. Pharmacol. 2004, 68, 2043–2052. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Shen, Z.L.; Zhai, S.; Xu, J.L.; Liang, H.; Shen, Q.; Li, Q.Y. Transport of curcumin derivatives in Caco-2 cell monolayers. Eur. J. Pharm. Biopharm. 2017, 117, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Prehm, P. Curcumin analogue identified as hyaluronan export inhibitor by virtual docking to the ABC transporter MRP5. Food Chem. Toxicol. 2013, 62, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Wahlstrom, B.; Blennow, G. A study on the fate of curcumin in the rat. Acta Pharmacol. Toxicol. 1978, 43, 86–92. [Google Scholar] [CrossRef]
- Ravindranath, V.; Chandrasekhara, N. Absorption and tissue distribution of curcumin in rats. Toxicology 1980, 16, 259–265. [Google Scholar] [CrossRef]
- Sharma, R.A.; McLelland, H.R.; Hill, K.A.; Ireson, C.R.; Euden, S.A.; Manson, M.M.; Pirmohamed, M.; Marnett, L.J.; Gescher, A.J.; Steward, W.P. Pharmacodynamic and pharmacokinetic study of oral curcuma extract in patients with colorectal cancer. Clin. Cancer Res. 2001, 7, 1894–1900. [Google Scholar] [PubMed]
- Kudo, C.; Yamakoshi, H.; Sato, A.; Nanjo, H.; Ohori, H.; Ishioka, C.; Iwabuchi, Y.; Shibata, H. Synthesis of 86 species of 1,5-diaryl-3-oxo-1,4-pentadienes analogs of curcumin can yield a good lead in vivo. BMC Pharmacol. 2011, 11, 4. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.P.C. Multidrug resistance protein 1 (MRP1, ABCC1), a “multitasking” ATP-binding cassette (ABC) transporter. J. Biol. Chem. 2014, 289, 30880–30888. [Google Scholar] [CrossRef] [PubMed]
- Martin-Broto, J.; Gutierrez, A.M.; Ramos, R.F.; Lopez-Guerrero, J.A.; Ferrari, S.; Stacchiotti, S.; Picci, P.; Calabuig, S.; Collini, P.; Gambarotti, M.; et al. Mrp1 overexpression determines poor prognosis in prospectively treated patients with localized high-risk soft tissue sarcoma of limbs and trunk wall: An isg/geis study. Mol. Cancer Ther. 2014, 13, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Evers, R.; Zaman, G.J.; van Deemter, L.; Jansen, H.; Calafat, J.; Oomen, L.C.; Oude Elferink, R.P.; Borst, P.; Schinkel, A.H. Basolateral localization and export activity of the human multidrug resistance-associated protein in polarized pig kidney cells. J. Clin. Investig. 1996, 97, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.P.; Deeley, R.G. Transport of glutathione and glutathione conjugates by MRP1. Trends Pharmacol. Sci. 2006, 27, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Lania-Pietrzak, B.; Michalak, K.; Hendrich, A.B.; Mosiadz, D.; Grynkiewicz, G.; Motohashi, N.; Shirataki, Y. Modulation of MRP1 protein transport by plant, and synthetically modified flavonoids. Life Sci. 2005, 77, 1879–1891. [Google Scholar] [CrossRef] [PubMed]
- Paszel-Jaworska, A.; Rubis, B.; Bednarczyk-Cwynar, B.; Zaprutko, L.; Rybczynska, M. Proapoptotic activity and ABCC1-related multidrug resistance reduction ability of semisynthetic oleanolic acid derivatives dioxol and himoxol in human acute promyelocytic leukemia cells. Chem. Biol. Interact. 2015, 242, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Durmus, S.; Sparidans, R.W.; Wagenaar, E.; Beijnen, J.H.; Schinkel, A.H. Oral availability and brain penetration of the B-RAFV600E inhibitor vemurafenib can be enhanced by the P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) inhibitor elacridar. Mol. Pharm. 2012, 9, 3236–3245. [Google Scholar] [CrossRef] [PubMed]
- Glavinas, H.; Krajcsi, P.; Cserepes, J.; Sarkadi, B. The role of ABC transporters in drug resistance, metabolism and toxicity. Curr. Drug Deliv. 2004, 1, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Litman, T.; Druley, T.E.; Stein, W.D.; Bates, S.E. From MDR to MXR: New understanding of multidrug resistance systems, their properties and clinical significance. Cell. Mol. Life Sci. 2001, 58, 931–959. [Google Scholar] [CrossRef] [PubMed]
- Qadir, M.; O’Loughlin, K.L.; Fricke, S.M.; Williamson, N.A.; Greco, W.R.; Minderman, H.; Baer, M.R. Cyclosporin a is a broad-spectrum multidrug resistance modulator. Clin. Cancer Res. 2005, 11, 2320–2326. [Google Scholar] [CrossRef] [PubMed]
- Robey, R.W.; Massey, P.R.; Amiri-Kordestani, L.; Bates, S.E. ABC transporters: Unvalidated therapeutic targets in cancer and the CNS. Anticancer Agents Med. Chem. 2010, 10, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Harpstrite, S.E.; Gu, H.; Natarajan, R.; Sharma, V. Interrogation of multidrug resistance (MDR1) P-glycoprotein (ABCB1) expression in human pancreatic carcinoma cells: Correlation of 99mTc-sestamibi uptake with western blot analysis. Nucl. Med. Commun. 2014, 35, 1067–1070. [Google Scholar] [CrossRef] [PubMed]
- Stefan, K.; Schmitt, S.M.; Wiese, M. 9-deazapurines as broad-spectrum inhibitors of the ABC transport proteins P-glycoprotein, multidrug resistance-associated protein 1, and breast cancer resistance protein. J. Med. Chem. 2017, 60, 8758–8780. [Google Scholar] [CrossRef] [PubMed]
- Myint, K.; Li, Y.; Paxton, J.; McKeage, M. Multidrug resistance-associated protein 2 (MRP2) mediated transport of oxaliplatin-derived platinum in membrane vesicles. PLoS ONE 2015, 10, e0130727. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; van der Linden, M.; de Haas, M.; Scheffer, G.L.; de Vree, J.M.; Smith, A.J.; Jansen, G.; Peters, G.J.; Ponne, N.; Scheper, R.J.; et al. Mrp3, an organic anion transporter able to transport anti-cancer drugs. Proc. Natl. Acad. Sci. USA 1999, 96, 6914–6919. [Google Scholar] [CrossRef] [PubMed]
- Norris, M.D.; Smith, J.; Tanabe, K.; Tobin, P.; Flemming, C.; Scheffer, G.L.; Wielinga, L.; Cohn, S.L.; London, W.B.; Marshall, G.M.; et al. Expression of multidrug transporter MRP4/ABCC4 is a marker of poor prognosis in neuroblastoma and confers resistance to irinotecan in vitro. Mol. Cancer Ther. 2005, 4, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Pratt, S.; Shepard, R.L.; Kandasamy, R.A.; Johnston, P.A.; Perry, W., 3rd; Dantzig, A.H. The multidrug resistance protein 5 (ABCC5) confers resistance to 5-fluorouracil and transports its monophosphorylated metabolites. Mol. Cancer Ther. 2005, 4, 855–863. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Compound | Target | Cytotoxicity (µM) | |||
|---|---|---|---|---|---|---|
| Ting-Chao Chou [49] | N3 | P-gp | CCRF-CEM: 13 | CCRF-CEM/VBL1000: 100 | ||
| Bin [50] | Compound 35 | P-gp | L292: >100 | MDA435/LCC6: >100 | MDA435/LCC6MDR: >100 | |
| Yang [45] | Compound 23 | P-gp | L292: >100 | MDA435/LCC6: >100 | MDA435/LCC6MDR: >100 | |
| Wang [51] | Compound 12 | P-gp | L292: >100 | MDA435/LCC6: >100 | MDA435/LCC6MDR: >100 | |
| Chen [53] | 5 Bromo-tetrandrine | P-gp | KB: 5.14 | KBv200: 6.17 | ||
| Sun [54] | W6 | P-gp | KB: ~2 | KBv200: ~4 | MCF-7: ~4.5 | MCF-7/DOX: ~5 |
| Zhu [55] | Compound 26 | P-gp | HepG2/ADR: >150 | MCF-7/ADR: >150 | ||
| Kraege [29] | GO-Y078 | BCRP | K562/BCRP: 0.31 | |||
| Wong [56] | Compound 51 | MRP1, P-gp and BCRP | L292: >100 | LCC6: >100 | LCC6MDR: >100 | |
| Wong [56] | 4e | MRP1 | L292: >100 | LCC6: >100 | LCC6MDR: >100 | |
| Compound | Target | Cellular Accumulation in MDR Cells (Relative-Fold) | MDR Reversal in MDR Cells (Relative-Fold) | |||||
|---|---|---|---|---|---|---|---|---|
| N3 [49] | P-gp | Vinblastine: 440 | ||||||
| Compound 35 [50] | P-gp | Doxorubicin 1 µM: 2.2 | Paclitaxel: 42.7 | |||||
| Compound 23 [45] | P-gp | Doxorubicin 1 µM: 2.4 | Paclitaxel: 48.0 | |||||
| Compound 12 [51] | P-gp | Doxorubicin 1 µM: 2.6 | Paclitaxel: 39.8 | |||||
| 5 Bromo-tetrandrine [53] | P-gp | Doxorubicin 1.5 µM: ~1.2 | Doxorubicin: 15.6 | Vincristine: 109.4 | Paclitaxel: 78.4 | Docetaxel: 57.8 | Epirubicin: 25.1 | |
| W6 [54] | P-gp | Doxorubicin KBv200 1 µM: 4 | Doxorubicin MCF-7/DOX 1 µM: 5.3 | KBv200 Doxorubicin: 27.8 MCF-7/DOX Doxorubicin: 30.3 | KBv200 Vincristine: 29.2 MCF-7/DOX Vincristine: 64.5 | KBv200 Paclitaxel:1049.6 MCF-7/DOX Paclitaxel: 99.3 | ||
| compound 26 [55] | P-gp | Rhodamine 123 HepG2/ADR 2 µM: 2.74 | HepG2/ADR 100 nM Doxorubicin: 71 | MCF-7/ADR 200 nM Doxorubicin: 36 | ||||
| GO-Y078 [29] | BCRP | Pheophorbide A 1 µM: >3 | SN-38: 1.18 | |||||
| compound 51 [56] | MRP1, P-gp and BCRP | Doxorubicin 2008/MRP1 1 µM: 2.6 | Doxorubicin HEK293/R2 (BCRP expressing) 1 µM: 10.4 | Paclitaxel: 31.4 | ||||
| 4e [56] | MRP1 | Doxorubicin 2008/MRP1 1 µM: 8.9 | ||||||
| 4e [57] | MRP1 | Doxorubicin 3 μM: 2.1 | 0.5 µM Doxorubicin: 13.7 | 0.5 µM Etoposide: 10.2 | ||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dantzic, D.; Noel, P.; Merien, F.; Liu, D.-X.; Lu, J.; Han, H.; McKeage, M.J.; Li, Y. The Effects of Synthetically Modified Natural Compounds on ABC Transporters. Pharmaceutics 2018, 10, 127. https://doi.org/10.3390/pharmaceutics10030127
Dantzic D, Noel P, Merien F, Liu D-X, Lu J, Han H, McKeage MJ, Li Y. The Effects of Synthetically Modified Natural Compounds on ABC Transporters. Pharmaceutics. 2018; 10(3):127. https://doi.org/10.3390/pharmaceutics10030127
Chicago/Turabian StyleDantzic, Daniel, Pawan Noel, Fabrice Merien, Dong-Xu Liu, Jun Lu, Haiyong Han, Mark J. McKeage, and Yan Li. 2018. "The Effects of Synthetically Modified Natural Compounds on ABC Transporters" Pharmaceutics 10, no. 3: 127. https://doi.org/10.3390/pharmaceutics10030127
APA StyleDantzic, D., Noel, P., Merien, F., Liu, D.-X., Lu, J., Han, H., McKeage, M. J., & Li, Y. (2018). The Effects of Synthetically Modified Natural Compounds on ABC Transporters. Pharmaceutics, 10(3), 127. https://doi.org/10.3390/pharmaceutics10030127